

Abstract
We previously demonstrated that tumor suppressor protein p53 augments plasminogen activator inhibitor-1 (PAI-1) expression in alveolar epithelial cells (AECs) during chronic cigarette smoke (CS) exposure-induced lung injury. Chronic lung inflammation with elevated p53 and PAI-1 expression in AECs and increased susceptibility to and exacerbation of respiratory infections are all associated with chronic obstructive pulmonary disease (COPD). We recently demonstrated that preventing p53 from binding to the endogenous PAI-1 mRNA in AECs by either suppressing p53 expression or blockading p53 interactions with the PAI-1 mRNA mitigates apoptosis and lung injury. Within this context, we now show increased expression of the C-X-C chemokines (CXCL1 and CXCL2) and their receptor CXCR2, and the intercellular cellular adhesion molecule-1 (ICAM-1), in the lung tissues of patients with COPD. We also found a similar increase in lung tissues and AECs from wild-type (WT) mice exposed to passive CS for 20 wk and in primary AECs treated with CS extract in vitro. Interestingly, passive CS exposure of mice lacking either p53 or PAI-1 expression resisted an increase in CXCL1, CXCL2, CXCR2, and ICAM-1. Furthermore, inhibition of p53-mediated induction of PAI-1 expression by treatment of WT mice exposed to passive CS with caveolin-1 scaffolding domain peptide reduced CXCL1, CXCL2, and CXCR2 levels and lung inflammation. Our study reveals that p53-mediated induction of PAI-1 expression due to chronic CS exposure exacerbates lung inflammation through elaboration of CXCL1, CXCL2, and CXCR2. We further provide evidence that targeting this pathway mitigates lung injury associated with chronic CS exposure.
Keywords: p53, PAI-1, COPD, passive cigarette smoke and lung inflammation
chronic obstructive pulmonary disease (COPD) affects up to 24 million people in the United States and remains the third leading cause of morbidity and mortality (21). Long-term cigarette smoke (CS), either due to active smoking or passive exposure, is a major risk factor for development of COPD (12). The chronic inflammation of the small airways and the lung parenchyma, leading to fixed narrowing of small airways and alveolar wall destruction (emphysema), typifies the pathogenesis of COPD (27). The pathogenic processes such as inflammation, alterations of cell growth, cellular apoptosis, abnormal cell repair, extracellular matrix destruction, and oxidative stress all contribute to development of COPD (11). The exaggerated accumulation of various inflammatory cells due to increased recruitment and reduced apoptosis and clearance of these cells leads to chronic inflammation in the lungs of patients with COPD (4). CS exposure triggers lung-resident macrophages and epithelial cells to produce chemokines leading to pulmonary infiltration of inflammatory immune cells (36). The activated immune cells release proteases leading to alveolar wall destruction and mucus hypersecretion (3, 11). Therefore, elucidation of the underlying mechanism(s) contributing to excess elaboration of chemokines will be valuable in regulating lung inflammation associated with exposure to CS.
Elevated levels of C-X-C and C-C chemokines have been detected in the bronchoalveolar lavage (BAL) fluids of COPD patients (39). Similarly, human bronchial airway cells exposed to CS release CXCL1 (also known as keratinocyte-derived chemokine) and CXCL2 (also known as macrophage inflammatory protein-2) (29). Chemokine production by alveolar epithelial cells (AECs) can be regulated by CS and neutrophil elastase (45). Epithelial cells specifically perpetuate neutrophil influx into the conducting airways of smokers via CXCL1 release (45). AECs exposed to CS shows increased expression of various chemokines (24), which are likely contribute to influx of inflammatory cells in the lungs.
Plasminogen activator inhibitor-1 (PAI-1) is a serine protease inhibitor that irreversibly inhibits urokinase-type plasminogen activator (uPA)-mediated plasminogen activation and promotes turnover of PAI-1-uPA-uPAR complexes (17, 20, 43). Furthermore, defective alveolar fibrinolysis due to a disproportionate increase in the expression of PAI-1 is common in lung diseases associated with chronic CS exposure such as COPD (8, 35). In our recent studies, AECs exposed to CS show augmented p53 and PAI-1 expression and that lung inflammation and apoptosis of AECs are facilitated by p53-mediated induction of PAI-1 expression. Previously it has been reported that PAI-1 activity is positively correlated with the level of CXCL1 (31). These observations suggest that p53-mediated induction of PAI-1 expression plays a pivotal role in the infiltration of immune cells and lung inflammation via regulating secretion of various chemokines in response to CS. However, the involvement of increased expression of PAI-1 induced by p53 in the control of chemokine secretion, immune cell infiltration, and lung inflammation in response to chronic exposure to CS has not been previously scrutinized.
In the present study, we show that exposure to CS augments CXCL1, CXCL2, and CXCR2, which is offset by a reduction in either p53 or PAI-1 expression. Furthermore, our study demonstrates that blockade of passive CS exposure induced p53 and PAI-1 expression in AECs by use of caveolin-1 scaffolding domain peptide (CSP) attenuates CXCL1, CXCL2, and CXCR2 expression. These changes were associated with a reduction in lung inflammation, evidenced by significant suppression in pulmonary myeloperoxidase (MPO) activity and intercellular adhesion molecule-1 (ICAM-1) expression. Our findings collectively for the first time demonstrate an intricate link between p53-mediated induction of PAI-1 expression and lung inflammation due to exacerbated chemokine expression. This study further suggests that targeting this pathway by using CSP may be a plausible means to treat inflammatory lung diseases associated with chronic exposure to CS.
MATERIALS AND METHODS
Human lung tissues.
Lung tissues from patients with a clinical diagnosis of COPD and control subjects without clinical diagnosis of COPD or histological evidence of emphysema or chronic bronchitis were obtained from Lung Tissue Research Consortium and from The Department of Pathology; University of Texas Health Science Center at Tyler (UTHSCT). The use of deidentified donor lung tissue samples from the patients with COPD or control subjects in the study were approved by the ethics committee of the UTHSCT under exempt protocol as we described elsewhere (23).
Isolation of AECs from mouse lungs.
All experiments involving mice were performed according to approved protocols under the guidelines of Animal Care and Use Committee of the UTHSCT. AECs were isolated from C57BL/6 mice following the method of Corti and colleagues (14) with minor modifications. The cells were plated on plastic culture dishes precoated with anti-CD-32 and anti-CD-45 antibodies for 2 h at 37°C. Nonadherent cells were collected, and the purity of AEC preparations was assessed by lithium carbonate staining for inclusion bodies.
Preparation of CSE for in vitro experiments.
Research cigarettes 2R4F were purchased from the Tobacco Health Research University of Kentucky (Lexington, KY). CS extracts were prepared by burning research cigarettes in a sidearm flask and the smoke generated was bubbled into phosphate-buffered saline at room temperature through an attached peristaltic pump as we described earlier (8). An absorbance of 1.0 at 230 nm is considered 100%. CS extract was filter sterilized by passing it through a 0.2-μm filter.
Passive CS exposure of mice.
Wild-type (WT) and p53- and PAI-1-deficient mice of C57BL/6 background were bred in our facilities or were purchased from Jackson Laboratories. These mice were exposed to passive CS from 40 research cigarettes over a 2-h period 5 days/wk for 20 wk (∼90 mg/m3 total solid particulates) by using a mechanical smoking chamber (Teague Enterprises, Davis, CA). Control mice were exposed to ambient air. Four weeks after initiation of passive CS exposure, the mice were administered an intraperitoneal injection of CSP or scrambled control peptide (CP) (18.75 mg/kg body wt) once a week for 4 wk. Exposure to passive CS was continued for another 12 wk as we described earlier (23). Mice were killed, and their lungs were used for further analyses (8).
Measurement of MPO activity.
Mouse lung MPO activity was determined as described previously (8). Briefly, lungs were suspended in 1 ml buffer (0.5% hexadecyltrimethyl ammonium bromide in 50 mM phosphate buffer, pH 6.0) and sonicated at 30 cycles twice for 30 s on ice. Homogenates were centrifuged at 12,000 rpm and MPO activity in supernatants measured by colorimetric method using o-dianisidine dihydrochloride and H2O2 in a 96-well plate. MPO activity levels in lung samples were determined from a standard curve generated with known amounts of purified MPO (Sigma, St. Louis, MO).
Immunoblotting.
Changes in CXCL1, CXCL2, CXCR2, ICAM-1, and PAI-1 expression levels were assessed by Western blotting of lung tissues homogenates or lysates of isolated AECs using human and mouse specific antibodies and enhanced chemiluminescence (Thermo, Rockford, IL) detection as described previously (23).
IHC analysis.
For immunohistochemical (IHC) analyses, paraffin-embedded lung tissues sections (5 μm) were deparaffinized with xylene and rehydrated by incubation with 100% and 95% alcohol. After 30 min of antigen retrieval with 10 mM sodium citrate buffer (pH 6.0), the sections were incubated with hydrogen peroxide for 30 min to quench endogenous peroxidase. The slides were incubated overnight with rabbit anti-mouse CXCL1, CXCL2, CXCR2, and ICAM-1 antibodies (Abcam, Cambridge, MA) in PBS containing 0.1% Tween 20, and further processed with the Lab Vision kit (Fremont, CA). The slides were later washed, mounted, and subjected to microscopic (BX 41; Olympus, Santa Valley, PA) examination as described (7). Two investigators independently obtained IHC scores (H-scores) following the method described earlier (26, 44). Briefly, the percentage of total lung cells with different staining intensities was visually scored as 1+ (for light staining), 2+ (for intermediate staining), and 3+ (for dark staining) with IHC ImageJ profiler. The H-score was calculated by integrating data related to the intensity of staining with the formula 1 × [percentage of cells staining weakly (1+)] + 2 × [percentage of cells staining moderately (2+)] + 3 × [percentage of cells staining strongly (3+)].
Immunofluorescence confocal microscopy.
Paraffin-embedded lung sections (5 μm) were subjected to immunofluorescent staining using combinations of CXCL1 and surfactant protein C (SPC) or CXCL2 and SPC or CXCR2 and SPC antibodies. SPC antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). CXCL1, CXCL2, and CXCR2 antibodies were purchased from Abcam. The sections were later incubated with Alexa Fluor 568-conjugated goat anti-mouse IgG (Invitrogen), Alexa Fluor 647-conjugated donkey anti-rabbit IgG (Invitrogen), or Alexa Fluor 568-conjugated donkey anti-mouse IgG secondary antibodies (Invitrogen). Stained slides were mounted with Prolong Gold (Invitrogen) and images were acquired on a Leica SP8 confocal microscope (Leica Microsystems). Image-capture parameters and minimum and maximum pixel intensity values were held constant for all samples.
Analysis of chemokines, CXCR2, and ICAM-1 mRNA expression.
Total RNA was isolated from the lung tissues of patients with COPD or control subjects or from the lungs of mice exposed to passive CS or kept in ambient air, or AECs treated with PBS or CS extracts by using TRI Reagent. The levels of CXCL1, CXCL2, CXCR2, ICAM-1, and β-actin mRNAs were quantitated with an aliquot of reverse transcribed total RNA and specific primers (Table 1) by real-time PCR as described earlier (33, 35).
Table 1.
List of primer sequences
| Primers | Sequence (5′>3′) |
|---|---|
| Human CXCL1 Forward | AACCGAAGTCATAGCCACAC |
| Human CXCL1 Reverse | GTTGGATTTGTCACTGTTCAGC |
| Human CXCL2 Forward | CGCCCAAACCGAAGTCATA |
| Human CXCL2 Reverse | TGCTCAAACACATTAGGCGC |
| Human CXCR2 Forward | AACATGGAGAGTGACAGCTTTG |
| Human CXCR2 Reverse | TCACATGGGGCGGCATC |
| Human ICAM-1 Forward | GGCCGGCCAGCTTATACAC |
| Human ICAM-1 Reverse | TAGACACTTGAGCTCGGGCA |
| Mouse CXCL1 Forward | TGTTTCCTGCCTCTGAAGC |
| Mouse CXCL1 Reverse | CTTCGTTTGTGATCCTCCG |
| Mouse CXCL2 Forward | GGCGTCACACTCAAGCTCT |
| Mouse CXCL2 Reverse | TCTGGCATGCCCTCTATTCTG |
| Mouse CXCR2 Forward | TCTGGCATGCCCTCTATTCTG |
| Mouse CXCR2 Reverse | AAGGTAACCTCCTTCACGTAT |
| Mouse ICAM-1 Forward | TGTTTCCTGCCTCTGAAGC |
| Mouse ICAM-1 Reverse | CTTCGTTTGTGATCCTCCG |
Statistical analysis.
The differences between two and multiple groups under various experimental conditions were analyzed by Student's t-test and one-way ANOVA, respectively. GraphPad 4.0 software was used to analyze the statistical difference.
RESULTS
We have recently reported that the tissues and AECs isolated from the lungs of patients with COPD show increased expression of p53 and PAI-1 expression (8). These changes were associated with a parallel increase in inflammation as evidenced by the presence of elevated levels of neutrophils and macrophages, and MPO activity. To directly test whether increased p53 and p53-mediated downstream induction of PAI-1 contribute to lung inflammation, we evaluated lung tissues and BAL fluids of WT mice exposed to 20 wk of passive CS. The responses were compared mice lacking p53 or PAI-1 expression exposed to CS as well as WT and p53- and PAI-1-deficent mice housed in ambient air. The lung tissues (Fig. 1A) and the BAL fluids (Fig. 1B) of WT mice exposed to chronic CS showed increased MPO activity, indicating inflammation. However, mice lacking either p53 or the p53 downstream target PAI-1 expression and exposed to CS failed to induce MPO activity. The resistance of p53- and PAI-1-deficient mice to CS-induced AEC injury (8) and lung inflammation suggests a crucial link between p53-mediated induction of PAI-1 expression due to chronic CS exposure and lung inflammation. To elucidate the underlying mechanism and establish pathological relevance, we initially subjected COPD lung sections to IHC to assess the expression of C-X-C chemokine (CXCL1 and CXCL2). As shown in Fig. 1C, both CXCL1 and CXCL2 antigen levels were markedly increased in lungs of patients with COPD. IHC analysis revealed that the CXCL1 and CXCL2 receptor, CXCR2 antigen expression, was also increased in COPD lung tissues along with augmented ICAM-1 expression, indicating lung inflammation. Analysis of total RNA from COPD lung tissues further demonstrated that CXCL1, CXCL2, CXCR2, and ICAM-1 mRNAs were significantly increased compared with their corresponding levels in control lung tissues (Fig. 1D).
Fig. 1.
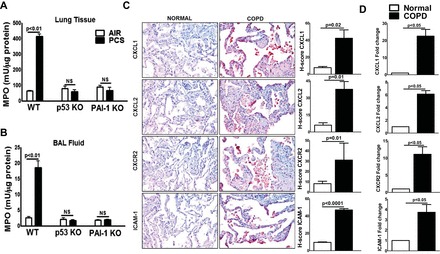
Induction of p53 and plasminogen activator inhibitor-1 (PAI-1) contributes to lung inflammation in mice exposed to passive cigarette smoke (CS) and patients with chronic obstructive pulmonary disease (COPD). Lung tissues (A) and bronchoalveolar lavage (BAL) fluids (B) collected from wild-type (WT) and p53- and PAI-1-deficient (KO) mice (n = 5/group) exposed to ambient air (AIR) or passive CS (PCS) for 20 wk were analyzed for myeloperoxidase (MPO) activity. MPO activity quantification is expressed as mean ± SD of 3 independent analyses. C: lung sections from patients with COPD and histologically “normal” donor subjects (n = 5) were subjected to immunohistochemical (IHC) analysis using CXCL1, CXCL2, CXCR2, and ICAM-1 antibodies. Images are representative of IHC staining pattern of 10 fields (×200 magnification) and graphs show IHC scores (H-scores). D: total RNA isolated from the lung tissues of patients with COPD or normal subjects (n = 5) were analyzed for CXCL1, CXCL2, CXCR2, and ICAM-1 mRNAs by real-time PCR. Bar represents mean ± SD of 3 independent analyses. NS, not significant.
Because COPD lung tissues showed increased levels of CXCL1 and CXCL2 antigen and mRNA, we analyzed the lung homogenates of WT and p53- and PAI-1-deficient mice exposed to CS for CXCL1 and CXCL2 expression. The responses were compared with control mice maintained in ambient air. Consistent with CXCL1 and CXCL2 expression in COPD lung tissues, immunoblotting (Fig. 2A) of whole lung homogenates of WT mice exposed to 20 wk of CS showed increased CXCL1 and CXCL2 expression. Interestingly, those deficient in p53 or PAI-1 exposed to chronic CS failed to induce CXCL1 or CXCL2 expression (Fig. 2A). This was further confirmed at the mRNA level by quantitative real-time PCR of total RNA extracted from whole lung tissues (Fig. 2B). IHC analysis of lung sections of WT mice exposed to 20 wk of passive CS also showed elevated levels of CXCL1 and CXCL2, which were significantly reduced in p53- and PAI-1-deficient mice (Fig. 2C).
Fig. 2.
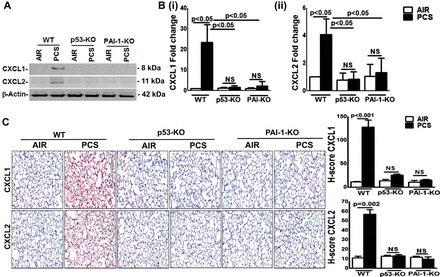
p53 and PAI-1 are prominently linked to PCS-induced CXCL1 and CXCL2 expression. A: lung homogenates from WT and p53- and PAI-1-deficient mice (n = 5/group) in ambient air or 20 wk of PCS were immunoblotted for CXCL1 and CXCL2. The same membranes were later stripped and analyzed for β-actin to assess comparable loading. Representative image from triplicate analyses is showed. B: total RNA isolated from the lungs of mice (n = 5/group) in air or exposed to PCS were analyzed for CXCL1 (i) and CXCL2 (ii) mRNA by real-time PCR and presented as fold change after normalization with the corresponding level of β-actin mRNA in the same sample. Bar represents mean ± SD of 3 independent analyses. C: lung sections of WT and p53- and PAI-1-deficient mice (n = 5) in air or exposed to PCS were subjected to IHC analysis to assess CXCL1 and CXCL2 antigens in situ. Images are representative of IHC staining pattern of 10 fields (×200 magnification) and graphs show H-scores.
We next analyzed whether CXCR2 expression is similarly induced in mice exposed to chronic CS. As showed in Fig. 3A, CS exposure increased the expression of CXCR2 proteins in the lungs of WT mice, whereas both p53- and PAI-1-deficient mice failed to respond and their responses were similar to mice kept in ambient air. Furthermore, analysis for CXCR2 mRNA confirmed the requirement of p53 and downstream PAI-1 expression (Fig. 3B). The IHC of lung sections also showed an increased CXCR2 antigen expression only in WT mice exposed to 20 wk of CS exposure (Fig. 3C).
Fig. 3.
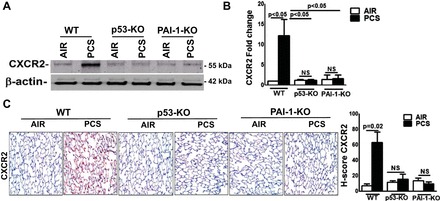
p53 and PAI-1 augment pulmonary CXCR2 expression in mice exposed to PCS. WT and p53- and PAI-1-deficient mice (n = 5/group) were exposed to ambient air or PCS for 20 wk. A: lung homogenates (n = 5/group) were immunoblotted for CXCR2 by use of anti-CXCR2 antibody. Representative blot from triplicate analyses is showed. B: total RNA from lung tissues (n = 5/group) was analyzed for CXCR2 mRNA by real-time PCR. Bar represents mean ± SD of 3 independent analyses. C: lung sections of the mice (n = 5/group) exposed to air or PCS were subjected to IHC analysis for CXCR2 antigens. Images are representative of IHC staining pattern of 10 fields (×200 magnification) and graph shows the H-scores.
Chronic CS exposure causes lung inflammation through a number of mechanisms including ICAM-1-mediated leukocyte adhesion to epithelial and endothelial cells and activation of macrophages and neutrophils (19). Therefore, we subjected lung homogenates to Western blotting to assess changes in ICAM-1 expression. As showed in Fig. 4A, exposure of WT mice to CS significantly increased ICAM-1 protein expression, which was resisted by both p53- and PAI-1-deficient mice. This was independently confirmed by measuring ICAM-1 mRNA by real-time PCR (Fig. 4B). The IHC analysis of lung sections (Fig. 4C) likewise revealed that increased expression of p53 and PAI-1 contributes to lung inflammation through increased level of ICAM-1 and chemokines by recruited inflammatory cells or activated AECs following to CS exposure.
Fig. 4.
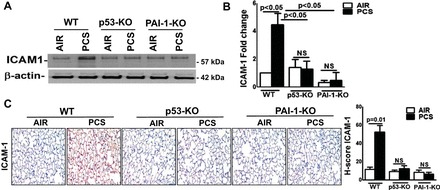
p53 and PAI-1 are prominently linked to PCS exposure-induced ICAM-1 expression. WT and p53- and PAI-1-deficient mice (n = 5/group) were kept in ambient air or exposed to PCS for 20 wk. A: lung homogenates prepared from these mice (n = 5) were immunoblotted for ICAM-1 expression. Representative image from triplicate analyses is showed. B: total RNA from the lung tissues of air or PCS mice (n = 5) were analyzed for ICAM-1 and β-actin mRNA. Bar graph represents mean ± SD of triplicate analyses. C: lung sections of air or PCS mice (n = 5) were subjected to IHC analysis for ICAM-1. Images are representative of IHC staining pattern of 10 fields (×200 magnification) and graph shows the H-scores.
CS augments p53 and PAI-1 expression in AECs in vitro and in vivo. p53- and PAI-1-deficient mice exposed to CS resist CXCL1, CXCL2, and CXCR2 expression or lung inflammation. These are otherwise increased in WT mice, indicating that CS-induced p53 and PAI-1 expression contributes to lung inflammation due to recruitment of inflammatory cells and through elaboration of CXCL1, CXCL2, and CXCR2. We previously reported that CSP treatment mitigates CS exposure-induced AEC injury both in vitro and in vivo and the process involves inhibition of p53 and p53-mediated downstream induction of PAI-1 expression in these cells. Therefore, we next treated WT mouse AECs exposed to CS extract with CSP and analyzed the lysates for CXCL1, CXCL2, and CXCR2 as well as ICAM-1 to assess changes in inflammation. Immunoblotting for CXCL1 and CXCL2 in the AEC lysates and conditioned media, or CXCR2 and ICAM-1 in the AEC lysates (Fig. 5A), showed that CS induces their expression in AECs in vitro. Further inhibition of CS induced p53 and downstream PAI-1 by treatment of AECs with CSP whereas treatment with the CP had no effect. Analysis of CXCL1, CXCL2, CXCR2, and ICAM-1 mRNAs also confirmed that CSP markedly reduced their expression in vitro (Fig. 5B). p53 induces PAI-1 expression through stabilization of PAI-1 mRNA transcript by binding to a 70 nucleotide 3′-untranslated region (3′UTR) determinant (23, 33). Inhibition of CS-induced p53 using CSP may affect multiple targets. To directly confirm that p53-mediated induction of PAI-1 expression contributes to increased lung inflammation, we transduced AECs with adenovirus expressing the p53-binding PAI-1 3′UTR sequence and later treated with CS extract as described earlier (7, 8, 35). The responses of competitive inhibition of CS-induced p53 from binding to endogenous PAI-1 mRNA were compared with naive control AECs or AECs exposed to adenovirus expressing nonbinding control sequences. As shown in Fig. 5C, the transduction of AECs with p53-binding PAI-1 mRNA 3′UTR sequences reduced CS-induced CXCL1 and CXCL2 expression, whereas control AECs exposed to CS extract still showed elevated levels of CXCL1 and CXCL2 in the culture media. Consistent with our earlier reports, we also found inhibition of PAI-1 expression, which was otherwise increased after treatment with CS extract in control AECs. Inhibition of p53-mediated induction of PAI-1 by treatment with CSP or PAI-1 3′UTR, however, failed to suppress CS-induced CCL2 (Fig. 5, A and C), indicating the specificity of inhibition. To directly confirm that CS-induced PAI-1 contributes to increased inflammation, we treated AECs from PAI-1-deficient mice with CS extract in vitro and analyzed for changes in CXCL1, CXCL2, CXCR2, and ICAM-1. We found that CS extract failed to induce CXCL1, CXCL2, CXCR2, and ICAM-1 in AECs lacking PAI-1 expression, whereas WT AECs responded as usual (Fig. 5D). To further support the idea that CS induces lung inflammation, we performed immunofluorescence analysis of lung sections. The lung section of WT-mice exposed to PCS showed marked increase in CXCL1-, CXCL2-, or CXCR2-positive cells, which express SPC (Fig. 5E). The result further reinforce that passive CS induces expression of CXCL1, CXCL2, or CXCR2 in AECs.
Fig. 5.
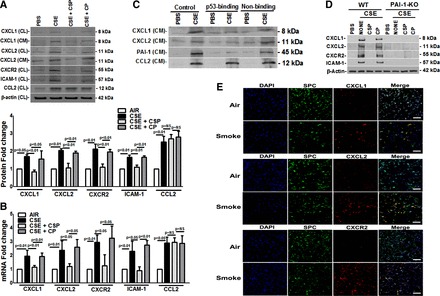
Inhibition of CS-induced CXCL1, CXCL2, CXCR2, and ICAM-1 by blocking p53-mediated induction of PAI-1 expression in alveolar epithelial cells (AECs). A: AECs isolated from WT mice were treated with PBS or 1.5% of CS extract (CSE; OD 1.00 at 260 nm = 100%) in PBS with or without 10 μM of caveolin-1 scaffolding domain peptide (CSP) or control peptide (CP) for 24 h in AEC culture media without antibiotics. The conditioned medium (CM) was analyzed for CXCL1 and CXCL2 and the AEC lysates (CL) were tested for CXCL1, CXCL2, CXCR2, CCL2, ICAM-1, and β-actin expression by Western blotting. Bar graph represents fold change in the densities of the CXCL1, CXCL2, CXCR2, and ICAM-1 bands in the CL after normalization with β-actin controls from 3 independent analyses. B: total RNA isolated from mouse AECs exposed to PBS or CSE (1.5%) or CSE+CSP or CSE+CP for 24 h were tested for PAI-1 and β-actin mRNA expression by real-time PCR. The results (mean ± SD) of triplicate analyses are presented as bar graph. C: AECs isolated from WT mice were treated with PBS or CSE or CSE with adenovirus constructs expressing p53-binding sequence to competitively block CS-induced p53 from binding endogenous PAI-1 mRNA. AECs infected with adenovirus construct harboring nonbinding PAI-1 3′-untranslated region (3′UTR) sequences were used as a control as described in materials and methods. Twenty-four hours later, the CM were analyzed for CXCL1, CXCL2, CCL2, and PAI-1 expression by Western blotting. Experiments were repeated at least 3 times, and representative results are illustrated. D: AECs isolated from WT and PAI-1-deficient mice were treated with PBS or 1.5% of CSE in PBS with or without CSP or CP as described in A. The AEC lysates were tested for CXCL1, CXCL2, CXCR2, ICAM-1, and β-actin expression by Western blotting. Representative image from triplicate analyses is showed. E: lung sections from WT mice (n = 5/group) kept in ambient air or exposed to PCS for 20 wk were treated with anti-CXCL1 and anti-surfactant protein C (SPC) or anti-CXCL2 and anti-SPC or anti-CXCR2 and anti-SPC antibodies, developed with fluorescent-labeled secondary antibodies and examined by confocal microscopy. Images are representative of immunofluorescence staining pattern of 10 fields (bars represent 50 μm). Green, red, and blue signals represent SPC, CXCL, and nucleus, respectively. Note colocalization of SPC and CXCL1 or CXCL2, respectively, in PCS samples in merge images, which are shown yellow.
Treatment of WT mice exposed to chronic CS but treated with CSP mitigates lung injury and the process involves inhibition of p53 and downstream PAI-1 expression in AECs in vivo (6). We therefore analyzed the lung homogenates of WT mice exposed to 20 wk of passive CS and treated with CSP for CXCL1 expression. Western blotting of lung homogenates revealed that CS exposure-induced CXCL1 and CXCL2 expressions were markedly suppressed in WT mice treated with CSP (Fig. 6A). However, mice exposed to CP failed to attenuate passive CS exposure-induced CXCL1 or CXCL2 expression. Consistent with the changes in CXCL1 and CXCL2 protein levels, a significant reduction in both CXCL1 (Fig. 6Bi) and CXCL2 (Fig. 6Bii) mRNA expression was found in WT mice exposed to CS and treated with CSP. This was further substantiated by the finding of IHC analysis of lung sections of WT mice exposed to 20 wk of passive CS and CSP (Fig. 6C).
Fig. 6.
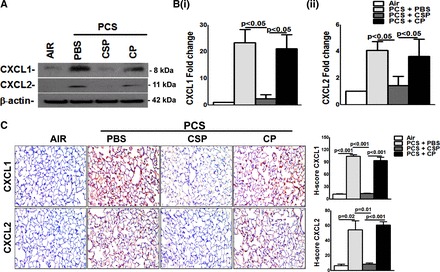
Inhibition of PCS exposure induced pulmonary CXCL1 and CXCL2 expression by CSP. WT mice were exposed to ambient air or PCS (n = 5/group) as described in materials and methods for 5 days/wk. After 4 wk of PCS exposure, mice exposed to PCS were intraperitoneally injected with or without 18.75 mg/kg body wt of CSP or CP once every week for 4 more wk. After 20 wk of PCS exposure, mice were euthanized. A: lung homogenates from these mice (n = 5/group) were immunoblotted for CXCL1 and CXCL2 expression. Representative blot from triplicate analyses is showed. B: total RNA from the lungs of these mice (n = 5/group) was tested for changes in the expression of CXCL1 (i) and CXCL2 (ii) and β-actin mRNA. Individual bars represent mean ± SD of triplicate analyses. C: lung sections from WT mice (n = 5/group) were subjected to IHC analysis by using anti-CXCL1 and CXCL2 antibody. Images are representative of IHC staining pattern of 10 fields (×200 magnification) and graph shows the H-scores.
Further analysis for CXCR2 protein (Fig. 7A) and mRNA (Fig. 7B) expression, by Western blotting of lung homogenates or real-time PCR of total RNA, revealed that CSP attenuates passive CS exposure-induced CXCR2 expression. Analysis of lung sections (Fig. 7C) likewise confirmed suppression of CXCR2 antigen levels in WT mice treated with CSP, which was otherwise induced following 20 wk of passive CS exposure.
Fig. 7.
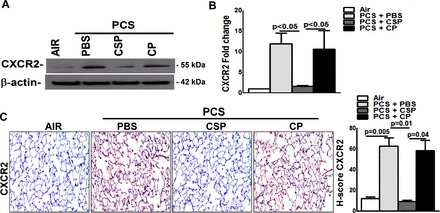
CSP inhibits PCS exposure induced pulmonary CXCR2 expression. WT mice were exposed to ambient air or PCS (n = 5/group) as described above. After 4 wk of PCS exposure, mice exposed to PCS were intraperitoneally injected with or without 18.75 mg/kg body wt of CSP or CP once every week for 4 more wk. After 20 wk of PCS exposure the mice were euthanized. A: lung homogenates (n = 5/group) were immunoblotted for CXCR2 and β-actin for loading equality. Representative image from triplicate analyses is showed. B: total RNA obtained from mice (n = 5/group) lung were analyzed for CXCR2 mRNA by RT-PCR as previously described. Bar graph represents mean ± SD of 3 independent analyses. C: lung sections from all groups of mice (n = 5/group) were subjected to IHC analysis using anti-CXCR2 antibody. Images are representative of IHC staining pattern of 10 fields (×200 magnification) and bar graph shows H-scores.
We next tested for deviations in ICAM-1 expression to assess changes in lung inflammation. Results of Western blotting (Fig. 8A) and real-time PCR (Fig. 8B) showed inhibition of both ICAM-1 protein and mRNA expression after treatment with CSP. This was also further verified by IHC analysis of lung sections (Fig. 8C). Thus we affirm that p53-mediated induction of PAI-1 expression due to chronic CS exposure may lead to lung inflammation through increased production of inflammatory chemokines, consistent with other classical inflammatory regulation. Further targeting this pathway by CSP suppresses lung inflammation, which was otherwise exacerbated by chronic CS exposure.
Fig. 8.
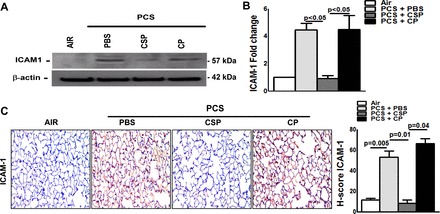
CSP-mediated inhibition of p53 and PAI-1 expression attenuates passive CS exposure-induced ICAM-1 in mouse lungs. WT mice were exposed to ambient air or PCS (n = 5/group) for 5 days/wk. After 4 wk of PCS exposure, mice exposed to PCS were intraperitoneally injected with or without 18.75 mg/kg body wt of CSP or CP once every week for 4 more wk. After 20 wk of PCS exposure, mice were euthanized. A: lung homogenates from mice (n = 5/group) were immunoblotted for ICAM-1 and β-actin protein. Representative image from triplicate analyses is showed. B: total RNA obtained from mice (n = 5/group) lung was analyzed for ICAM-1 mRNA by real-time PCR. Bar represents mean ± SD of 3 independent analyses. C: lung sections from all groups of mice (n = 5/group) were subjected to IHC analysis using anti-ICAM-1 antibody. Images are representative of IHC staining pattern of 10 fields (×200 magnification) and bar graph shows H-scores.
DISCUSSION
Chronic CS exposure is a major risk factor lung diseases such as COPD and cancer, cardiovascular diseases, and atherosclerosis, to name a few. Lung diseases associated with chronic CS exposure alone are estimated to cause 2 million or more deaths worldwide on an annual basis (25). Inflammation and proteolysis play a pivotal role in the initiation and progression of lung diseases associated with chronic CS exposure including COPD (28). At present, there is no effective treatment that actually alleviates lung inflammation or reverses lung injury caused by exposure to CS. This is mainly due to a lack of in-depth understanding of the mechanisms that protect or conversely that contribute to the deleterious effects of CS. Therefore, elucidation of the underlying mechanisms that contribute innate and inflammatory responses may identify new targets and may lead to effective means to arrest exacerbation lung inflammation or reverse lung disorders associated with chronic CS exposure such as COPD.
PAI-1 levels are markedly elevated during local and systemic inflammation (41). PAI-1, besides being a key inhibitor of fibrinolysis, has also been implicated in inflammatory processes in various organs. We previously reported that p53 induces PAI-1 expression in injured lung epithelial cells (34, 35). p53 and PAI-1 as well as inflammatory mediators are also expressed by macrophages, neutrophils, and other recruited cells and will likely contribute lung inflammation during lung injury, including CS exposure injury. However, in the present study, we show that p53-mediated induction of PAI-1 expression occurs in AECs after chronic exposure to CS. These changes appear to associate with parallel increase in lung inflammation through production of chemokines. Furthermore, targeting this pathway by using CSP or the p53-binding PAI-1 mRNA 3′UTR sequences, which inhibit CS-induced p53 from interacting with PAI-1 mRNA, suppressed expression of PAI-1 with parallel reduction in CXC chemokines, CXCR2, and lung inflammation. These responses appear to correlate with the effects of p53 or PAI-1 deficiency in terms of lung inflammation after long exposure to CS. The mechanism by which CSP inhibits CS exposure-induced p53 and downstream PAI-1 in AECs is not clear. We earlier found that CS induces Src activation and activated Src in turn inactivates protein phosphatase 2A by phosphorylation of tyrosine residue 307 in the catalytic subunit. PP2A inactivates ATM kinase, leading to suppression of serine 15 phosphorylation of p53 protein and subsequent degradation of p53 by mdm2-mediated ubiquitination. We found that CSP inactivates Src activation by inhibitory phosphorylation leading to suppression of serine 15 phosphorylated and total p53, and downstream PAI-1 expression (23). CXCR2 ligands CXCL1 and CXCL2 have been shown significantly elevated in BAL fluids of mice exposed CS and serve as chemokines (10). Acrolin is effective in inducing IL-8 releases in lung cells and acrolin level is higher in side stream than mainstream CS (42). Similarly, increased expression of CXCR2 mRNA has been previously reported in the lungs of mice exposed to CS, and CXCR2 is required for neutrophilic inflammation (46), indicating an intricate link between chronic CS exposure induced p53 and PAI-1 expression and lung inflammation. This is further supported by increased expression of ICAM-1 by AECs following exposure CS.
Proteases, particularly elastases, from both neutrophils and macrophages have been implicated in tissue destruction associated with chronic CS exposure including COPD. Alpha1 anti-trypsin deficiency heightens the risks of COPD (37), whereas genetic polymorphism in the MMP-12 (macrophage elastase) promoter region has been shown to reduce the risk of COPD (2). MMP-12 degrades alpha1 anti-trypsin, suggesting the importance of proteases produced by the inflammatory cells in development of COPD (13, 32). Besides chronic inflammation, COPD is frequently associated with loss of both matrix and cellular elements of the lung, leading to impaired gas exchange between the alveolar space and the capillary blood, a feature implicated in the development of emphysema (22). In recent years, excessive apoptosis and senescence of AECs have been recognized as major mechanisms contributing to the development of emphysema (40). Since lung inflammation has been implicated in both AEC apoptosis and senescence in response to chronic CS exposure (16), the identification of molecular mediators that trigger lung responses such as inflammation offers the potential for development of novel interventions. Previous studies indicated that CS extract induces CXCL1 induction through mRNA stabilization and the process is regulated through the TLR4/MyD88 pathway in bronchial epithelial cells (38). We found that mice deficient in p53 or PAI-1 expression resist chronic CS-induced CXCL1 and CXCL2 and their receptor CXCR2 expression and these mice also resist induction of ICAM-1 expression.
The relative contribution of AECs in chronic CS exposure-induced pulmonary accumulation of inflammatory cells and lung inflammation is difficult to discern. However, increased expression of CXCL1, CXCL2, and their receptor CXCR2 by AECs exposed to CS extract in vitro suggests that they play a pivotal role at least in part in attracting inflammatory cells. Inactivation of neutrophils and macrophages resulting from CSP-mediated suppression of p53 and PAI-1 expression could contribute to the anti-inflammatory responses. Furthermore, inhibition of CXCL1 attenuates neutrophil accumulation in the lungs after intratracheal administration of LPS (15), indicating the importance of CXCL1 in lung inflammation. Recent reports suggest that Th17 cell differentiation and interleukin-17A (IL-17A) production are mediated by CXCL1 in sepsis-induced lung injury (1). IL-17A was significantly elevated in the peripheral lung tissues of patients with severe to very severe COPD (GOLD stage III/IV) (30), as well as in mice exposed to chronic CS (47). Increased CXCL2 expression may be mediated by elevated local CXCL1 expression as reported earlier (9). Our results provide a strong correlation in terms of CXCL1/CXCL2 and ICAM-1 expression and we know that ICAM-1 plays a major role in lung inflammation through recruit of neutrophils (5, 18). Earlier studies revealed the requirement of neutrophil elastase in neutrophil and monocytes recruitment and for the activation of MMP-12. Our findings set the stage for future investigations to explore whether targeting p53-mediated PAI-1 expression via CSP can be used effectively to treat lung diseases such as COPD, in which chronic inflammation plays a principal role.
GRANTS
This study was supported in part by a Flight Attendant Medical Research Institute Clinical Innovator Award (FAMRI-ID-123010) and by the American Heart Association (15GRNT25800004).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.T., A.S.M., and Y.T. performed experiments; N.T., A.S.M., and Y.T. analyzed data; N.T., M.I., and S.S. interpreted results of experiments; N.T. and A.S.M. prepared figures; N.T. and S.S. drafted manuscript; N.T., M.I., J.F., and S.S. edited and revised manuscript; N.T., A.S.M., J.F., and S.S. approved final version of manuscript; S.S. conception and design of research.
REFERENCES
- 1.Aujla SJ, Dubin PJ, Kolls JK. Th17 cells and mucosal host defense. Semin Immunol 19: 377–382, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes PJ. Molecular genetics of chronic obstructive pulmonary disease. Thorax 54: 245–252, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD 1: 59–70, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 22: 672–688, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Basit A, Reutershan J, Morris MA, Solga M, Rose CE, Ley K. ICAM-1 and LFA-1 play critical roles in LPS-induced neutrophil recruitment into the alveolar space. Am J Physiol Lung Cell Mol Physiol 291: L200–L207, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Bhandary YP, Shetty SK, Marudamuthu AS, Gyetko MR, Idell S, Gharaee-Kermani M, Shetty RS, Starcher BC, Shetty S. Regulation of alveolar epithelial cell apoptosis and pulmonary fibrosis by coordinate expression of components of the fibrinolytic system. Am J Physiol Lung Cell Mol Physiol 302: L463–L473, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhandary YP, Shetty SK, Marudamuthu AS, Ji HL, Neuenschwander PF, Boggaram V, Morris GF, Fu J, Idell S, Shetty S. Regulation of lung injury and fibrosis by p53-mediated changes in urokinase and plasminogen activator inhibitor-1. Am J Pathol 183: 131–143, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhandary YP, Shetty SK, Marudamuthu AS, Midde KK, Ji HL, Shams H, Subramaniam R, Fu J, Idell S, Shetty S. Plasminogen activator inhibitor-1 in cigarette smoke exposure and influenza A virus infection-induced lung injury. PloS One 10: e0123187, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai S, Batra S, Lira SA, Kolls JK, Jeyaseelan S. CXCL1 regulates pulmonary host defense to klebsiella infection via CXCL2, CXCL5, NF-κB, and MAPKs. J Immunol 185: 6214–6225, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther 121: 55–68, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J 31: 1334–1356, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol 294: L612–L631, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-α release. Am J Respir Crit Care Med 167: 1083–1089, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Corti M, Brody AR, Harrison JH. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 14: 309–315, 1996. [DOI] [PubMed] [Google Scholar]
- 15.Gong Q, Xu JF, Yin H, Liu SF, Duan LH, Bian ZL. Protective effect of antagonist of high-mobility group box 1 on lipopolysaccharide-induced acute lung injury in mice. Scand J Immunol 69: 29–35, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol 81: 289–296, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Juhan-Vague I, Moerman B, De Cock F, Aillaud MF, Collen D. Plasma levels of a specific inhibitor of tissue-type plasminogen activator (and urokinase) in normal and pathological conditions. Thromb Res 33: 523–530, 1984. [DOI] [PubMed] [Google Scholar]
- 18.Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol 24: 25–29, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Krüger K, Dischereit G, Seimetz M, Wilhelm J, Weissmann N, Mooren FC. Time course of cigarette smoke-induced changes of systemic inflammation and muscle structure. Am J Physiol Lung Cell Mol Physiol 309: L119–L128, 2015. [DOI] [PubMed] [Google Scholar]
- 20.Kruithof EK, Gudinchet A, Bachmann F. Plasminogen activator inhibitor 1 and plasminogen activator inhibitor 2 in various disease states. Thromb Haemost 59: 7–12, 1988. [PubMed] [Google Scholar]
- 21.Mannino DM. COPD: epidemiology, prevalence, morbidity and mortality, and disease heterogeneity. Chest 121, Suppl 5: 121S–126S, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet 370: 765–773, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Marudamuthu AS, Bhandary YP, Shetty SK, Fu J, Sathish V, Prakash Y, Shetty S. Role of the urokinase-fibrinolytic system in epithelial-mesenchymal transition during lung injury. Am J Pathol 185: 55–68, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moodie FM, Marwick JA, Anderson CS, Szulakowski P, Biswas SK, Bauter MR, Kilty I, Rahman I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-κB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J 18: 1897–1899, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 364: 613–620, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Pirker R, Pereira JR, von Pawel J, Krzakowski M, Ramlau R, Park K, de Marinis F, Eberhardt WE, Paz-Ares L, Störkel S, Schumacher KM, von Heydebreck A, Celik I, O'Byrne KJ. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol 13: 33–42, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 154: 1055–1060, 1996. [DOI] [PubMed] [Google Scholar]
- 28.Rangasamy T, Misra V, Zhen L, Tankersley CG, Tuder RM, Biswal S. Cigarette smoke-induced emphysema in A/J mice is associated with pulmonary oxidative stress, apoptosis of lung cells, and global alterations in gene expression. Am J Physiol Lung Cell Mol Physiol 296: L888–L900, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, Ley K. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest 116: 695, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roos A, Bjermer L, Stampfli M, Erjefält J. Elevated IL-17A in very severe COPD is localized to mast cells and correlates with lung function decline. Eur Respir J 44: 1722, 2014. [Google Scholar]
- 31.Rothenbacher D, Müller-Scholze S, Herder C, Koenig W, Kolb H. Differential expression of chemokines, risk of stable coronary heart disease, and correlation with established cardiovascular risk markers. Arterioscler Thromb Vasc Biol 26: 194–199, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Shapiro SD, Ingenito EP. The pathogenesis of chronic obstructive pulmonary disease: advances in the past 100 years. Am J Respir Cell Mol Biol 32: 367–372, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Shetty SK, Bhandary YP, Marudamuthu AS, Abernathy D, Velusamy T, Starcher B, Shetty S. Regulation of airway and alveolar epithelial cell apoptosis by p53-induced plasminogen activator inhibitor-1 during cigarette smoke exposure injury. Am J Respir Cell Mol Biol 47: 474–483, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shetty S, Padijnayayveetil J, Tucker T, Stankowska D, Idell S. The fibrinolytic system and the regulation of lung epithelial cell proteolysis, signaling, and cellular viability. Am J Physiol Lung Cell Mol Physiol 295: L967–L975, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Shetty S, Shetty P, Idell S, Velusamy T, Bhandary YP, Shetty RS. Regulation of plasminogen activator inhibitor-1 expression by tumor suppressor protein p53. J Biol Chem 283: 19570–19580, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevenson CS, Coote K, Webster R, Johnston H, Atherton HC, Nicholls A, Giddings J, Sugar R, Jackson A, Press NJ, Brown Z, Butler K, Danahay H. Characterization of cigarette smoke-induced inflammatory and mucus hypersecretory changes in rat lung and the role of CXCR2 ligands in mediating this effect. Am J Physiol Lung Cell Mol Physiol 288: L514–L522, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Stoller JK, Fromer L, Brantly M, Stocks J, Strange C. Primary care diagnosis of alpha-1 antitrypsin deficiency: issues and opportunities. Cleve Clin J Med 74: 869, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Thorley AJ, Tetley TD. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2: 409–428, 2007. [PMC free article] [PubMed] [Google Scholar]
- 39.Traves SL, Culpitt SV, Russell REK, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROα and MCP-1 in sputum samples from patients with COPD. Thorax 57: 590–595, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tuder RM, Yoshida T, Arap W, Pasqualini R, Petrache I. State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc 3: 503–510, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ueda S, Nishio K, Minamino N, Kubo A, Akai Y, Kangawa K, Matsuo H, Fujimura Y, Yoshioka A, Masui K, Doi N, Murao Y, Miyamoto S. Increased plasma levels of adrenomedullin in patients with systemic inflammatory response syndrome. Am J Respir Crit Care Med 160: 132–136, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Vardavas CI, Panagiotakos DB. The causal relationship between passive smoking and inflammation on the development of cardiovascular disease: a review of the evidence. Inflamm Allergy Drug Targets 8: 328–333, 2009. [DOI] [PubMed] [Google Scholar]
- 43.Vassalli JD, Sappino AP, Belin D. The plasminogen activator/plasmin system. J Clin Invest 88: 1067, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venkatasubramanian S, Tripathi D, Tucker T, Paidipally P, Cheekatla S, Welch E, Raghunath A, Jeffers A, Tvinnereim AR, Schechter ME, Andrade BB, Mackman N, Idell S, Vankayalapati R. Tissue factor expression by myeloid cells contributes to protective immune response against Mycobacterium tuberculosis infection. Eur J Immunol 46: 464–479, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Witherden IR, Vanden Bon EJ, Goldstraw P, Ratcliffe C, Pastorino U, Tetley TD. Primary human alveolar type II epithelial cell chemokine release: effects of cigarette smoke and neutrophil elastase. Am J Respir Cell Mol Biol 30: 500–509, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 87: 1047–1082, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X, Zheng H, Zhang H, Ma W, Wang F, Liu C, He S. Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provoked by cigarette smoke. Cytokine 56: 717–725, 2011. [DOI] [PubMed] [Google Scholar]