

Abstract
Earlier work from this laboratory showed that autocrine generation of angiotensin II and c-Jun-NH2-terminal kinase phosphorylation (p-JNK) are both required events in alveolar epithelial cell (AEC) apoptosis. Although earlier data showed that angiotensin-(1–7) [ANG-(1–7)] protects against AEC apoptosis, the pathways by which ANG-(1–7)/mas activation prevent JNK phosphorylation and apoptosis are poorly understood. Therefore, in the current study, it was theorized that ANG-(1–7) activates a mitogen-activated protein kinase phosphatase (MKP) and thereby reduces JNK phosphorylation to inhibit apoptosis and promote cell survival. This hypothesis was evaluated in the human A549 and mouse MLE12 AEC lines and primary cultures of human AECs. Cells were transfected with small-interfering RNAs, antisense oligonucleotides, or inhibitors specific for MKP-2 or mas, and were then assayed for phospho-JNK, caspase-9, loss of mitochondrial membrane potential, and nuclear fragmentation. Silencing of MKP-2 significantly prevented the blockade of all apoptotic markers by ANG-(1–7). Knockdown or blockade of mas receptor by antisense oligonucleotides or by the receptor antagonist A779, respectively, caused significant decreases in MKP-2, and simultaneously increased the apoptotic markers of caspase-9 activation and nuclear fragmentation. These data show that the ANG-(1–7)/mas pathway constitutively prevents JNK phosphorylation and apoptosis of AECs by maintaining activation of the JNK-selective phosphatase MKP-2, and further demonstrate the critical role of the ANG-(1–7) receptor mas in AEC survival.
Keywords: type II pneumocyte, angiotensin peptides, c-Jun-NH2-terminal kinase phosphorylation, mitogen-activated protein kinase phosphatase
it is well established that alveolar epithelial cell (AEC) apoptosis contributes to the pathogenesis of several types of lung disease (20). Therefore, understanding the underlying signaling mechanisms of AEC apoptosis is critical to determine the pathogenesis of lung injury. Blockade of apoptosis by broad-spectrum caspase inhibitors or genetic deletion of apoptotic genes prevented lung injury in animal models (32). In recent years, activation of a local angiotensin system (ANG) in the lung has shown to play a major role in AEC apoptosis and subsequent lung injury (33). Previous work from this laboratory demonstrated that inducers of apoptosis generate angiotensinogen (AGT), the 58-kDa protein that, after enzymatic cleavage generates the effector octapeptide angiotensin II (ANG II; see Refs. 21, 39, 40). Moreover, it was shown that autocrine generation of ANG II is required in AEC apoptosis, through experiments that blocked apoptosis by either antisense oligonucleotides against AGT mRNA, angiotensin type 1 (AT1) receptor antagonists, or by neutralizing antibodies against ANG II itself (34). Subsequent in vitro studies showed that binding of ANG II to its receptor AT1 causes phosphorylation of c-Jun-NH2-terminal kinase (JNK), a member of the mitogen-activated protein kinase (MAPK) family, which is required for AEC apoptosis (36).
Angiotensin-converting enzyme-2 (ACE-2) degrades ANG II by removal of a single amino acid to generate the heptapeptide angiotensin-(1–7) [ANG-(1–7)]. Recent work showed that ACE-2 is protective against experimental lung injury. Lung tissues from both experimental mouse and rat models treated with bleomycin showed significantly reduced levels of ACE-2 mRNA, protein, and enzymatic activity, suggesting that loss of ACE-2 contributes to accumulation of ANG II causing AEC apoptosis and subsequent lung injury (19). Accordingly, in both pulmonary and nonpulmonary systems ANG-(1–7) has shown to counteract detrimental effects of ANG II through the mas receptor (17). Radioligand binding studies have provided evidence that ANG-(1–7) binds to its receptor mas, which is distinct from the AT1 and AT2 receptor subtypes (26).
Experimental studies in this laboratory demonstrated that ANG-(1–7) inhibits ANG II- or bleomycin-induced JNK phosphorylation in AECs (37). Furthermore, ANG-(1–7) also inhibited caspase activation and apoptosis, which were blocked by the mas receptor antagonist A779 {d-Ala7-[ANG-(1–7)]}, which has very low affinity for the AT1 or AT2 receptors. Although the exact downstream signaling mechanisms of the ANG-(1–7)/mas pathway are currently unclear, several groups have shown the activation of a phosphatase in different cell types (3, 11). Mitogen-activated protein kinase phosphatases (MKPs) belong to the family of dual-specificity phosphatases (DUSPs), which are important negative regulators of MKPs through dephosphorylating the Thr-X-Tyr motif of MKPs (7). A recent publication by Uhal et al. showed that at baseline (without stimulation) ANG-(1–7) is more abundant than ANG II in the cell culture media bathing primary AECs and that ANG-(1–7) dephosphorylates p-JNK as a cell survival mechanism (37). Therefore, it was theorized that the ANG-(1–7)/mas pathway activates a JNK-selective mitogen-activated protein kinase phosphatase-2 (MKP-2) to reduce p-JNK levels, thus promoting cell survival. We report here the findings that silencing MKP-2 (DUSP 4) prevents the blockade of JNK phosphorylation and apoptosis (ANG II signaling) by ANG-(1–7) in AECs. Furthermore, we also report that silencing mas decreases MKP-2 and promotes apoptosis in these cells.
MATERIALS AND METHODS
Reagents and materials.
ANG II and ANG-(1–7) were purchased from Sigma-Aldrich (St. Louis, MO). Mas receptor antagonist A779 was purchased from GenScript (Piscataway, NJ). Antibodies for the detection of MKP-2, Mas receptor as well as MKP-2-specific small-interfering RNAs (siRNA) and control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for the detection of p-JNK, active forms of caspase-9, total JNK, and β-actin were all obtained from Cell Signaling Technology (Boston, MA). Antisense oligonucleotides against the mas receptor and the control antisense were obtained from Genemed Synthesis (San Antonio, TX). 3,3′-Dihexyloxacarbocyanine iodide (DiOC6) was obtained from Life Technologies (Carlsbad, CA). All other materials were of reagent grade and were purchased from Sigma Aldrich.
Cell culture.
The human type II epithelial cell-derived cell line (A549) was obtained from American Type Culture Collection (ATCC, Manassas, VA) and was grown in F-12 medium containing 10% serum. Primary human AECs were obtained from ScienCell Research Laboratories (Carlsbad, CA) and were cultured in specific AEC medium formulated by the supplier. The mouse lung epithelial cell line (MLE12), a generous gift from the laboratory of Dr. J. Whitsett, University of Cincinnati, was grown in complete HITES media. All cells were grown in 6-, 12-, or 24-well culture plates and were analyzed at subconfluent densities. The primary cells were treated at day 2 of culture, a stage at which they are type II cell-like by accepted morphological and biochemical criteria (38). All subsequent incubations with ANG-(1–7) and/or A779 (mas receptor antagonist) were performed in serum-free medium. In all studies cells were exposed to ANG-(1–7) (10−7 M) for 40 min and/or A779 (10−7 M) for 30 min before exposure to ANG II (10−7 M) for 5 min to 20 h as indicated. Exposure to siRNA and control siRNA was done before treatment with ANG-(1–7) and ANG II.
Gene knockdown.
Antisense oligonucleotides against human mas were designed using the antisense design tool from Integrated DNA Technologies (IDT, Coralville, IA) and were synthesized as phosphorothioated 20-mers. A549 cells or primary human AECs were transfected with antisense oligonucleotides or control antisense (final concn 0.1 μM) by using Lipofectamine2000 reagent (Life Technologies, Grand Island, NY) at 2 μl/ml as the vehicle dissolved in F-12 media without any serum or antibiotics. After transfections, the cells were incubated at 37°C with 5% CO2 for 6 h followed by addition of normal growth medium with three times the normal serum and antibiotic concentration (3× normal growth medium). At 24 h, the transfection reagents were removed and replaced by complete F-12 media for an additional 24 h. Afterwards, the cells were serum starved overnight, and immediately thereafter ANG-(1–7) (10−7 M) were added for 40 min to 12 h as indicated.
The siRNA against human MKP-2 were commercially synthesized and purchased from Santa Cruz Biotechnologies. The siRNA-to-lipofectine ratio was optimized to yield effective knockdown, which was confirmed by Western blotting. A549s were transfected (final concn 0.1 μM) similarly as described above and were treated with ANG-(1–7) (10−7 M) followed by ANG II (10−7 M). A scrambled siRNA of the same sequence was used as a negative control. The MKP-2 siRNA is a pool of three different siRNA duplexes: 1) sense 5′-GAAGGACACUAUCAGUACAtt-3′ and antisense 5′-UGUACUGAUAGUGUCCUUCtt-3′, 2) sense 5′-GGACUCCGAAUACAUAAUAtt-3′ and antisense 5′-UAUUAUGUAUUCGGAGUCCtt-3′, and 3) sense 5′-CACAGAUCCUAGCAAAUGUtt-3′ and antisense 5′-ACAUUUGCUAGGAUCUGUGtt-3′.
Detection of apoptosis.
Apoptotic cells were detected by nuclear fragmentation assay using propidium iodide (PI) as described earlier (21) after enzymatic digestion of ethanol-fixed cells with DNase-free RNase in PBS containing 5 μg/ml PI. During fixation with 70% ethanol, detached cells were retained by centrifugation of the 24-well culture plates. Cells with discrete nuclear fragments with condensed chromatin were counted as apoptotic using epifluorescence microscopy. Apoptotic cells were scored over a minimum of four separate microscopic fields from each of at least three culture vessels per treatment group. As in earlier publications from this laboratory, the induction of apoptosis is verified by in situ end labeling (ISEL) of fragmented DNA conducted by a modification of the method of Mundle et al. (35). Briefly, the cells were washed with distilled water for 10 min followed by an incubation with 0.23% periodic acid for 10 min. After five washes with 0.15 M PBS, cells were incubated with saline-sodium citrate solution at 80°C for 20 min followed by four washes with 0.5 M PBS and three times with 50 mM Tris·HCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, and 0.005% BSA in water (buffer A). Next, cells were incubated with an ISEL solution (0.001 mM biotin-dUTP, 0.01 mM of each dATP, dCTP, and dGTP, 20 U/ml DNA polymerase I in buffer A) for 2 h at 20°C. Afterwards, cells were washed three times in buffer A and with 0.5 M PBS five times. Next, the cells were incubated with a Vectastain ELITE solution that contained avidin and biotin-peroxidase solution dissolved in 1% BSA and 0.5% Tween 20 in 0.5 M PBS. After 30 min, cells were washed with PBS followed by an incubation with 0.25 mg/ml diaminobenzidine solution in 0.05 M Tris·HCl containing 0.01% H2O2 to detect end labeling. The active forms of caspase-9 were detected by Western blotting using antibodies specific to the cleaved forms.
Estimation of mitochondrial membrane potential.
The mitochondrial membrane potential (MMP) in A549 cells transfected with MKP-2 siRNA as described above was assessed with the lipophilic probe DiOC6. After treatment with ANG II (10−7 M) for 8 h in the presence or absence of ANG-(1–7) (10−7 M), the cells were incubated with PBS containing 500 nM DiOC6 for 15 min at 37°C followed by an assay in a fluorescence plate reader (BioTek, Winooski, VT) at 360 nm excitation and 420 nm emission. To determine the total DNA, cells were fixed with 70% ethanol for 30 min followed by an incubation with 10 μm Hoechst 33342 dye dissolved in PBS for 10 min. Next, the cells were reanalyzed at the same wavelengths for quantitation of total cellular DNA. Data were then normalized.
Western blotting.
Cells were lysed either with a modified lysis buffer for phosphoproteins containing 50 mM HEPES, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 M EGTA, 1.5 mM MgCl2, 100 μM sodium orthovanadate, and the protease inhibitor cocktail (Complete Mini; Roche, Nutley, NJ) or with a Nonidet P-40-based lysis buffer containing protease inhibitors (for MKP-2, mas, and caspase-9). After being harvested, proteins were run on polyacrylamide gels and transferred to polyvinylidene difluoride membranes. Next, the bands were visualized by chemiluminescent substrate West Femto detection systems (Pierce, Rockford, IL).
RESULTS
To investigate the downstream signaling of the mas receptor in type II AECs, antisense oligonucleotides were used to knock down the mas receptor, which significantly silenced the receptor compared with the controls incubated with a random sequence (Fig. 1A). In Fig. 1B, treatment of A549 cells with mas antisense oligonucleotides (final concn 0.1 μM), in the presence of ANG-(1–7) (10−7 M), induced the activated form of caspase-9. Similarly, nuclear fragmentation assessed with PI was significantly increased after the mas receptor was knocked down (Fig. 1C). Figure 2A shows antisense oligonucleotide-mediated knockdown of the mas receptor alone (at baseline, i.e., without additional stimuli) significantly increased p-JNK levels compared with the control that received a random sequence oligonucleotide. In Fig. 2B, caspase-9 (active form) was increased in the cells that were treated with mas antisense oligonucleotides.
Fig. 1.

Mas knockdown induces caspase-9 activation and apoptosis in alveolar epithelial cells (AECs). A: A549 cells were treated with antisense oligonucleotides (0.1 μM final concn) against mas receptor to verify mas knockdown. At 48 h, cells were harvested for Western blotting. B: A549 cells were transfected as in A in the presence of ANG-(1–7) (10−7 M) followed by cell harvesting for Western blotting. Bars are means + SE of n = 4 over two experiments. *P < 0.05 vs. control (CTL) and ***P < 0.001 vs. ANG-(1–7) by ANOVA and Tukey-Kramer multiple-comparison test. C: A549 cells were treated with antisense oligonucleotides for 48 h as described above. Cells were subjected to propidium iodide (PI) assay (see materials and methods) after 12 h treatment with ANG-(1–7) (10−7 M). Bars are means + SE for n ≥ 3 over two experiments. ***P < 0.001 vs. CTL and ANG-(1–7) by ANOVA and Student-Newman-Keuls post hoc analysis.
Fig. 2.
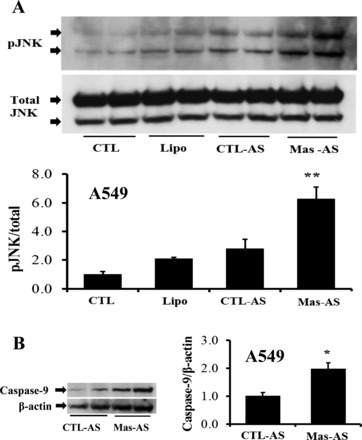
Mas knockdown increases basal c-Jun-NH2-terminal kinase (JNK) phosphorylation and basal caspase-9 activation. A: A549 cells were treated with antisense oligonucleotides as described in Fig. 1A. At 18 h (posttransfection), cells were harvested for Western blotting to detect basal levels of phospho (p)-JNK. Bars are means + SE of n = 4 over two experiments. **P < 0.01 vs. CTL-antisense (AS) by Tukey-Kramer multiple-comparison test. B: the same samples in A were subjected to caspase-9 (active form) detection. Bars are means + SE of n = 4 over two experiments. *P < 0.05 vs. CTL-AS by Mann-Whitney Test.
Figure 3A shows that blocking the action of the ANG-(1–7) produced endogenously by AECs, using the specific mas receptor blocker A779 (at 10−7 M added in the final 30 min of a 12-h serum starvation), induces p-JNK compared with the controls with no added A779. Figure 3, B and C, shows that when A779 (10−7 M) was applied in a similar manner as in Fig. 3A, but for 30 min only, MKP-2 protein was reduced in both mouse (MLE12, Fig. 3B) and human (A549, Fig. 3C) lung epithelial cells.
Fig. 3.

A mas receptor blocker increases basal JNK phosphorylation and decreases basal MKP-2 protein. After 12 h serum starvation, mouse lung epithelial cell line (MLE12) cells were challenged with the mas receptor antagonist A779 (10−7 M) for 30 min [without removing the endogenous ANG-(1–7) that had accumulated in 12 h] to block the ANG-(1–7)/mas pathway. Control cells did not receive A779 but received serum-free media + vehicle. A: Western blotting for phospho- and total JNK (see materials and methods). Bars are means + SE of n = 4 over two experiments. *P < 0.05 vs. CTL by Mann-Whitney test. B: MLE12 cells were treated as in A and were then harvested for Western blotting for detection of mitogen-activated protein kinase phosphatase-2 (MKP-2). Bars are means + SE of n = 4 over two experiments. *P < 0.01 vs. CTL by unpaired t-test. C: A549 cells were treated as described in A and were analyzed for MKP-2 levels as in B. Bars are means + SE of n = 4 over two experiments. *P < 0.05 vs. CTL by unpaired t-test.
In Fig. 4, A and B, A549 or MLE12 cells that received exogenous ANG-(1–7) showed increased MKP-2 protein compared with the controls. The induction of MKP-2 by ANG-(1–7) was blocked by preincubating the cells with the mas receptor antagonist A779 (Fig. 4B). Moreover, antisense-mediated knockdown of mas also prevented the increase in MKP-2 induced by ANG-(1–7) (Fig. 4C), which confirms the involvement of mas on the induction of MKP-2 by ANG-(1–7) (in both MLE12 and A549 cells). As shown in Fig. 4D, treatment with ANG-(1–7) (10−7 M) and/or A779 (10−7 M) does not have any effect on the other JNK-selective dual specific phosphatase MKP-1 and -5.
Fig. 4.
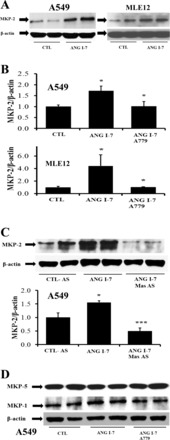
ANG-(1–7) induces MKP-2 in lung epithelial cells through mas but does not influence MKP-5 or -1. A: A549 or MLE12 cells were serum starved for 12 h, after which the media were removed and cells were challenged with freshly prepared ANG-(1–7) (10−7 M) for 40 min before harvesting for Western blotting for MKP-2. The control group did not receive ANG-(1–7) but media were changed similarly. Bars are means + SE of n = 4 over two experiments. *P < 0.05 vs. CTL by Mann-Whitney test. B: A549 or MLE12 cells were preincubated with ANG-(1–7) (10−7 M) for 40 min in the presence or absence of A779 (10−7 M), the mas receptor antagonist, followed by harvesting for Western blotting of MKP-2. Bars are means + SE of at least 6 separate cell cultures over two or three experiments. *P < 0.05 vs. CTL and *P <0.05 vs. ANG-(1–7) by ANOVA and Student-Newman-Keuls post hoc analysis. C: A549 cells were treated with antisense oligonucleotides against mas as in Fig. 1A, in the presence of ANG-(1–7) (10−7 M) for 30 min followed by harvesting. Bars are means + SE of n = 4 over two separate experiments. *P < 0.05 vs. CTL and ***P < 0.001 vs. ANG-(1–7) by ANOVA and Tukey-Kramer multiple-comparison test. D: A549 cells treated as in B were subjected to detection of MKP-5 and -1; no significant changes were found by densitometry.
In Fig. 5A, treatment with siRNA against MKP-2 silenced the MKP-2 protein compared with the negative control with a scrambled sequence (also compared with transfecting reagents only). Figure 5B shows that the MKP-2 silencing is specific and does not interfere with the expression of the two other known JNK-selective phosphatases MKP-3 and MKP-7, which were unaffected. Earlier work showed that JNK phosphorylation is a required event in AEC apoptosis; to determine whether silencing MKP-2 induces JNK phosphorylation, A549 cells were treated with MKP-2 siRNA in the presence or absence of ANG-(1–7) (10−7 M) and/or ANG II (10−7 M) for 40 and 5 min, respectively. Figure 5, C and D, shows that the silencing of MKP-2 prevented ANG-(1–7) blockade of JNK phosphorylation. In a study of basal JNK phosphorylation (Fig. 6), treatment with MKP-2 siRNA alone (without additional stimuli) induced p-JNK significantly (Fig. 6A), and further Fig. 6B shows the induction of basal caspase-9 (active form) when MKP-2 is silenced compared with the control RNA sequence.
Fig. 5.
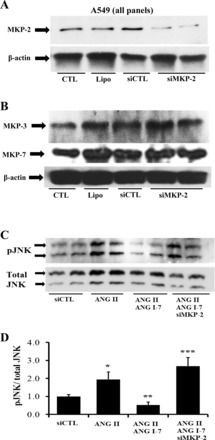
Specific knockdown of MKP-2 by small-interfering RNA (siRNA) prevents inhibition of JNK phosphorylation by ANG-(1–7). A: A549 cells were transfected with either MKP-2 siRNA or a scrambled RNA sequence as described in materials and methods. After 48 h cells were harvested for Western blotting of MKP-2. B: the same samples in A were subjected to Western blotting for MKP-7 and -3. C: A549 cells were transfected with MKP-2 siRNA for 48 h, followed by an incubation with ANG-(1–7) (10−7 M) and ANG II (10−7 M) for an additional 40 min and 5 min, respectively. Next, the cells were harvested for Western blotting for phospho- and total JNK. D: densitometry of C and additional replicate experiments; bars are means ± SE of n ≥ 4 over three separate experiments. *P < 0.05 vs. CTL, **P < 0.05 vs. ANG II, and ***P < 0.01 vs. ANG II/ANG-(1–7) by ANOVA and Student-Newman-Keuls post hoc analysis.
Fig. 6.

Silencing of MKP-2 increases basal JNK phosphorylation and basal caspase-9 activation. A: A549 cells were treated with MKP-2 siRNA or a scrambled sequence as described in Fig. 5A. After 48 h cells were serum starved and incubated for an additional 12 h. Next, cells were harvested for Western blotting to detect basal levels of p-JNK. Bars are means + SE of n ≥ 4 over two experiments. *P < 0.05 vs. siCTL by ANOVA and Student-Newman-Keuls post hoc analysis. B: the same samples in A were subjected for caspase-9 (active form) detection. Bars are means + SE of n ≥ 3 over two experiments. *P < 0.05 vs. siCTL by Mann-Whitney test.
ANG II leads to loss of MMP (8). In Fig. 7A, treatment of A549 cells with the lipophilic dye DiOC6 demonstrates that the loss of MMP induced by ANG II was prevented by ANG-(1–7). Furthermore, the blockade by ANG-(1–7) was inhibited by MKP-2 knockdown with siRNA (densitometry showed that siMKP-2 was not significantly different from ANG II alone). To assess the possibility that DiOC6 measurements might reflect changes in the number of mitochondria per cell rather than changes in MMP, the 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) assay was conducted to measure total mitochondrial function. MTT analyses showed that mitochondrial function per cell is constant across all treatment groups in Fig. 7A (data not shown). Given that knockdown of MKP-2 increased both p-JNK and caspase-9 (Fig. 6), it was of high interest to determine whether silencing MKP-2 would also inhibit the ability of ANG-(1–7) to prevent apoptosis in A549 cells. In Fig. 7, B–D, the silencing of MKP-2 significantly prevented the ability of ANG-(1–7) to inhibit generation of the cleaved form of caspase-9 (Fig. 7B), to inhibit nuclear fragmentation assessed by PI assay (Fig. 7C), or to inhibit DNA fragmentation measured by ISEL assay (Fig. 7D).
Fig. 7.
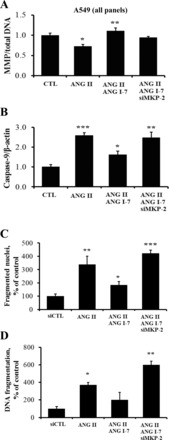
Silencing of MKP-2 prevents ANG-(1–7) rescue of mitochondrial membrane potential (MMP), caspase-9 activation, nuclear fragmentation, and DNA fragmentation in A549 cells. A: A549 cells were treated in the same way as in Fig. 6 with MKP-2 siRNAs, scrambled control RNAs, and/or ANG-(1–7) (10−7 M for 40 min), followed by ANG II (10−7 M) for an additional 8 h. Next, cells were incubated with the lipophilic probe 3,3′-dihexyloxacarbocyanine iodide (DiOC6) for 15 min at 37°C for the estimation of MMP. Bars are means + SE of n ≥ 3 in two or more cell cultures. *P < 0.05 vs. CTL and **P < 0.01 vs. ANG II by ANOVA and Tukey-Kramer multiple-comparison test. B: A549 cells were treated similarly to A but were harvested for Western blotting for caspase-9 (active form) and densitometry; bars are means + SE of n = 4 over two experiments. ***P < 0.001 vs. CTL, *P < 0.05 vs. ANG II, and **P < 0.05 vs. ANG II/ANG-(1–7) by ANOVA and Tukey-Kramer multiple-comparison test. C: A549 cells were transfected with MKP-2 or scrambled siRNAs followed by an incubation with ANG-(1–7) (10−7 M) for an additional 40 min. Thereafter, the cells were exposed to ANG II (10−7 M) for 20 h followed by microscopic quantitation of nuclear fragmentation (see materials and methods). Bars are means + SE of n = 3; **P < 0.01 vs. CTL, *P < 0.01 vs. ANG II, and ***P < 0.01 vs. ANG II/ANG-(1–7) by ANOVA and Student-Newman-Keuls post hoc analysis. D: after ethanol fixation of the above cells, cultures were subjected to the in situ end labeling (ISEL) procedure for the detection of fragmented DNA by the 3,3′-diaminobenzidine (DAB) detection method (21). Bars are means + SE of n = 3; *P < 0.05 vs. CTL and **P < 0.01 vs. ANG II/ ANG-(1–7) by ANOVA and Student-Newman-Keuls post hoc analysis.
In accord with the experiments conducted in Fig. 3, the ability of A779 to block MKP-2 upregulation was assessed in cultures of primary human AECs. In Fig. 8A, blockade of endogenous ANG-(1–7) by treatment with A779 alone (10−7 M) reduced MKP-2 significantly compared with the control that did not receive A779. To determine if silencing MKP-2 could inhibit ANG-(1–7) antiapoptotic signaling in the primary cells as it did in A549 cells, primary cultures of human AECs were challenged with ANG II (10−7 M) for 20 h and were examined by nuclear fragmentation assay (21). Figure 8B illustrates that silencing of MKP-2 significantly prevented the ANG-(1–7) inhibition of nuclear fragmentation induced by ANG II in human AEC primary cultures.
Fig. 8.
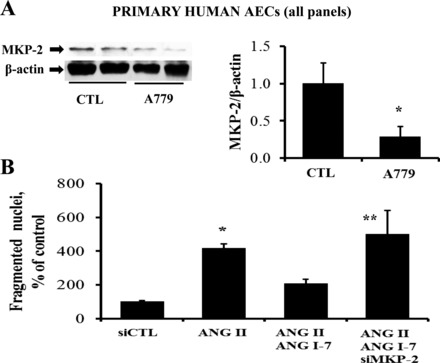
The mas/MKP-2 pathway is functional in primary cultures of human AECs. A: primary human AECs were cultured to 70–80% confluence and then serum starved for 12 h before challenging with A779 (10−7 M) for 30 min [without removing the endogenous ANG-(1–7) that accumulated in 12 h]. Cells were then harvested for Western blotting. Bars are means + SE of n = 3. *P = 0.05 vs. CTL by Mann-Whitney test. B: primary human AECs were treated with MKP-2 siRNA as in Fig. 7C and were exposed to ANG II (10−7 M) for an additional 20 h, followed by microscopic quantitation of nuclear fragmentation (see materials and methods). Bars are means + SE of n = 3; *P < 0.05 vs. CTL and **P < 0.05 vs. ANG II/ANG-(1–7) by ANOVA and Student-Newman-Keuls post hoc analysis.
Figure 9 depicts a summary of MKP-2 and mas receptor functions in AEC survival. ANG II induces p-JNK and apoptosis through the AT1 receptor. Both JNK phosphorylation and apoptosis are blocked by the ANG-(1–7)-mas axis. Moreover, the blockade by ANG-(1–7) is prevented by silencing MKP-2. The mechanisms by which ANG-(1–7)/mas upregulates MKP-2 are yet to be elucidated.
Fig. 9.

Role of MKP-2 in AEC survival. Earlier work demonstrated blockade of ANG II/angiotensin type 1 receptor (AT1R)-induced p-JNK and apoptosis by the ANG-(1–7)/mas pathway (37). ACE-2 limits the accumulation of ANG II by degrading the octapeptide ANG II to the heptapeptide ANG-(1–7). In the current study, the data support that ANG-(1–7) upregulates MKP-2 through mas receptor, sustaining low p-JNK levels to promote AEC survival. Blockade or knock down of mas by the receptor antagonist A779 or antisense oligonucleotides prevented the induction of MKP-2 by ANG-(1–7). The intermediate signaling mechanism(s) by which ANG-(1–7)/mas activates MKP-2 are currently unknown (??).
DISCUSSION
The exact downstream signaling mechanisms of the ANG-(1–7)/mas pathway are currently of high interest to investigators seeking to understand the regulation of AEC apoptosis and its contribution to lung diseases. Past studies from this laboratory have shown the involvement of the ANG system in AEC apoptosis and subsequent lung injury (9). It is well known that, in many organ systems, detrimental effects of the ANG II/AT1 pathway are counteracted by the opposing axis of the ANG-(1–7)/mas signaling pathway (15). Recently, Uhal et al. (37) demonstrated that JNK phosphorylation is a required event in AEC apoptosis in response to binding of ANG II to the AT1 receptor. These authors also showed that bleomycin- or ANG II-induced JNK phosphorylation and apoptosis were blocked by ANG-(1–7) through its binding to mas receptor (37).
The enzyme ACE-2 functions as a monocarboxypeptidase and is one of the enzymes that could degrade the proapoptotic ANG II to form the antiapoptotic ANG-(1–7) (13). Lentiviral overexpression of ACE-2 has been shown to protect against experimental lung injury and cardiac fibrosis in response to bleomycin and ANG II, respectively (27, 14). Similarly, infusion of ANG-(1–7) subcutaneously in C57BL/6 mice attenuated lung injury, and, moreover, treatment with the specific mas blocker A779 aggravated collagen deposition and lung tissue remodeling (5). In nonpulmonary cell types, ANG-(1–7) has shown physiological responses that are opposite to those of ANG II. The heptapeptide ANG-(1–7) has been shown to inhibit ANG II-induced MKP signaling in cardiac myocytes (31), endothelial cells (24), smooth muscle cells (10), and renal proximal tubular cells (28). Furthermore, cellular responses to ANG-(1–7) were blocked by pretreatment with the mas selective blocker A779. It was shown that mas knockout mice exhibit impaired cardiac function in vivo and in vitro, which demonstrated the physiological significance of mas receptor (25). Likewise, the data shown here demonstrate a similar role for the ANG-(1–7)/mas pathway in AECs; antisense oligonucleotide-mediated mas knockdown induced the activated form of caspase-9 and nuclear fragmentation (Fig. 1). Moreover, knockdown of the mas receptor, in the absence of ANG II/ANG-(1–7), shows an induction of p-JNK and caspase-9 (Fig. 2) and thus points to a critical role for mas in preventing AEC apoptosis. These data confirm the involvement of mas in the survival of human AECs and, moreover, are consistent with previously published data showing that blockade of the mas receptor with A779 in the mouse lung epithelial cell line (MLE12) prevented the inhibition of apoptosis by ANG-(1–7) (37).
Several studies have shown that imbalance in the levels of extracellular ANG II and ANG-(1–7) contribute to the pathogenesis of lung injury and defects in other organs (41). A recent study of AECs by Uhal et al. (36) showed extracellular ANG-(1–7) levels in serum-free cell culture media are much higher than extracellular ANG II levels under unstimulated conditions, which was interpreted as a mechanism to maintain cell survival. In the same study it was found that JNK phosphorylation is a required event in AEC apoptosis. Consistent with those findings, in the present study blocking the action of the endogenous ANG-(1–7) with the mas antagonist A779 significantly increased JNK phosphorylation (Fig. 3). Other research groups have shown that, in nonpulmonary cells, ANG-(1–7) inhibits ANG II-induced signaling through activation of a phosphatase. Given that p-JNK was increased in response to the mas blocker (Fig. 3), it was of high interest to determine whether blockade of mas could reduce MKP-2 protein; this result was observed (Fig. 3B). Furthermore, removing the endogenous ANG-(1–7) and adding freshly prepared exogenous ANG-(1–7) significantly induced MKP-2 (Fig. 4A), demonstrating that ANG-(1–7) regulates AEC survival by upregulating a MAPK-selective phosphatase and dephosphorylating JNK. The further finding that the induction of MKP-2 by ANG-(1–7) was prevented by the mas antagonist A779 and mas antisense oligonucleotides (Fig. 4, B and C) showed that ANG-(1–7) induces MKP-2 through its receptor mas. These results are consistent with earlier work from this laboratory that demonstrated mas-mediated blockade of ANG II- or bleomycin-induced signaling by ANG-(1–7) (37). Together, those data and the results from Fig. 4 strongly suggested that MKP-2 mediates the action of the receptor mas in AEC apoptosis. The data showing that antisense oligonucleotides against the mas receptor significantly prevented the ANG-(1–7)-induced increase in MKP-2 protein levels (Fig. 4C) provide further support for this concept.
Together, these data are consistent with experimental results that several other research groups have obtained by studying nonpulmonary cell types. In proximal tubular cells, ANG-(1–7) activated a tyrosine phosphatase and thereby prevented high glucose-stimulated phosphorylation of p38 (12). Furthermore, in studies of cardiac myocytes, it was found that ANG II stimulated the phosphorylation of ERK1/ERK2, but this was reduced by cotreatment with ANG-(1–7) (22). Moreover, the same authors found that ANG-(1–7) induces mitogen-activated protein kinase phosphatase-1 (MKP-1) and, additionally, attenuates cardiac remodeling (22). Consistent with these findings, transgenic mice with constitutive overexpression of MKP-1 did not activate JNK, p38, or ERK1/ERK2 in the heart, and, furthermore, catecholamine-induced hypertrophy was prevented by overexpression of MKP-1 (2). Those data showed that DUSPs, primarily MKP-1, are important in counterregulating MKPs in cardiac cells. Activation of MKP-1 was also demonstrated in vascular smooth muscle cells (VSMCs), and this activation antagonized ANG II/AT1-mediated vascular injury (29). However, the data presented herein strongly demonstrate the upregulation of MKP-2 in AECs as a cell survival mechanism, and treatment with ANG-(1–7) and/or A779 did not change MKP-1 or MKP-5 as shown in Fig. 4D. These differences could be due to organ- or cell type-specific postreceptor signaling mechanisms.
In light of the data implicating MKP-2 in AEC survival, it was of high interest to determine the functional effects of MKP-2 silencing in AECs with siRNA (Figs. 5 and 6). As illustrated here, knockdown of MKP-2 increased basal levels of p-JNK and caspase-9 and demonstrated the importance of MKP-2 in AEC survival (Fig. 6). The siRNA-mediated knockdown of MKP-2 caused a blockade of the ability to ANG-(1–7) to inhibit ANG II-induced JNK phosphorylation (Fig. 5), caspase-9 activation (Fig. 6), and DNA fragmentation and apoptosis measured by nuclear fragmentation (Fig. 7). Consistent with this finding, silencing MKP-2 in primary human AECs significantly prevented ANG-(1–7) blockade of ANG II-induced nuclear fragmentation (Fig. 8B). Moreover, inhibition of the mas receptor by A779 decreased MKP-2 levels in primary cultures of human AECs (Fig. 8A). This finding is in agreement with earlier work from this laboratory (38) suggesting a critical role of mas in primary human AECs. The data presented here further extend those findings and demonstrate that MKP-2 is a critical mediator of the ANG-(1–7)/mas pathway in primary cultures of human AECs. These data are also in line with findings by Gava et al. (12) who showed that blockade of a tyrosine phosphatase by the inhibitor phenylarsine oxide reversed the effects of ANG-(1–7) (12). Overexpression of MKP-2 in human endothelial cells prevented tumor necrosis factor-α-induced apoptosis by preventing JNK phosphorylation, and the induction of apoptotic markers by these cells was also reversed by overexpressing MKP-2 (1).
A recent publication from this laboratory demonstrated endoplasmic reticulum stress-induced mitochondrial dysfunction in AECs was blocked by ANG-(1–7) (38). In the present study, ANG-(1–7) significantly prevented ANG II-induced reduction of the MMP (Fig. 7A), and, moreover, the prevention by ANG-(1–7) was blocked in the absence of MKP-2. Even though MKP-2 knockdown did not bring the MMP value significantly below the control value, blockade of caspase-9 (Fig. 7B) by ANG-(1–7) was eliminated by silencing MKP-2. This could be explained by multiple signaling factors in apoptosis and that MMP is not the only factor involved in induction of nuclear fragmentation. Cadalbert et al. (4) showed that MKP-2 protects against stress-induced apoptosis in human embryonic kidney cells 293, and, moreover, the authors showed specificity of MKP-2 to dephosphorylate JNK in vivo (4). The data from these groups and our current data suggest that ANG-(1–7) activates different phosphatases and causes multiple different biological effects in different cell types.
To date, only a few studies have investigated the downstream signaling of the ANG-(1–7)/mas pathway. It was shown that, in rat neurons, ANG-(1–7) induces phosphatase and tensin homolog, which dephosphorylates membrane-phosphorylated lipids to prevent recruitment of Akt (23). By contrast, in isolated adult myocytes, ANG-(1–7) increased nitric oxide (NO) production associated with induction in endothelial NO synthase and Akt signaling, which were all blocked by treatment with A779 (6). These different mechanisms of ANG-(1–7) action could be due to cell type specificity. Tallant and Clark (30) demonstrated that ANG-(1–7) stimulates prostacyclin and stimulated cAMP production in rat VSMCs. However, the molecular mechanisms by which the ANG-(1–7)/mas pathway stimulates MKP-2 are currently unknown. In vascular smooth muscle cells, blockade of the NO/cGMP pathway prevented the induction of MKP-1 (16). Along similar lines, Lara et al. (18) demonstrated that activation of Na+-ATPase in response to ANG II was blocked by ANG-(1–7) through the cAMP/protein kinase A (PKA)-mediated pathway (18). Therefore, it will be of high interest to determine whether cAMP/PKA or cGMP pathways are involved in the induction of MKP-2 in AECs. It is also a possibility that p-JNK might be a substrate for other phosphatases, but the results of the siRNA knockdowns shown here suggest that MKP-2 is uniquely responsible for maintaining AEC survival. Although studies have demonstrated the activation of phosphatases by ANG-(1–7) in different organs, transcriptional regulation and mRNA stability of the various phosphatases are poorly understood and need to be investigated.
In summary, the experimental studies herein showed that the ability of ANG-(1–7) to block ANG II-induced p-JNK, caspase-9, MMP, DNA fragmentation, and apoptosis is abolished if MKP-2 is silenced. These data support the concept that ANG-(1–7) upregulates the phosphatase MKP-2 through mas and thereby maintains low p-JNK levels to promote AEC survival. Blockade or knockdown of the mas receptor by the antagonist A779 or antisense oligonucleotides attenuated the induction of MKP-2 by ANG-(1–7) and confirmed that mas acts through MKP-2. Collectively, these studies showed that upregulation of MKP-2 by the ANG-(1–7)/mas pathway constitutively dephosphorylates JNK and prevents apoptosis. These signaling mechanisms suggest the potential for pharmacological manipulation of AEC apoptosis and related pathogenic conditions in the lung through mas and MKP-2.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-45136 to B. D. Uhal.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.G. and B.D.U. conception and design of research; I.G. performed experiments; I.G. and B.D.U. analyzed data; I.G. and B.D.U. interpreted results of experiments; I.G. prepared figures; I.G. drafted manuscript; I.G. and B.D.U. edited and revised manuscript; I.G. and B.D.U. approved final version of manuscript.
REFERENCES
- 1.Al-Mutairi M, Al-Harthi S, Cadalbert L, Plevin R. Over-expression of mitogen-activated protein kinase phosphatase-2 enhances adhesion molecule expression and protects against apoptosis in human endothelial cells: MKP-2 over-expression in human endothelial cells. Br J Pharmacol 161: 782–798, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bueno OF, De Windt LJ, Lim HW, Tymitz KM, Witt SA, Kimball TR, Molkentin JD. The dual-specificity phosphatase mkp-1 limits the cardiac hypertrophic response in vitro and in vivo. Circ Res 88: 88–96, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Burgun C, Esteve L, Humblot N, Aunis D, Zwiller J. Cyclic AMP-elevating agents induce the expression of MAP kinase phosphatase-1 in PC12 cells. FEBS Lett 484: 189–193, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Cadalbert L, Sloss CM, Cameron P, Plevin R. Conditional expression of MAP kinase phosphatase-2 protects against genotoxic stress-induced apoptosis by binding and selective dephosphorylation of nuclear activated c-jun N-terminal kinase. Cell Signal 17: 1254–1264, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Chen Q, Yang Y, Huang Y, Pan C, Liu L, Qiu H. Angiotensin-(1–7) attenuates lung fibrosis by way of Mas receptor in acute lung injury. J Surg Res 185: 740–747, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Dias-Peixoto MF, Santos RA, Gomes ER, Alves MN, Almeida PW, Greco L, Rosa M, Fauler B, Bader M, Alenina N, Guatimosim S. Molecular mechanisms involved in the angiotensin-(1–7)/mas signaling pathway in cardiomyocytes. Hypertension 52: 542–548, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci 119: 4607–4615, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin ii-mediated mitochondrial dysfunction linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 102: 488–496, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Fine A, Janssen-Heininger Y, Soultanakis RP, Swisher SG, Uhal BD. Apoptosis in lung pathophysiology. Am J Physiol Lung Cell Mol Physiol 279: L423–L427, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Freeman EJ, Chisolm GM, Ferrario CM, Tallant EA. Angiotensin-(1–7) inhibits vascular smooth muscle cell growth. Hypertension 28: 104–108 1996. [DOI] [PubMed] [Google Scholar]
- 11.Gallagher PE, Ferrario CM, Tallant EA. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am J Physiol Cell Physiol 295: C1169–C1174, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gava E, Samad-Zadeh A, Zimpelmann J, Bahramifarid N, Kitten GT, Santos RA, Touyz RM, Burns KD. Angiotensin (1–7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol Dial Transplant 24: 1766–1773, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamming I, Cooper M, Haagmans BI, Hooper NM, Korstanje R, Osterhaus A, TimensW, Turner AJ, Navis G, van Goor H. The emerging role of ACE2 in physiology and disease. J Pathol 212: 1–11, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, Raizada MK. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp Physiol 90: 783–790, 2005. [DOI] [PubMed] [Google Scholar]
- 15.Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1–7)-Mas receptor axis. Hypertens Res 32: 533–536, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacob A, Molkentin JD, Smolenski A, Lohmann SM, Begum N. Insulin inhibits PDGF-directed VSMC migration via NO/cGMP increase of MKP-1 and its inactivation of MKPs. Am J Physiol Cell Physiol 283: C704–C713, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Jiang F, Yang J, Zhang Y, Dong M, Wang S, Zhang Q, Liu F, Zhang K, Zhang C. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat Rev Cardiol 11: 413–426, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lara LS, Vives D, Correa JS, Cardozo FP, Marques-Fernades M, Lopes AG, Caruso-Neves C. PKA-mediated effect of MAS receptor in counteracting angiotensin II-stimulated renal Na+-ATPase. Arch Biochem Biophys 496: 117–122, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Molina-Molina M, Abdul-Hafez A, Uhal V, Xaubet A, Uhal BD. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol 295: L178–L185, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Shu R, Filippatos G, Uhal BD. Apoptosis in lung injury and remodeling. J Appl Physiol 97: 1535–1542, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Zhang H, Soledad-Conrad V, Zhuang J, Uhal BD. Bleomycin-induced apoptosis of alveolar epithelial cells requires angiotensin synthesis de novo. Am J Physiol Lung Cell Mol Physiol 284: L501–L507, 2003. [DOI] [PubMed] [Google Scholar]
- 22.McCollum LT, Gallagher PE, Tallant EA. Angiotensin-(1–7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. Am J Physiol Heart Circ Physiol 302: H801–H810, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Modgil A, Zhang Q, Pingili A, Singh N, Yao F, Ge J, Guo L, Xuan C, O'Rourke ST, Sun C. Angiotensin-(1–7) attenuates the chronotropic response to angiotensin II via stimulation of PTEN in the spontaneously hypertensive rat neurons. Am J Physiol Heart Circ Physiol 302: H1116–H1122, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sampaio WO, Castro CH, Santos RA, Schiffrin EL, Touyz RM. Angiotensin-(1–7) counterregulates angiotensin ii signaling in human endothelial cells. Hypertension 50: 1093–1098, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, Paula RD, Cruz JS, Ramos AS, Rosa KT, Irigoyen MC, Bader M, Alenina N, Kitten GT, Ferreira AJ. Impairment of in vitro and in vivo heart function in angiotensin-(1–7) receptor MAS knockout mice. Hypertension 47: 996–1002, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SB, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos M, Schultheiss HP, Speth R, Walther T. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA 100: 8258–8263, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, Jun J, Sriramula S, Mariappan N, Pourang D, Venugopal CS, Francis J, Reudelhuber T, Santos RA, Patel JM, Raizada MK, Katovich MJ. The angiotensin-converting enzyme 2/angiogenesis-(1–7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med 182: 1065–1072, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su Z, Zimpelmann J, Burns KD. Angiotensin-(1–7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int 69: 2212–2218, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Takeda-Matsubara Y, Nakagami H, Iwai M, Cui T, Shiuchi T, Akishita M, Nahmias C, Ito M, Horiuchi M. Estrogen activates phosphatases and antagonizes growth-promoting effect of angiotensin II. Hypertension 39: 41–45, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Tallant EA, Clark MA. Molecular mechanisms of inhibition of vascular growth by angiotensin-(1–7). Hypertension 42: 574–579, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Tallant EA, Ferrario CM, Gallagher PE. Angiotensin-(1–7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am J Physiol Heart Circ Physiol 289: H1560–H1566, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Uhal BD. The role of apoptosis in pulmonary fibrosis. Eur Respir Rev 17: 138–144, 2008. [Google Scholar]
- 33.Uhal BD. Epithelial apoptosis in the initiation of lung fibrosis. Eur Respir J Suppl 44: 7s–9s, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Uhal BD. Apoptosis in lung fibrosis and repair. Chest 122: 293S–298S, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol Lung Cell Mol Physiol 275: L1192–L1199, 1998. [DOI] [PubMed] [Google Scholar]
- 36.Uhal BD, Li X, Piasecki CC, Molina-Molina M. Angiotensin signalling in pulmonary fibrosis. Int J Biochem Cell Biol 44: 465–468, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhal BD, Li X, Xue A, Gao X, Abdul-Hafez A. Regulation of alveolar epithelial cell survival by the ACE-2/angiotensin 1–7/Mas axis. Am J Physiol Lung Cell Mol Physiol 301: L269–L274, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uhal BD, Nguyen H, Dang M, Gopallawa I, Jiang J, Dang V, Ono S, Morimoto K. Abrogation of ER stress-induced apoptosis of alveolar epithelial cells by angiotensin 1–7. Am J Physiol Lung Cell Mol Physiol 305: L33–L41, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang R, Alam G, Zagariya A, Gidea C, Pinillos H, Lalude O, Choudhary G, Oezatalay D, Uhal BD. Apoptosis of lung epithelial cells in response to TNF-α requires angiotensin II generation de novo. J Cell Physiol 185: 253–259, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Wang R, Zagariya A, Ang E, Ibarra-Sunga O, Uhal BD. Fas-induced apoptosis of alveolar epithelial cells requires ANG II generation and receptor interaction. Am J Physiol Lung Cell Mol Physiol 277: L1245–L1250, 1999. [DOI] [PubMed] [Google Scholar]
- 41.Wösten-van Asperen RM, Bos AP, Bem RA, Dierdorp BS, Dekker T, van Goor H, Kamilic J, van der Loos CM, van den Berg E, Bruijn M, van Woensel JB, Lutter R. Imbalance between pulmonary angiotensin-converting enzyme and angiotensin-converting enzyme 2 activity in acute respiratory distress syndrome. Pediatr Crit Care Med J Soc Crit Care Med World Fed Pediatr Intensive Crit Care Soc 14: e438–441, 2013. [DOI] [PubMed] [Google Scholar]