

Abstract
Both increased and decreased fatty acid (FA) availability contribute to control of food intake. For example, it is well documented that intestinal FA reduces feeding by triggering enterondocrine secretion of satietogenic peptides, such as cholecystokinin (CCK) and glucagon-like peptide-1 (GLP-1). In contrast, mechanisms by which decreased FA availability increase feeding are not well understood. Over the past three decades substantial research related to FA availability and increased feeding has involved use of the orexigenic compound mercaptoacetate (MA). Because MA reportedly inhibits FA oxidation, it has been assumed that reduced FA oxidation accounts for the orexigenic action of MA. Recently, however, we demonstrated that MA antagonizes G protein-coupled receptor 40 (GPR40), a membrane receptor for long and medium chain FA. We also demonstrated that, by antagonizing GPR40, MA inhibits GLP-1 secretion and attenuates vagal afferent activation by FA. Because both vagal afferent activation and GLP-1 inhibit food intake, we postulated that inhibition of GPR40 by MA might underlie the orexigenic action of MA. We tested this hypothesis using male and female GPR40 knockout (KO) and wild-type (WT) mice. Using several testing protocols, we found that MA increased feeding in WT, but not GPR40 KO mice, and that GPR40 KO mice gained more weight than WT on a high-fat diet. Metabolic monitoring after MA or saline injection in the absence of food did not reveal significant differences in respiratory quotient or energy expenditure between treatment groups or genotypes. These results support the hypothesis that MA stimulates food intake by blocking FA effects on GPR40.
Keywords: mercaptoacetate, GPR40, fatty acid, lipoprivic feeding
mercaptoacetate (MA) is an orexigenic compound (31) reported previously to reduce fatty acid (FA) oxidation by blocking mitochondrial acyl-CoA dehydrogenases (1, 2). The orexigenic effects of MA have been attributed to blockade of FA oxidation, leading to the concept of a “lipoprivic” control of feeding (20, 21, 25). Although it is reasonable to assume that reduction of this important metabolic fuel would provide a stimulus for feeding, this assumption has been difficult to prove with regard to MA. This is due in part to the fact that, with the exception that MA-induced feeding is known to require vagal sensory neurons (4, 28, 29), site of action of MA has remained obscure.
We recently discovered a novel mechanism whereby MA may stimulate feeding (9). Using in vitro approaches, we found that that MA antagonizes the G protein-coupled receptor (GPR) for long and medium chain FAs (GPR40). In cultured nodose neurons, linoleic acid (LA) and caprylic acid (long and medium chain FAs, respectively) and GW9508, a synthetic agonist of GPR40 (5), increased cytosolic Ca2+ levels ([Ca2+]i), whereas MA reversibly blocked those effects without altering responses to agents that increase [Ca2+]i by mechanisms not involving GPR40. We also confirmed the expression of GPR40 by nodose neurons using RT-PCR (8).
GPR40 mediates a number of responses through which FAs alter food intake and metabolic homeostasis, including FA-induced secretion of glucagon-like peptide-1 (GLP-1) (10) and cholecystokinin (CCK) (23), both of which have well-established satietogenic actions (35, 37). GPR40 also mediates FA-induced potentiation of glucose-stimulated insulin secretion (40), which may also exert satietogenic effects (38). Therefore, since MA blocks GPR40 and presumably also these GPR40-mediated satietogenic effects, we hypothesized that GPR40 may contribute to or mediate the orexigenic actions of MA.
In the present experiment we use transgenic male and female mice in which the GPR40 receptor has been deleted to directly test the role of GPR40 in MA-induced feeding. We compared the feeding responses to MA in male and female GPR40 knockout (KO) and wild-type (WT) mice in several testing paradigms. We also measured MA-induced changes in respiratory quotient, energy expenditure, and activity in the absence of food. We also examined body weight over a 26-wk period beginning at weaning in WT and KO mice maintained on high-fat diet (HFD) or chow. We found that GPR40 deletion eliminates MA-induced feeding in mice and increases body weight gain compared with WT controls in adult mice on a HDF. Importantly, MA did not alter metabolic measures in either genotype. We conclude that GPR40 contributes to body weight homeostasis and is a required substrate for MA-induced feeding.
RESEARCH DESIGN AND METHODS
Animals.
Male and female mice were maintained in an animal care facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care. The GPR40-targeted KO mice were developed by replacing the GPR40 coding region with a 21-nucleotide DNA fragment encoding genes for 9 amino acids of influenza hemagglutinin antigen eGFP and neomycin (23). Offspring were maintained in our vivarium where they were crossed with C57BL/6N mice. At least three generations of heterozygous breedings were used to generate homozygous GPR40 KO and WT mice used in our experiments. With one exception (see Feeding tests and results), WT and KO offspring of heterozygous parents were used as subjects. Mice were housed after being weaned in groups of three and maintained on a 12-h light/12-h dark cycle, under controlled temperature conditions (22°C), with ad libitum access to pelleted rodent chow (no. 5001, LabDiet, St. Louis, MO) or a high-fat diet (HFD; no. D12451, Research Diets, New Brunswick, NJ) and tap water, as described in Measurement of body weight gain. All experimental procedures were approved by Washington State University Institutional Animal Care and Use Committee, which conforms to National Institutes of Health guidelines.
Genotyping.
Both male and female WT and KO littermates from pairs of heterozygous mice were used in the experiments. Genotyping PCR was performed using genome DNA isolated from ear punches, with primers specific for the WT (5-CTG TTC CCA AGT AGC CAG TGA CCA G-3′ and 5-GGA GGC TTC CTA GCT GCT CTC AGC-3,′ and 565 bp product size) and for mutant alleles (5′-CAC AGC TCT CCT TCG CTC TCT A-3′ and 5′-TAG CGG CTG AAG CAC TGC A-3,′ and 289 bp product size). The cycles of PCR were 2 min at 94°C for initial denature and 35 cycles of amplification (30 s at 94°C, 30 s at 60°C, and 60 s at 68°C), and then 7 min at 68°C for final extension. Single bands for each pair of primers were confirmed after gel separation of PCR products.
Measurement of body weight gain.
Body weight was measured between 3 and 29 wk of age in groups of male and female mice maintained, beginning at 5 wk of age, on rodent chow (3.36 kcal/g; 13.5% calories from fat) or HFD (4.73 kcal/g; 45.0% calories from fat). Male mice on chow were not monitored after 17 wk of age, because they were used in other experiments that required changes in diet.
Feeding tests.
All feeding tests were conducted using HFD because a HFD is frequently used for MA-induced feeding and has been reported to produce larger responses than a low-fat diet (34). Mice raised on rodent chow were switched to HFD at least 3 days before the first feeding test and were maintained on that diet until feeding tests were complete. Food intake was tested using three different protocols. In each case, mice were used as their own controls for the MA and saline tests. Except for the first protocol listed here, tests were conducted using WT and KO offspring of heterozygous parents. For the first protocol, female and male offspring of homozygous KO mice and offspring of the WT mice were tested in groups of 3 mice/cage. Groups were composed of mice that were cage mates under daily housing conditions. Cages were modified by insertion of a wire mesh floor and bedding was removed before the test to facilitate accurate measurement of spillage. Spillage was collected on paper towels beneath the cage floor and was air-dried before measurement. β-Mercaptoacetate (68 mg/kg/2 ml saline ip) was prepared for injection just before use, and total food intake per cage was measured in 4-h tests following MA or saline control injection. MA was administered intraperitoneally, as has been typical in previous feeding studies. Recent work has shown that vascular, but not luminal, stimulation of GPR40 activates secretion of GLP-1 from intestinal L cells (8). Tests were separated by at least 3 rest days. Second, the same protocol using groups of 3 mice/cage was used to test male and female KO and WT offspring from heterozygous parents. Third, male mice from heterozygous parents were singly housed and tested in suspended wire mesh cages, with spillage collected as described for the group-housed mice. Finally, food intake and other parameters were measured in male WT and KO mice (from heterozygous parents) housed individually in an automated monitoring chamber (Promethion, Sable Systems International, Las Vegas, NV) in which feeding events were recorded every second.
Twenty-four hour spontaneous intake of HFD and chow were also measured in WT and KO male mice using the Promethion automated meal-monitoring system.
Metabolic parameters.
Metabolic measures were taken in male KO and WT mice maintained on HFD before the test. Tests were conducted using the Promethion system after 3 days of adaptation to the apparatus. KO and WT mice were injected with MA or saline control, as in the feeding test, but food was withheld during this test so that the effects of MA could be observed in the absence of the confounding effect of food. Measurements were taken during the 2 h following MA and saline injections, corresponding to the time of peak feeding activity in the MA tests. Respiratory quotient (RQ, the ratio of the volume of carbon dioxide generated and the volume of oxygen consumed), energy expenditure (EE), and activity (Y-axis beam breaks) were recorded. Values were calculated after application of algorithms using macros provided with the analysis software ExpeData.
Statistical analysis.
Results are presented as means ± SE. For statistical analysis of data, we used t-test when only two groups were compared (24 h food intake) and one-way ANOVA for multiple comparisons (body weight, MA-induced feeding, and metabolic data). After significance was determined by ANOVA, multiple comparisons between individual groups were tested using the Student-Newman-Keuls post hoc test. P < 0.05 was considered to be statistically significant.
RESULTS
Body weight.
GPR40 KO mice gained more weight than WT on a HFD. Figure 1 shows body weight gain in KO and WT male (Fig. 1A) and female (Fig. 1B) mice when fed a chow diet or a HFD. For both males and females, body weight did not differ in KO and WT when fed a chow diet. However, both males and female mice gained more body weight on the HFD, but KO mice gained significantly more than WT on the HFD. Diet-induced differences in body weight began to emerge as mice passed the age of puberty (6–8 wk), but were not significant until about 17 wk of age.
Fig. 1.
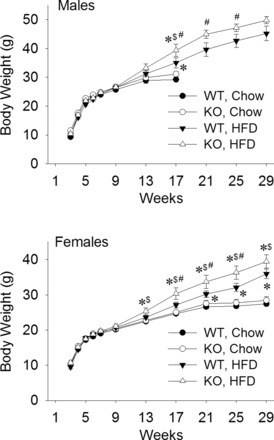
Body weight gain of male and female mice fed on chow or high-fat diet (HFD). Male and female GPR40 knockout (KO) and wild-type (WT) mice were fed on chow or HFD beginning at 5 wk of age. Chow-fed males were removed from the experiment after 17 wk of age. Adult male and female KO mice gained significantly more weight than WT mice on HFD. WT and KO mice maintained on chow did not differ in body weight. *P < 0.05 vs. WT, Chow; $P < 0.05 vs. KO, Chow; #P < 0.05 vs. WT, HFD, at the same age, respectively; Student-Newman-Keuls test after one-way ANOVA (or t-test for males during 21–29 wk). N = 13–20 mice per group.
Food intake.
GPR40 KO mice did not increase food intake in response to MA. Figure 2 shows 4-h intake of HFD in response to MA and saline injections in male and female mice using different testing protocols. Figure 2, A and B, shows food intake in male and female KO mice, respectively, from homozygous parents and the WT background strain tested in groups of 3 per cage. WT, but not KO mice, increased food intake in response to MA. Figure 2, C and D, shows male and female KO and WT mice, respectively, derived from heterozygous parents and housed and tested in groups of 3 per cage. MA increased feeding in WT but not in KO mice. Figure 2E shows food intake in male mice housed and tested individually in wire mesh cages. Figure 2F shows food intake of male mice tested individually in the Promethion automated meal-monitoring apparatus. As in group-housed mice, individually housed WT mice increased feeding in response to MA but KO mice did not. Thus, in all cases, regardless of the sex of the mice or the protocol utilized for the test, MA stimulated feeding significantly in WT mice but not in KO mice. The maintenance diet (HFD or chow) before adaptation to the test diet (HFD) did not appear to alter the responsiveness to MA; e.g., compare Fig. 2, E (maintained on chow) and C (maintained on HFD). Saline-treated KO mice tended to eat more in 4-h feeding tests than saline-treated WT mice (P values < 0.05, in Fig. 2, A–C, and E). However, in some cases, food intake in saline-treated KO and WT mice did not differ (P values > 0.6, in Fig. 2, D and F).
Fig. 2.
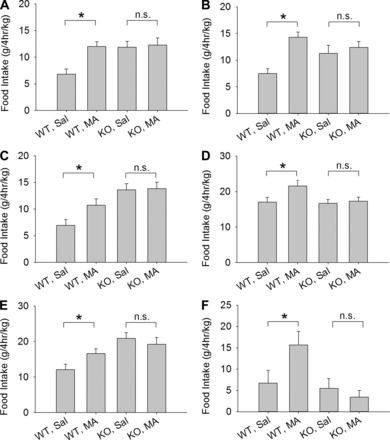
Mercaptoacetate (MA)-induced feeding in male and female GPR40 KO and WT mice. Mice were housed and tested under different conditions using cages designed for accurate measurement of spillage. Male (A) and female (B) KO mice derived from homozygous parents and WT mice of the background strain C57BL/6N were housed and tested in groups of 3 per cage. Male (C) and female (D) KO and WT mice derived from heterozygous parents were tested in groups of 3 per cage. Male KO and WT mice were tested individually in wire mesh cages (E) or in the Promethion system (F). All bars represent total intake of HFD in a 4-h test beginning immediately after MA (68 mg·kg−1·2 ml−1 ip) or saline injection. Data are corrected for body weight of group or individual/cage. Under all conditions and in both sexes, MA stimulated feeding in WT, but not in KO mice. *P < 0.05 WT, MA vs. WT, Sal; n.s., not significant, KO, MA vs. KO, Sal. Student-Newman-Keuls test after one-way ANOVA. N = 7–12 cages/group for A–D, and N = 7–8 mice/group for E and F.
Twenty-four-hour food intake, RQ, and EE did not differ between KO and WT mice after MA or saline. Figure 3 shows 24 h spontaneous intake of chow (Fig. 3A) and HFD (Fig. 3B) and intakes plotted as total calories (Fig. 3C) in male KO and WT mice, as measured in the Promethion system. Food intake of the two genotypes did not differ on either diet in total amount or circadian distribution. Twenty-four-hour food intake was also measured in female mice. In females, as in males, intake did not differ between genotypes (data not shown). Similarly metabolic measures in male KO and WT mice, also shown in Fig. 3, did not differ between groups. Figure 3D shows RQ mapped in 30-min bins over the 120 min following drug or saline injection. MA treatment did not produce a significant change in RQ compared with saline in either KO or WT mice. Figure 3B shows that EE, like RQ in the same mice, did not differ by genotype or drug treatment. Figure 3C shows activity of the four groups during the test. The activity of the MA-injected groups (KO and WT) was significantly higher 60 min after the injection than in saline-treated groups. Activity remained significantly higher only in the MA-treated WT mice at the 90-min time point. Total activity during the 2-h test, however, did not differ between treatment groups or genotypes (see inset in Fig. 3C).
Fig. 3.
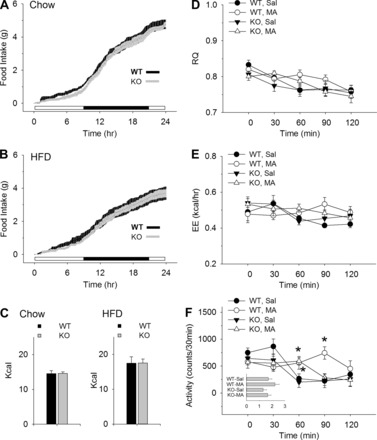
Food intake, metabolic, and activity measures in male mice. Cumulative 24-h food intake in adult WT and KO male mice maintained on chow (A) or HFD (B) and total caloric intake during the same measurement period (C). Changes in respiratory quotient (RQ), energy expenditure (EE), and activity in WT and KO mice during the 2-h period after an MA (68 mg·kg−1·2 ml−1 ip) or saline (2 ml/kg) injection are shown in D, E, and F, respectively. In D–F, mice were maintained on HFD, but food was removed during the test. KO and WT mice did not differ in the circadian distribution or caloric value of their daily feeding. RQ and EE did not differ between groups, but differences were present, as indicated, in activity measures. Inset in F: total activity counts (×1,000) during the 2-h test after injection of MA or saline. WT-MA mice had slightly, but not significantly, higher overall activity levels. N = 8/group (A–F), *P < 0.05 vs. WT, Sal; Student-Newman-Keuls test after one-way ANOVA at each time point.
DISCUSSION
Our results reveal for the first time that deletion of the gene for the long and medium chain FA receptor GPR40 eliminates increased feeding in response to MA in both male and female mice. Mice were tested using several different protocols. They were group housed and individually housed during the test and also were tested using an automated meal-monitoring apparatus. In all protocols, MA significantly increased feeding in WT, but not in KO mice. Results indicate that the orexigenic effect of MA requires the GPR40 FA receptor. These results are consistent with our previous finding that MA blocks GPR40-mediated effects of linoleic acid, a long chain FA, in cultured nodose neurons (9), and with our recently reported data showing that MA blocks FA-induced stimulation of GLP-1 in vivo in rats (22). Finally, our results are consistent with results from other groups, discussed previously, showing that GPR40 controls secretion of peptides that mediate satiation (10, 23, 35, 37).
Previously it was shown that MA blocks FA oxidation by inhibiting mitochondrial acyl Co-A dehydrogenases (1, 2). Over the last three decades, it has been assumed that the orexigenic activity of MA is a response to an antimetabolic effect. Results of experiments reported here, however, do not support this hypothesis. When tested in the absence of food, but otherwise under the same dietary and testing conditions used to measure MA-induced feeding, RQ did not reveal any detectable reduction of fat oxidation following MA administration in either KO or WT animals. In addition, there is no available evidence to suggest that GPR40 is involved in transmembrane transport of FA, which could potentially provide a mechanism for altering FA oxidation. If MA had blocked FA oxidation, we would have expected that effect to increase RQ in both WT and KO mice in our experiment, reflecting an increase in utilization of carbohydrates in response to decreased metabolism of FA. It is possible that MA blocks FA oxidation only in a very restricted group of cells, without producing systemic changes that are detectable using our method of metabolic measurement. Therefore, we cannot completely exclude the possibility that MA blocks fat oxidation. However, since deletion of the GPR40 gene completely eliminated MA-induced feeding, any effects of MA on FA oxidation must not contribute significantly to the feeding response to MA.
The fact that MA-induced increase in food intake is independent of reduced FA oxidation does not rule out inhibition of FA oxidation as a mechanism by which other compounds increase food intake. For example, both methyl palmoxirate (11, 12) and etomoxir (15, 18) are reported to reduce FA oxidation by blocking carnitine palmatoyltransferase 1, thus interfering with delivery of FA to mitochondrial oxidation sites. Administration of either of these compounds is reported to increase food intake, although by comparison to the effect of MA, feeding responses to methyl palmoxirate and etomoxir are quite small. Nevertheless, we recently reported that etomoxir, in contrast to MA, did not suppress GLP-1 or insulin secretion and had no effect on blood glucose concentration following olive oil gavage, suggesting that etomoxir does not antagonize GPR40 (23). Additional work will be required to clarify the differences in the mechanisms of action between these drugs and MA.
GPR40 is one of several FA receptors expressed by mammals. Recently, we reported that, in addition to antagonizing GPR40, MA also antagonizes the long chain FA receptor GPR120 (22). Specifically, we found that linoleic acid and the synthetic GPR120 partial agonist grifolic acid increased [Ca2+]i in STC-1 cells. MA antagonized Ca2+ responses to both agents, although its antagonism at GPR120 was less robust than at GPR40. It is possible that GPR120 contributes to the control of food intake by FA. However, MA antagonism at GPR120 alone is not sufficient to increase food intake because the feeding response to MA was completely absent in GPR40 KO mice, even though these mice do exhibit normal levels of GPR120 expression.
Although GPR40 deletion eliminated MA-induced feeding, WT and KO mice did not differ in their 24-h food intake or in the circadian distribution of their feeding behavior over the 24-h period. In addition, mice on a low-fat diet did not differ with respect to body weight. These results are similar to results of a study in which daily injection of DC260126, a small molecule antagonist of GPR40, failed to alter food intake or body weight measured periodically over an 8-wk period in adult female Zucker rats maintained on a 15% fat diet (42). However, our results differ in that both male and female KO mice did gain more weight than WT on a high-fat (45%) diet. Although daily food intake was not measured over the 26-wk period of our study, the fact that KO mice gained more weight on a HFD than WT mice suggests that they also consumed slightly more calories than WT. The tendency of KO mice to eat more than WT in the saline baseline tests can be seen in male mice in the “home cage” feeding tests where the KO controls (mice given saline) in some cases ate more than WT controls.
Paradoxically, male KO mice ate significantly less after either saline (baseline) or MA injection when they were tested in the Promethion metabolic chambers than when they were tested under the same conditions in suspended wire cages. Because mice were adapted to the Promethion system and spillage was carefully measured in both testing environments, we cannot explain this difference in feeding behavior. We attribute it tentatively to somewhat reduced accessibility of the food for the Promethion testing apparatus. The fact remains, however, that MA increased food intake in WT mice and failed to increase intake in the KO mice, regardless of the test environment that was used. Thus the relative differences between WT and KO response to MA remains robust and reproducible across different test environments.
In addition to the clear relevance of GPR40 to MA effects on food intake, it is noteworthy that in a number of cases we observed that baseline food intake was elevated in GPR40 KO mice relative to baseline intake by WT mice. This observation suggests that, without pharmacological manipulation, absence of GPR40 signaling increases food intake, possibly reflecting a role for constitutive activity of GPR40 in maintaining basal metabolic tone.
Our previous in vitro experiments revealed that MA acts directly on vagal afferent GPR40 receptors to prevent vagal activation by long chain FA (9). MA-induced feeding is dependent on intact vagal afferent neurons (4, 28, 29). Therefore, direct MA action at vagal GPR40 may contribute to the orexogenic action of MA. On the other hand, GPR40 is expressed by a variety of tissues at which MA also could act to enhance food intake. For example, GPR40 activation mediates FA-stimulated secretion of CCK (23) from enteroendocrine I cells and GLP-1 secretion (10) from enteroendocrine L cells. Because both CCK and GLP-1 inhibit food intake, antagonism of GPR40 on I cells or L cells might be expected to increase food intake. In this regard, it is significant that we recently reported that MA blocks GLP-1 secretion in rats following olive oil gavage and also attenuates GLP-1 release by enteroendocrine STC-1 cells in vitro (22). Similarly, because pancreatic insulin also has satietogenic properties, the fact that FA-induced activation of GPR40 on pancreatic β cells potentiates glucose-stimulated insulin secretion (40) (for review see Ref. 16) raises the possibility that MA antagonism of islet cell GPR40 could increase food intake by attenuating insulin secretion. Indeed, our previously reported finding that MA attenuates insulin secretion is consistent with this possibility. Finally, GPR40 is expressed by taste bud cells, where it has been implicated in gustatory fat sensation (7, 13, 32). The possibility that blockade of gustatory GPR40 might enhance food intake is an additional avenue of MA action that awaits further investigation. In summary, any or all of the above responses to GPR40 activation are consistent with potential GPR40 membrane receptor contributions to reduction of food intake and, thus, are supportive of our hypothesis that MA-induced blockade of GPR40 is the critical mechanism by which MA increases food intake.
Although GPR40 is widely expressed throughout the brain (19, 24, 30, 41), the GPR40 receptors responsible for the effects of MA on feeding most likely are located peripherally. Compelling evidence has shown that vagotomy and selective lesion of vagal sensory neurons abolish MA-induced feeding (3, 28, 29), indicating a peripheral site of action for MA. Furthermore, MA has not been found to be effective in stimulating feeding when administered directly into the brain. However, several brain sites activated by peripherally administered MA have been described. These include the nucleus of the solitary tract, the lateral parabrachial nucleus, the central nucleus of the amygdala (26), and the lateral hypothalamus (33). Correspondingly, lesions of these areas abolish MA-induced feeding (6, 27, 29). Because vagotomy blocks activation of these sites by systemic MA administration (27), as indicated by measurements of c-Fos expression, their involvement in MA-induced food intake is likely to be downstream of the peripheral GPR40-mediated effects of MA on secretion and circulating levels of centrally acting gastrointestinal peptides. However, identification of GPR40 expression in brain sites activated by systemic MA would suggest that the possibility of a direct central action of MA should be reevaluated using site-specific injections.
Two effects mediated by GPR40 that have attracted considerable attention are the secretion of the incretin hormones GLP-1 and GIP. Incretin hormones stimulate insulin secretion and reduce blood glucose, actions of obvious value in the treatment of Type 2 diabetes. Consequently, GPR40 is under intense scrutiny for its potential value in translational medicine. Indeed, GPR40 agonists have been introduced for diabetes therapy in humans (16, 17). Nevertheless, the GPR40 receptor is complex and not yet fully understood. It has been reported recently that when activated by certain agonists, GPR40 can signal through both Gq and the IP3 signaling cascade, but also through Gs and the cAMP cascade and, in addition, that double agonists are more effective than single agonists in stimulating incretin effects (14). Furthermore, recent studies in GPR40 and GPR120 double knockout mice indicate that these two FA receptors may function cooperatively in conditioning flavor preferences. GPR40/GPR120 double knockout mice exhibited deficits in conditioning of flavor preferences by postoral fat, while deletion of the genes individually did not have this effect (32). Mechanisms such as synergism and allosteric modulation of FA receptors at the same GPR40 receptive sites have been reported and may contribute to these interactive effects (36, 39). Thus the effort to understand this receptor is of considerable importance and MA, as a GPR40 antagonist, may contribute to this effort.
Perspectives and Significance
We show here for the first time that the FA receptor GPR40 mediates the orexigenic effect in MA in male and female mice. This result, in combination with our finding that MA reduces GPR40-mediated stimulation of GLP-1 secretion in vivo and in vitro and suppresses insulin secretion (22), indicates that the orexigenic effect of MA arises from a widespread reduction of activity at the multiple (largely peripheral) GPR40 receptor sites and reduction of their multiple functions. These results also highlight the converse, that simultaneous activation of this single receptor type at its multiple expression sites by circulating fatty acids is an effective mechanism for reduction of food intake.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK040498 and DK097437 to S. Ritter.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.-J.L. and S.R. conception and design of research; A.-J.L., M.F.W., and Q.W. performed experiments; A.-J.L. analyzed data; A.-J.L. and S.R. interpreted results of experiments; A.-J.L. prepared figures; A.-J.L. and S.R. edited and revised manuscript; A.-J.L., M.F.W., Q.W., S.A.W., and S.R. approved final version of manuscript; S.R. drafted manuscript.
ACKNOWLEDGMENTS
We thank Christine Pedrow for assistance with mouse testing and genotyping.
REFERENCES
- 1.Bauche F, Sabourault D, Giudicelli Y, Nordmann J, Nordmann R. 2-Mercaptoacetate administration depresses the beta-oxidation pathway through an inhibition of long-chain acyl-CoA dehydrogenase activity. Biochem J 196: 803–809, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauche F, Sabourault D, Giudicelli Y, Nordmann J, Nordmann R. Inhibition in vitro of acyl-CoA dehydrogenases by 2-mercaptoacetate in rat liver mitochondria. Biochem J 215: 457–464, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt K, Arnold M, Geary N, Langhans W, Leonhardt M. Beta-adrenergic-mediated inhibition of feeding by mercaptoacetate in food-deprived rats. Pharmacol Biochem Behav 85: 722–727, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Brandt K, Arnold M, Geary N, Langhans W, Leonhardt M. Vagal afferents mediate the feeding response to mercaptoacetate but not to the beta (3) adrenergic receptor agonist CL 316,243. Neurosci Lett 411: 104–107, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, McCoy DC, Kenakin TP, Andrews JL, Ammala C, Fornwald JA, Ignar DM, Jenkinson S. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol 148: 619–628, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calingasan NY, Ritter S. Lateral parabrachial subnucleus lesions abolish feeding induced by mercaptoacetate but not by 2-deoxy-d-glucose. Am J Physiol Regul Integr Comp Physiol 265: R1168–R1178, 1993. [DOI] [PubMed] [Google Scholar]
- 7.Cartoni C, Yasumatsu K, Ohkuri T, Shigemura N, Yoshida R, Godinot N, le Coutre J, Ninomiya Y, Damak S. Taste preference for fatty acids is mediated by GPR40 and GPR120. J Neurosci 30: 8376–8382, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen LW, Kuhre RE, Janus C, Svendsen B, Holst JJ. Vascular, but not luminal, activation of FFAR1 (GPR40) stimulates GLP-1 secretion from isolated perfused rat small intestine. Physiol Rep 3: e12551, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darling RA, Zhao H, Kinch D, Li AJ, Simasko SM, Ritter S. Mercaptoacetate and fatty acids exert direct and antagonistic effects on nodose neurons via GPR40 fatty acid receptors. Am J Physiol Regul Integr Comp Physiol 307: R35–R43, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57: 2280–2287, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman MI, Harris RB, Ji H, Ramirez I, Tordoff MG. Fatty acid oxidation affects food intake by altering hepatic energy status. Am J Physiol Regul Integr Comp Physiol 276: R1046–R1053, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Friedman MI, Tordoff MG. Fatty acid oxidation and glucose utilization interact to control food intake in rats. Am J Physiol Regul Integr Comp Physiol 251: R840–R845, 1986. [DOI] [PubMed] [Google Scholar]
- 13.Gilbertson TA, Khan NA. Cell signaling mechanisms of oro-gustatory detection of dietary fat: advances and challenges. Prog Lipid Res 53: 82–92, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Hauge M, Vestmar MA, Husted AS, Ekberg JP, Wright MJ, Di Salvo J, Weinglass AB, Engelstoft MS, Madsen AN, Luckmann M, Miller MW, Trujillo ME, Frimurer TM, Holst B, Howard AD, Schwartz TW. GPR40 (FFAR1) - Combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol Metab 4: 3–14, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horn CC, Ji H, Friedman MI. Etomoxir, a fatty acid oxidation inhibitor, increases food intake and reduces hepatic energy status in rats. Physiol Behav 81: 157–162, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Huang H, Dai MH, Tao YX. Physiology and therapeutics of the free fatty acid receptor GPR40. Prog Mol Biol Transl Sci 121: 67–94, 2014. [DOI] [PubMed] [Google Scholar]
- 17.Ichimura A, Hasegawa S, Kasubuchi M, Kimura I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front Pharmacol 5: 236, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahler A, Zimmermann M, Langhans W. Suppression of hepatic fatty acid oxidation and food intake in men. Nutrition 15: 819–828, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Khan MZ, He L. The role of polyunsaturated fatty acids and GPR40 receptor in brain. Neuropharmacology In press. [DOI] [PubMed] [Google Scholar]
- 20.Langhans W, Egli G, Scharrer E. Regulation of food intake by hepatic oxidative metabolism. Brain Res Bull 15: 425–428, 1985. [DOI] [PubMed] [Google Scholar]
- 21.Langhans W, Scharrer E. Role of fatty acid oxidation in control of meal pattern. [DOI] [PubMed] [Google Scholar]
- 22.Li AJ, Wang Q, Dinh TT, Simasko SM, Ritter S. Mercaptoacetate blocks fatty acid-induced GLP-1 secretion in male rats by directly antagonizing GPR40 fatty acid receptors. Am J Physiol Regul Integr Comp Physiol 310: R714–R722, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liou AP, Lu X, Sei Y, Zhao X, Pechhold S, Carrero RJ, Raybould HE, Wank S. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140: 903–912, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamoto K, Nishinaka T, Sato N, Mankura M, Koyama Y, Kasuya F, Tokuyama S. Hypothalamic GPR40 signaling activated by free long chain fatty acids suppresses CFA-induced inflammatory chronic pain. PLos One 8: e81563, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ritter S, Calingasan N. Neural Substrates for Metabolic Control of Feeding. In: Appetite and Body Weight Regulation: Sugar, Fat, and Macronutrient Substitutes, edited by Fernstrom JD, and Miller GD. Boca Raton, FL: CRC, 1994. [Google Scholar]
- 26.Ritter S, Dinh TT. 2-Mercaptoacetate and 2-deoxy-D-glucose induce Fos-like immunoreactivity in rat brain. Brain Res 641: 111–120, 1994. [DOI] [PubMed] [Google Scholar]
- 27.Ritter S, Hutton B. Mercaptoacetate-induced feeding is impaired by central nucleus of the amygdala lesions. Physiol Behav 58: 1215–1220, 1995. [DOI] [PubMed] [Google Scholar]
- 28.Ritter S, Taylor JS. Capsaicin abolishes lipoprivic but not glucoprivic feeding in rats. Am J Physiol Regul Integr Comp Physiol 256: R1232–R1239, 1989. [DOI] [PubMed] [Google Scholar]
- 29.Ritter S, Taylor JS. Vagal sensory neurons are required for lipoprivic but not glucoprivic feeding in rats. Am J Physiol Regul Integr Comp Physiol 258: R1395–R1401, 1990. [DOI] [PubMed] [Google Scholar]
- 30.Sartorius T, Drescher A, Panse M, Lastovicka P, Peter A, Weigert C, Kostenis E, Ullrich S, Haring HU. Mice lacking free fatty acid receptor 1 (GPR40/FFAR1) are protected against conjugated linoleic acid-induced fatty liver but develop inflammation and insulin resistance in the brain. Cell Physiol Biochem 35: 2272–2284, 2015. [DOI] [PubMed] [Google Scholar]
- 31.Scharrer E, Langhans W. Control of food intake by fatty acid oxidation. Am J Physiol Regul Integr Comp Physiol 250: R1003–R1006, 1986. [DOI] [PubMed] [Google Scholar]
- 32.Sclafani A, Zukerman S, Ackroff K. GPR40 and GPR120 fatty acid sensors are critical for postoral but not oral mediation of fat preferences in the mouse. Am J Physiol Regul Integr Comp Physiol 305: R1490–R1497, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sergeyev V, Broberger C, Gorbatyuk O, Hokfelt T. Effect of 2-mercaptoacetate and 2-deoxy-D-glucose administration on the expression of NPY, AGRP, POMC, MCH and hypocretin/orexin in the rat hypothalamus. Neuroreport 11: 117–121, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Singer-Koegler LK, Magluyan P, Ritter S. The effects of low-, medium-, and high-fat diets on 2-deoxy-D-glucose- and mercaptoacetate-induced feeding. Physiol Behav 60: 321–323, 1996. [DOI] [PubMed] [Google Scholar]
- 35.Smith GP, Gibbs J. Cholecystokinin: a putative satiety signal. Pharmacol Biochem Behav 3: 135–138, 1975. [PubMed] [Google Scholar]
- 36.Tanaka H, Yoshida S, Minoura H, Negoro K, Shimaya A, Shimokawa T, Shibasaki M. Novel GPR40 agonist AS2575959 exhibits glucose metabolism improvement and synergistic effect with sitagliptin on insulin and incretin secretion. Life Sci 94: 115–121, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Tang-Christensen M, Larsen PJ, Goke R, Fink-Jensen A, Jessop DS, Moller M, Sheikh SP. Central administration of GLP-1-(7–36) amide inhibits food and water intake in rats. Am J Physiol Regul Integr Comp Physiol 271: R848–R856, 1996. [DOI] [PubMed] [Google Scholar]
- 38.Woods SC, Porte D Jr. The role of insulin as a satiety factor in the central nervous system. Adv Metab Disord 10: 457–468, 1983. [DOI] [PubMed] [Google Scholar]
- 39.Xiong Y, Swaminath G, Cao Q, Yang L, Guo Q, Salomonis H, Lu J, Houze JB, Dransfield PJ, Wang Y, Liu JJ, Wong S, Schwandner R, Steger F, Baribault H, Liu L, Coberly S, Miao L, Zhang J, Lin DC, Schwarz M. Activation of FFA1 mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Mol Cell Endocrinol 369: 119–129, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Yashiro H, Tsujihata Y, Takeuchi K, Hazama M, Johnson PR, Rorsman P. The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets. J Pharmacol Exp Ther 340: 483–489, 2012. [DOI] [PubMed] [Google Scholar]
- 41.Zamarbide M, Etayo-Labiano I, Ricobaraza A, Martinez-Pinilla E, Aymerich MS, Luis Lanciego J, Perez-Mediavilla A, Franco R. GPR40 activation leads to CREB and ERK phosphorylation in primary cultures of neurons from the mouse CNS and in human neuroblastoma cells. Hippocampus 24: 733–739, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X, Yan G, Li Y, Zhu W, Wang H. DC260126, a small-molecule antagonist of GPR40, improves insulin tolerance but not glucose tolerance in obese Zucker rats. Biomed Pharmacother 64: 647–651, 2010. [DOI] [PubMed] [Google Scholar]