

Abstract
Gonadotrophin-releasing hormone (GnRH) is important in reproduction, although some of the mechanisms for its synthesis and release remain elusive. Progress in understanding the GnRH neurone has been hampered by the limited number and diffuse distribution of the neurone in the mammalian brain. Several stable GnRH-expressing cell lines have been developed using in vivo expression of the simian virus 40 T Antigen (TAg), and they have been helpful for the study of gene expression and neuronal function. However, expression of an immortalising gene may interfere with normal cellular function. We developed a novel GnRH-secreting cell line transgenic mouse model suitable for targeted transformation in post-pubertal mice using a tetracycline-regulated TAg transgene. This clonal cell line, GRT, expresses neuronal markers and GnRH. GRT cells grown in medium containing tetracycline-free serum express increasing mRNA levels of GnRH associated with declining levels of TAg expression. The novelty and ultimately the usefulness of this cell line is that TAg expression, which could affect the GnRH neuronal phenotype, can be regulated by tetracycline.
Keywords: LHRH, GnRH, transgenic mice, cell lines, T antigen oncogene
Hypothalamic gonadotrophin-releasing hormone (GnRH) neurones originate outside the central nervous system, in the olfactory placode, and thereafter migrate into the forebrain during prenatal development (1, 2). After reaching their final destination in the septum and preoptic area of the hypothalamus, GnRH neurones project axons to the median eminence to make contacts with the pituitary portal vessels. It is this population of GnRH neurones that is responsible for reproductive function, becoming integral members of the hypothalamic-pituitary-gonadal axis postnatally. This migratory pattern has been well documented in man and other species, and is fundamental for the development of normal reproductive function. Impaired migration of GnRH neurones is the pathogenic factor responsible for hypogonadotrophic hypogonadism that occurs in patients with Kallmann’s syndrome (1, 3–5). Mice with an interrupted migration of GnRH neurones have been shown to develop hypogonadotrophic hypogonadism, resulting in lack of pubertal development, underdeveloped gonads and accessory sex organs and infertility (6).
The difficulty in studying the GnRH neurone has been hampered by its scarce and scattered distribution in the hypothalamus (1, 7). Thus, in recent years, many investigators performing in vitro studies have relied on using the GN and GT1 immortalised GnRH neuronal cell lines, GT1-7 (8) and GN or NLT (6, 9). These both cell lines were obtained from tumours derived from mice in which oncogenesis of the simian virus 40 T Antigen (TAg) was targeted to the GnRH neurone. For both the GN and GT1 cell lines, it is not. These cell lines, derived by TAg immortalisation in transgenic mice, have played a prominent role in characterising the GnRH neurone because of their ability to propagate as a homogenous cell mass in vitro. However, the precise developmental stage that the GnRH neurones were transformed in these cell lines remains unknown. In addition, expression of an immortalising gene may interfere with the normal cellular function of the GnRH neurone. Thus, these constitutively expressing TAg cell lines may not reflect the differentiated nature of the intact GnRH neurone. These considerations potentially limit the usefulness of these cell lines in studying the function of the GnRH neurone.
In the present study, we report the development of a novel cell line derived from a tetracycline-inducible TAg transgene expression transgenic mouse model suitable for targeted transformation of the GnRH neurone in a post-pubertal mouse. This clonal cell line expresses neuronal markers and GnRH. The novelty, and ultimately the usefulness, of these cells lies in the fact that TAg and GnRH expression can be regulated by tetracycline.
Materials and methods
Transgenic mice
The GnRH-TAgTeR transgene was produced in three steps. The GnRH-CRETeR plasmid was constructed by cloning the murine GnRH promoter into the CRETeR vector (10). The CRETeR vector was digested with Bg/II to release the CMV promoter but retaining part of the 2 × Tet operator in the proximal promoter. The proximal portion of the mGnRH promoter was modified by polymerase chain reaction (PCR) amplification of the −3446/+28 mGnRH promoter (generous gift of Dr. Donald DeFranco) to contain the remainder of the 2 × Tet operator and a BamHI site on the 3′ end, and a SgrAI restriction site flanked by a BamHI site on the 5′ end. The BamHI fragment of the PCR product was inserted into the Bg/II sites of the CRETeR vector. The GnRH-TAgTeR vector was then produced by excising the CRE recombinase gene by digesting with MscI and NotI. The SV40 Large TAg oncogene was PCR amplified from the GnRH-TAg vector (6) with a NotI site added to the 5′ side and an MscI site added to the 3′ side. The amplified TAg gene was then inserted into the GnRH-CRETeR vector, essentially replacing the CRE gene. The vector was linearised with SgrAI, which removed much of the vector backbone. The targeting construct (Fig. 1) was injected into pronuclei of fertilised one-cell embryos from CD-1 females by the University of Chicago Transgenic Mouse/Embryonic Stem Cell Facility. Offspring were screened by using PCR of genomic DNA from a phenol/chloroform extraction obtained from tail snips. The DNA was then used in a standard PCR reaction with a primer set to detect mouse TAg (sense: 5′-AACAGAGAGGAATCTTTGCAGC-3′; antisense: 5′-GAGCAAAACAGGTTTTCCTC-3′). These primers span the intron in the TAg fragment and amplify a 648-bp band. PCR products were analysed by gel electrophoresis. Southern blot analysis using a probe against the TAg gene (6) was used to confirm the results of the PCR analysis. Three founder mice were identified that contained the transgene, but only one founder transmitted the gene to F2 generation. F2 offspring were screened for the transgene, and positive siblings were mated. Transgene containing F3 offspring at 4 months of age were treated with a 5 mg slow-release doxycycline (a long acting tetracycline analogue) pellet (Innovative Research of America, Sarasota, FL, USA), with a release time over 21 days of 0.7 mg/day (11). Pellets were inserted subcutaneously into transgenic mice as directed by the manufacturer. At 5 months of age, dissection of the hypothalamus was performed, followed by tissue dissociation using 10 mg/ml collagenase and 10 μg/ml deoxyribonuclease (12).
Fig. 1.

Schematic Illustrating the Strategy Used for Producing the GRT Cells. The GnRH-TAgTeR transgene contains the )3446/+28 bp murine gonadotrophin-releasing hormone (GnRH) promoter fragment modified in the proximal promoter to insert two tetracycline operators (TetO2). The GnRH promoter lies upstream of the large T Antigen (TAg) oncogene. A Kozak consensus site is included at the translation start site. In the same transgene lies the Tet repressor (TetR) gene, derived from the Tn10 transposon, regulated by the CMV promoter. Constitutive expression of TetR results in suppression of GnRH promoter activity, and thus TAg expression, due to TetR binding (TetR ovals) at the tetracycline operators. Doxycycline (black circles) can bind to the TetR protein and cause it to be released from the tetracycline operators resulting in GnRH promoter driven TAg expression.
Cell culture and reagents
Cells were plated onto plastic culture dishes in Dulbecco’s modified Eagle’s medium (DMEM; Cellgro, Hernden, VA, USA) with 10% fetal calf serum (FCS) (Hyclone, Logan, UT, USA) supplemented with 10 mM l-glutamine (Invitrogen, Carlsbad, CA, USA) and antibiotic/antimytotic (Invitrogen). Cell lines were derived through limited-dilution cloning. These cells have been in continuous culture for over 10 months. Because commercial serum products contain tetracycline-derived contaminants that can dramatically affect the inducible regulation of tetracycline systems, experiments to measure the effects of TAg induction in GRT cells were performed in 10% Tet-System Approved FCS (BD Biosciences, San Jose, CA, USA). To observe the effects of stimulating the GRT cells on GnRH gene expression and GnRH secretion, cells were incubated in 56 mM KCl (Sigma-Aldrich, St Louis, MO, USA) for 60 min. To observe the effects of doxycycline, GRT cells were grown for either 24 or 48 h in Doxycycline Hyclate (Sigma-Aldrich) diluted in water to a final concentrations of 2 μg/ml.
GnRH radioimmunoassay
Cells were also grown in six-well plates with 1 ml of medium per well. Medium was collected after 16 h. Radioimmunoassay for GnRH was performed at the Ligand Assay Core of the Baltimore-Chicago Cooperative Centre in Reproductive Research at Northwestern University (Dr Jon Levine, Director). The assay exhibits an inter- and intra-assay coefficient of variation of < 10%. The lower limit of sensitivity of the assay is 0.1 pg/100 μl.
Cell counting
GRT cells were seeded at 1 × 106 cells/plate and were cultured for 30–48 h of growth (until exponential growth achieved) collected with trypsin (Sigma-Aldrich) and counted with a hemacytometer (Hausser Scientific, Horsham, PA, USA). 2 × 106 Cells were used for RNA extractions.
Standard curve and copy number
Mouse GnRH cDNA obtained from a cloned plasmid was used to produce a standard curve by quantitative real-time PCR (qRT-PCR). Quantities ranging from 10−10–10−15 g of GnRH cDNA were used. Absolute copy number of standards was derived by dividing the molecular mass of the GnRH cDNA plasmid (g/mol) into the mass of each standard (g). These values were then multiplied by Avogadro’s number (mol−1) to derive a standard curve of threshold cycle versus copy number. The copy number of GnRH transcript in unknown PCR products were then determined from their cycle threshold (Ct) values on the standard curve.
qRT-PCR
qRT-PCR was performed as previously described (13, 14). Total RNA was harvested from GRT cells by Trizol (Invitrogen) extraction. Two micrograms of RNA was reverse transcribed (iScript cDNA Synthesis Kit; Bio-Rad, Hercules, CA, USA) to produce cDNA. cDNA representing 50 ng of starting RNA was used in each reaction. Twenty-five microlitre PCR reactions were performed using the IQ SybrGreen supermix (Bio-Rad). Reactions were measured using the MyiQ quantitative real time PCR machine (Bio-Rad). Primer sets for mouse GnRH (sense 5′-CCCTTTGACTTTCACATCC-3′ and antisense 5′-GGGTTCTGCCATTTGATCCAC-3′), TAg (sense 5′-GATGCAACTGAGATTCCAACCT-3′ and antisense 5′-GCAATTCTGAAGGAAAGTCC-3′), and a ribosomal 18S control (sense 5′-TGGTTGATCCTGCCAGTAG-3′ and antisense 5′-CGACCAAAGGAACCATAACT-3′) were used. PCR conditions were optimised to generate > 95% PCR efficiency and only those reactions with between 95% and 105% efficiency were included in subsequent analysis. Ct was obtained for each sample. A corrected Ct (DCt) was calculated by subtracting the 18S Ct from the unknown sample Ct for each sample. Relative differences from the control sample were calculated using the formula: fold change = 2^(control ΔCt minus sample ΔCt). PCR products were also analysed by gel electrophoresis.
Immunocytochemistry
GRT cells (2 × 104) were grown on Fibronectin (Sigma-Aldrich) coated 12-mm glass coverslips in a 24-well plate for 24 h, rinsed with phosphate-buffered saline (PBS), and fixed with 3.7% formaldehyde in PBS (pH 7.4) for 30 min, rinsed twice with PBS and placed in blocking solution consisting of 0.1% Triton X-100, 1% bovine serum albumin (BSA) (Sigma-Aldrich) in PBS for 15 min at room temperature. Cells were incubated with GnRH, TAg (Chemicon International Inc., Temecula, CA, USA) or NeuN (Chemicon International Inc.) primary antibodies (1 : 50 000, 1 : 500 and 1 : 250 dilutions respectively) in blocking solution for 1 h at room temperature. The rabbit antibody to GnRH (LR-5) was obtained from Dr Robert Benoit (McGill University Health Center, Montreal, Canada). The antigenic determinants consist of amino acids 2–4 and 7–10 of GnRH (2). GnRH neurones were stained with rabbit polyclonal antibody, followed by Cy3 conjugated secondary antibody. Both TAg and NeuN were stained with mouse monoclonal antibody, followed by anti-mouse Cy2 antibody. Images were captured on a Zeiss microscope Axiovert-200 (× 40 objective) (Carl Zeiss, Thornwood, NY, USA).
Immunohistochemistry
Animals were perfused transcardially with normal saline followed by buffered 4% paraformaldehyde. After fixation, all brains were sunk in 30% sucrose in phosphate buffer and sectioned on a cryostat at 30-lm thickness. Sections were rinsed in PBS (pH 7.4), treated with 0.5% H2O2/1 × PBS solution, rinsed, and blocked with 2% Normal Goat Serum (Vector Laboratories, Burlingame, CA, USA) in PBS with 0.3% Triton X-100 and 1% BSA incubated with anti-GnRH (LR-5) in PBS with 0.3% Triton X-100 and 1% BSA, for 18 h at 4 °C. After rinsing, the tissue was incubated for 1 h at room temperature in biotinylated goat anti-rabbit IgG (heavy and light chains; Vector Laboratories) at a concentration of 1 : 250 in PBS with 0.1% Triton X-100 and 0.3% BSA, rinsed, and incubated for 1 h in avidin-biotin complex solution (‘elite’ ABC kit, 4.5 μl each per ml incubation mixture; Vector Laboratories). Visualisation was performed using DAB substrate kit for peroxidase (Vector Laboratories) according to the company’s protocol.
Statistical analysis
All quantitative RT-PCR studies were replicated at least three times with a triplicate determination of each individual sample. Tukey’s multiple comparison test or a two-way ANOVA with repeated measures was used to determine significance as indicated.
Results
Development of transgenic mice
Four transgenic founder mice carrying the GnRH-TAgTeR gene were obtained. All founder, F1 and F2 mice were fertile, suggesting that the transgene was not expressed in the absence of doxycycline. Previous studies using the promoter of GnRH fused to TAg resulted in tumourigenesis, which interfered with normal sexual maturation and infertility (6, 8). In one study, tumourigenesis occurred with migratory arrest of GnRH neurones (6). None of the mice in the present study displayed evidence of a brain tumour. The anatomical distribution and morphology of the GnRH neurones was normal in the non-induced GnRH-TAgTeR mice (Fig. 2). Four weeks after introduction of the doxycycline pellet, the hypothalamus of one animal was dissected for tissue culture.
Fig. 2.
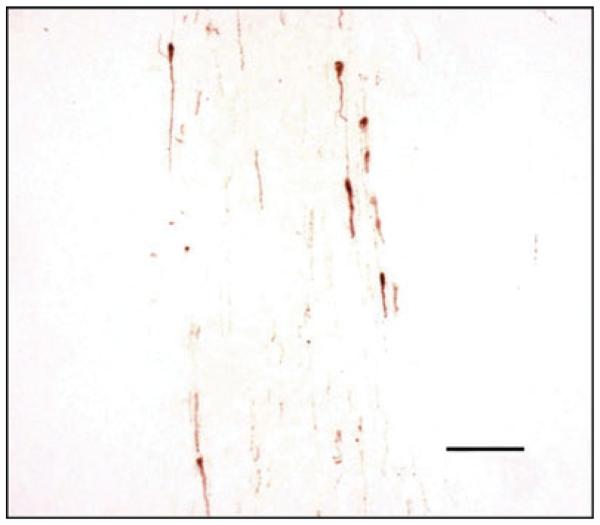
Gonadotrophin-releasing hormone (GnRH) neurones exhibit normal anatomical distribution and morphology in non-induced GnRH-TAgTeR mice. Coronal section at the level of the diagonal band of Broca. GnRH neurones are visualised with DAB and appear brown. Seven GnRH neurones are observed in this representative section. Scale bar = 100 μM.
Derivation of the GRT cell line and characterisation by immunohistochemistry
The dispersed hypothalamus included neural and glial phenotypes that were continually cultured for 4 months by repeated passage on plastic culture dishes using limiting-dilution cloning to separate cell types. GRT cell cultures were established by continuous propagation for 10 months with no change in phenotype. GRT cells attached to culture dishes exhibited a distinct homogeneous neuronal morphology, including the extension of multiple lengthy neurites that were seen contacting distant cells or end in growth cones (Fig. 3A).
Fig. 3.

Immunostaining of GRT cells with neuronal marker (NeuN), T Antigen (TAg) and gonadotrophin-releasing hormone (GnRH) antibody. GRT cells were grown on fibronectin coated coverslips. After washing and permeablising, cells were stained with (A) NeuN, (B) TAg and (C) GnRH primary antibody. (D) Nuclei were stained with DAPI. Cy2 or Cy3 were used as second antibody as described in the Materials and methods.
Cells were immunostained for GnRH, TAg or the neuronal marker, NeuN
GnRH was localised to the cytoplasm and in the neurites of cells (Fig. 3C). TAg immunostaining was nuclear except where shown in dividing cells (Fig. 3B). Neuronal staining was observed in nuclei, cell bodies and neurites (Fig. 3A). Negative staining of cells was observed with normal rabbit serum followed by a Cy3 conjugated secondary antibody. Nuclei stained blue after DAPI staining. These cells didot show nonspecific Cy3 staining (Fig. 3D).
GRT cells express GnRH and TAg
qRT-PCR of total RNA from GRT cells, as well as the other two established cell lines, GT1-7 and GN11, was performed using exonic primers designed to include part of exon 1, exon 2, which codes for the GnRH decapeptide, and part of exon 3. This primer pair spans introns 1 and 2 (Fig. 4A). As confirmation for the specificity of the PCR reaction, melting curve analysis was performed and the PCR product was run on an agarose gel, which demonstrated the presence of GnRH mRNA in the three cell lines with a corresponding band of 193 bp (Fig. 4B). As a negative control, RNA that was not reverse transcribed was used. An expression level of GnRH from these cell lines was measured from a standard curve using known quantities of a GnRH cDNA containing plasmid. The data are shown as a threshold cycle for the detection of product from the three cell types (Fig. 4C) and tabulated as copy number derived from representative molar quantities (Fig. 4D). GnRH gene expression in GRT cells is approximately eight-fold higher in GRT cells than GN cells. Radioimmunoassay was performed to detect GnRH secretion from GRT cells. Secretion levels were compared with the GT1-7 and GN11 cell lines (Fig. 4E). To further demonstrate neurone-like behaviour. GRT cells were stimulated with 56 mM KCl. qRT-PCR of cellular RNA from GRT cells was performed as described above. The baseline value is represented by cells grown without KCl treatment. GnRH gene expression levels increased three-fold after 60 min (Fig. 4F). Additionally, GnRH secretion from the cells was increased two-fold relative to untreated cells within 1 h (Fig. 4F).
Fig. 4.

Gonadotrophin-releasing hormone (GnRH) gene expression and secretion in GnRH-expressing neuronal cell lines. (A) Location of primers on the GnRH gene designed to amplify GnRH cDNA (193 bp) in the GT1-7, GN11, and GRT cell lines. (B) Agarose gel products from a quantitative real-time polymerase chain reaction using primers for the GnRH gene in the respective cell lines. A 193-bp band is shown in all cell lines. RNA that was not reverse transcribed was used as a negative control. (C) Logarithmic graph charting quantities of GnRH mRNA expression in GT1-7, GN11 and GRT cells (triplicate black circles). A standard curve was produced using known quantities of GnRH cDNA contained in a pGEMTeasy plasmid (Green line). (D) Copy number of GnRH in GT1-7, GN11 and GRT cells derived by calculating molar quantities of mRNA levels of each cell type. RNA that was not reverse transcribed was used as a negative control. (E) GnRH secretion (pg/ml) in GT1-7, GRT and GN11 cells. (F) GnRH gene expression and secretion in GRT cells treated with 56 mM KCl for 60 min.
GRT cells grown in serum after withdrawal of tetracycline exhibit decreased TAg expression and cell growth
To determine the effects of tetracycline on TAg gene expression, cultured cells were split into dishes and maintained in either DMEM supplemented with tetracycline-free FCS (10% Tet System Approved FCS) or standard FCS. Growth rates for the cells varied from a doubling time of 15 h for cells grown in tetracycline free serum to 10 h for cells grown in media in the presence of tetracycline. After exposure to tetracycline, cell morphology demonstrated a more spread out shape and granular appearance compared to cells grown in the absence of tetracycline (Fig. 5A). A spindle-like neuronal phenotype with extension of multiple neurites contacting distal cells is characteristic of GRT cells grown in tetracycline-free serum (Fig. 5B). The cells were maintained for 2 weeks without tetracycline with no signs of apoptosis as evaluated by light microscopic analysis.
Fig. 5.

Morphology of GRT cells. Dark field images of GRT cells grown in (A) serum containing tetracycline, (B) tetracycline free media [10% Tet System Approved fetal calf serum (FCS)] and (C) serum with the addition of 2 μg/ml doxycycline. DMEM, Dulbecco’s modified Eagle’s medium.
To demonstrate TAg expression in GRT cells, cells were grown in serum with or without tetracycline. qRT-PCR of cellular RNA was performed using an intron spanning sense primer of the TAg gene and a 3′ antisense primer (Fig. 6A). The presence of TAg mRNA was observed in GRT cells and in the GT1-7 and GN11 cell lines, which were all grown in media containing tetracycline, with a corresponding band of 192 bp (Fig. 6B). As a negative control, RNA that was not reverse transcribed was used. The expression level of TAg from the cell lines was measured using qRT-PCR. There was a 54% decrease in TAg mRNA by 8 days and a 57% decrease by 14 days after media containing standard FCS was replaced with media devoid of tetracycline. This difference was significantly different from TAg expression of cells grown in standard serum conditions (*P < 0.01 versus baseline; Fig. 7A). The data shown are expressed as the mean ± SEM (n = 3).
Fig. 6.
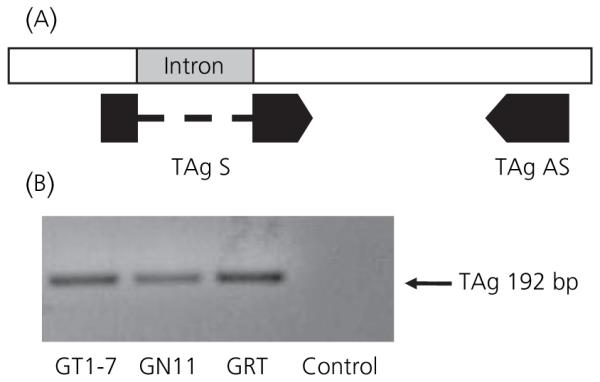
T Antigen (TAg) gene expression in GRT cells. (A) Location of primers on the TAg gene designed for qRT-PCR to amplify the TAg fragment (192 bp) in GT1-7 GN11, and GRT cell lines. Primers were designed to span the intron of the TAg fragment to avoid amplification of possible genomic DNA contamination of RNA extractions. (B) Agarose gel products from a quantitative real-time polymerase chain reaction using primers utilising primers for the TAg gene in the respective cell lines. RNA that was not reverse transcribed was used as a negative control.
Fig. 7.

T Antigen (TAg) and gonadotrophin-releasing hormone (GnRH) gene expression in GRT cells grown in media containing tetracycline-free fetal serum or with the addition of doxycycline. GRT cells were thawed and grown separately in either Dulbecco’s modified Eagle’s medium (DMEM) with tet-free fetal calf serum (FCS) or DMEM with standard FCS. Quantitative real-time polymerase chain reaction (qRT-PCR) was then performed to detect TAg and GnRH gene expression. (A) Time course of TAg gene expression in GRT cells grown in DMEM with tet-free FCS or DMEM with standard FCS. The data shown are expressed as the mean ± SEM (n = 3) (*P < 0.01 versus baseline). (B) During the same time period, GnRH gene expression is similarly quantitated. GnRH gene expression in GRT cells grown in media containing tetracycline-free serum versus serum containing tetracycline. The data shown are expressed as the mean ± SEM (n = 3). (*P < 0.005 versus baseline). (C) GRT cells were grown in 2 μg/ml doxycycline for 48 h and TAg and GnRH mRNA measured by qRT-PCR. RNA was harvested by Trizol extraction, followed by qRT-PCR. The baseline value is represented by the control plate that was grown without doxycycline treatment. Forty-eight hours of doxycycline treatment displayed a four-fold increase in TAg gene expression. The data shown are expressed as the mean ± SEM (n = 3). (*P < 0.05 versus baseline, **P < 0.001 versus baseline). As a negative control, RT-PCR was performed without reverse transcriptase and no product was detected (data not shown).
As TAg expression decreases, GnRH expression increases in GRT cells grown in media devoid of tetracycline
To demonstrate the effects of tetracycline on expression of GnRH, measurements of GnRH mRNA by qRT-PCR were performed from cellular RNA retrieved in the previous experiment. GnRH gene expression revealed a concomitant 50% increase at 8 days and a 95% increase at 14 days (*P < 0.005 versus baseline; Fig. 7B). This experiment was performed in triplicate and data are shown as the mean ± SEM (n = 3).
Doxycycline induces TAg expression
To demonstrate the ability of doxycycline to induce TAg expression; cells were treated with 1 μg/ml and 2 μg/ml doxycycline for 24 or 48 h. The baseline value is represented by the control plate that was grown without doxycycline treatment. There was no change at the lower dose of doxycycline (data not shown) but a four-fold increase in TAg mRNA after treatment with 2 μg/ml doxycycline for 48 h (Fig. 7C). As a control, qRT-PCR was performed without reverse transcriptase and no product was detected (*P ≤ 0.05, **P ≤ 0.001; Fig. 7C). GnRH secretion did not change in response to the addition of doxycycline to the cells.
Discussion
GnRH-secreting cell lines were developed by targeting expression of a regulated GnRH gene promoter in transgenic mice. In the present study, transgenic mice were developed in which two copies of the Tet operator replaced the transcriptional start site of GnRH. Binding of a constitutively expressed Tet repressor (TetR) blocks GnRH promoter activity, and hence, TAg expression. Tetracycline addition causes release of TetR and permits GnRH-specific gene expression of TAg. This genetically engineered animal achieved induced GnRH neuronal expression in vivo from which a cell line was derived. GnRH neuronal expression was accomplished by using a promoter that had previously been shown to target GnRH neurones (15). The expression of TAg, controlled by the absence or addition of tetracycline in vivo, was also found in the resultant cultured cells.
Several studies have identified cell-specific elements in mouse, rat and human GnRH promoters (15–20). However, cell type specific immortalisation of GnRH neurones using constitutive expression of the TAg transgene has been shown to have deleterious effects on neuronal migration and phenotypic hypogonadism (6). Both the GN and GT cell lines were harvested from mice containing a transgene that consisted of a GnRH promoter fragment fused directly to the TAg gene. Thus, the timing of transformation could have been at any point from embryonic day 10.5 when GnRH expression commences until weeks, or days, before the date of harvesting of the tumour. For the GN cell line, the tumour was identified in the olfactory region, indicating that the tumour formation had interfered with GnRH neuronal migration and thus transformation occurred during embryonic life. For the GT cell line, the tumour was harvested after signs of neurological distress from a hypothalamic tumour. It is unclear from the original report (8) at what age this occurs, but there is mention that the mice were infertile, and indeed had not undergone puberty. Therefore it is assumed that the GnRH neurones were transformed pre-pubertally, but following neuronal migration. To circumvent these problems, we developed a system in which expression of TAg is kept silent for an extended period of time (21), induced at a desired point in time, and then resilenced. Other promoters have been used to regulate gene expression in vivo. Inducible promoters, such as heat shock, metallothionein and murine mammary tumour virus, which are regulated by temperature, zinc, or dexamethasone, respectively, are frequently associated with a high basal level of expression variability and often relatively low levels of induction of the transgene and potential cellular toxicity (22–24). Conditional gene expression in vivo using a CRE/LoxP recombination has also been used to delete endogenous genes in a tissue specific and time specific manner (25). However, this system leads to irreversible effects after induction. Drug or ligand-dependent systems have been developed involving the use of a chimeric transcriptional activator that reversibly activates a target gene in response to the administration of the inducing agent. The Escherichia coli tetracycline resistance operon has been used to generate cell lines with tightly regulated gene expression in response to tetracycline (11, 23, 26, 27). This system has been used effectively in transgenic mouse strains bearing the SV40 TAg under the control of a Tet operator and the tetracycline-responsive transcriptional activator (tTA) under the control of a cell-specific promoter. In the absence of tetracycline, tTA activates the TAg gene through binding to promoter sequence (27–31). We have modified this system in the development of a single mouse strain bearing both control elements (2 × Tet operator) and the control protein (TetR). Treatment with doxycycline releases the TetR from the GnRH promoter and permits TAg expression. A major advantage of this vector is that all of the genetic components necessary for targeting, regulation and selection are incorporated into a single plasmid. The strict regulation of the recombinant genes is simply achieved by the addition or removal of tetracycline or doxycycline.
The expression of immortalising genes has been reported to strongly influence cellular characteristics and to activate a tremendous number of genes in various cell systems(32–35), including an estimated 389 genes in a murine fibroblast model(32). Genes that were regulated in the murine fibroblast include those important for controlling cell proliferation, nucleotide metabolism, cell cycle and other processes (32). GRT cells are valuable because they are a product of immortalisation using a vector that allows conditional expression of TAg and the additional advantage of reversibility. The induction of the immortalising gene, TAg, leads not only to immortalisation and cell proliferation, but also may influence cell morphology and physiology in undesirable ways (32). Controlling TAg expression solves this problem because it reverses the proliferative phenotype in a reproducible manner. Although, in the repressed state of doxycycline withdrawal, a residual TAg level is observed, it does significantly decrease the proliferative ability of the cell. However, we cannot exclude the possibility that this low basal level of TAg expression deregulates the expression of some genes. Under the induction conditions, GRT cells exhibit properties similar to those of constitutively immortalised cells. With the withdrawal of the inducing agent by growth of the cells in tetracycline free media, GRT cell proliferation decreases with a concomitant increase in GnRH expression (Fig. 7A and B). When cells are treated with doxycycline, there was a significant increase of TAg expression at 24 and 48 h above that seen in normal serum (Fig. 7C); however, this was not associated with a reduction in GnRH expression (Fig. 7B).
The GRT cell line has characteristics of a differentiated cell. It exhibits a neuronal morphology with spontaneous extension of neurites, expresses specific neural markers and expresses GnRH. The cell line maintains a highly differentiated phenotype in long-term standard culture conditions. Thus, this conditionally immortalised cell line, GRT, has advantageous properties over constitutively immortalised cells and will allow for new studies of GnRH neurones in culture where the potentially undesirable effect of TAg expression will be minimised. Moreover, the presence of a GnRH-inducible TAg transgene in these mouse lines will potentially allow for expression of TAg during a specific developmental stage using the appropriate timed administration of doxycycline.
Acknowledgements
The authors are much indebted to Dr Donald DeFranco for providing the murine GnRH promoter. The authors would also like to thank Dr Pamela Mellon for providing the GT1-7 cell line. The technical assistance of Lan Su is also acknowledged. Finally, the assistance of George Park and Daniel Diaczok with the assembly and critical analysis of the manuscript is greatly appreciated.
References
- 1.Wray S, Grant P, Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci USA. 1989;86:8132–8136. doi: 10.1073/pnas.86.20.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–164. doi: 10.1038/338161a0. [DOI] [PubMed] [Google Scholar]
- 3.Tsai PS, Gill JC. Mechanisms of disease: insights into X-linked and autosomal-dominant Kallmann syndrome. Nat Clin Pract Endocrinol Metab. 2006;2:160–171. doi: 10.1038/ncpendmet0119. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Martinez D, Hu Y, Bouloux PM. Ontogeny of GnRH and olfactory neuronal systems in man: novel insights from the investigation of inherited forms of Kallmann’s syndrome. Front Neuroendocrinol. 2004;25:108–130. doi: 10.1016/j.yfrne.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Bhagavath B, Podolsky RH, Ozata M, Bolu E, Bick DP, Kulharya A, Sherins RJ, Layman LC. Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertil Steril. 2006;85:706–713. doi: 10.1016/j.fertnstert.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 6.Radovick S, Wray S, Lee E, Nicols DK, Nakayama Y, Weintraub BD, Westphal H, Cutler GB, Jr, Wondisford FE. Migratory arrest of gonadotropin-releasing hormone neurons in transgenic mice. Proc Natl Acad Sci USA. 1991;88:3402–3406. doi: 10.1073/pnas.88.8.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King JC, Tobet SA, Snavely FL, Arimura AA. LHRH imminopositive cells and their projections to the median eminence and organum vasculosum of the lamina teminalis. J Comp Neurol. 1982;209:287–300. doi: 10.1002/cne.902090307. [DOI] [PubMed] [Google Scholar]
- 8.Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-e. [DOI] [PubMed] [Google Scholar]
- 9.Zhen S, Dunn IC, Wray S, Liu Y, Chappell PE, Levine JE, Radovick S. An alternative gonadotropin-releasing hormone (GnRH) RNA splicing product found in cultured GnRH neurons and mouse hypothalamus. J Biol Chem. 1997;272:12620–12625. doi: 10.1074/jbc.272.19.12620. [DOI] [PubMed] [Google Scholar]
- 10.Naik K, Pittman IVI, Wolfe A, Miller RS, Radovick S, Wondisford FE. A novel technique for temporally regulated cell type-specific Cre expression and recombination in the pituitary gonadotroph. J Mol Endocrinol. 2006;37:63–69. doi: 10.1677/jme.1.02053. [DOI] [PubMed] [Google Scholar]
- 11.Schultze N, Burki Y, Lang Y, Certa U, Bluethmann H. Efficient control of gene expression by single step integration of the tetracycline system in transgenic mice. Nat Biotechnol. 1996;14:499–503. doi: 10.1038/nbt0496-499. [DOI] [PubMed] [Google Scholar]
- 12.Ben-Johnathon N, Peleg E, Hoefer MT. Optimization of culture conditions for short-term pituitary cell culture. Methods Enzymol. 1983;103:249–257. doi: 10.1016/s0076-6879(83)03016-5. [DOI] [PubMed] [Google Scholar]
- 13.Divall SA, Radovick S, Wolfe A. Egr-1 binds the GnRH promoter to mediate the increase in gene expression by insulin. Mol Cell Endocrinol. 2007;270:64–72. doi: 10.1016/j.mce.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HH, Divall SA, Deneau RM, Wolfe A. Insulin regulation of GnRH gene expression through MAP kinase signaling pathways. Mol Cell Endocrinol. 2005;242:42–49. doi: 10.1016/j.mce.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Kim HH, Wolfe A, Smith GR, Tobet SA, Radovick S. Promoter sequences targeting tissue-specific gene expression of hypothalamic and ovarian gonadotropin-releasing hormone in vivo. J Biol Chem. 2002;277:5194–5202. doi: 10.1074/jbc.M110535200. [DOI] [PubMed] [Google Scholar]
- 16.Wolfe AM, Wray S, Westphal H, Radovick S. Cell-specific expression of the human gonadotropin-releasing hormone gene in transgenic animals. J Biol Chem. 1995;271:20018–20023. doi: 10.1074/jbc.271.33.20018. [DOI] [PubMed] [Google Scholar]
- 17.Wolfe A, Kim HSD, Tobet SA, Radovick S. Identification of a discrete promoter region of the human GnRH gene that is sufficient for directing GRT GnRH expressing cell line 1037 neuron specific expression: a role for POU homeodomain transcription factors. Mol Endocrinol. 2002;16:435–449. doi: 10.1210/mend.16.3.0780. [DOI] [PubMed] [Google Scholar]
- 18.Lawson MA, Whyte DB, Eraly SA, Mellon PL. Hypothalamus-specific regulation of gonadotropin-releasing hormone gene expression. Recent Prog Horm Res. 1995;50:459–463. doi: 10.1016/b978-0-12-571150-0.50037-8. [DOI] [PubMed] [Google Scholar]
- 19.Kepa JK, Wang C, Neeley CI, Raynolds MV, Gordon DF, Wood WM, Wierman ME. Structure of the rat gonadotropin-releasing hormone (rGnRH) gene promoter and functional analysis in hypothalamic cells. Nucleic Acid Res. 1992;20:1393–1399. doi: 10.1093/nar/20.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kepa JK, Spaulding AJ, Jacobsen BM, Fang Z, Xiong X, Radovick S, Wierman ME. Structure of the distal human gonadotropin releasing hormone (hGnRH) gene promoter and functional analysis in Gt1-7 neuronal cells. Nucleic Acids Res. 1996;24:3614–3620. doi: 10.1093/nar/24.18.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fedorov LM, Tyrsin OY, Krenn V, Chernigovskaya EV, Rapp UR. Tet-system for the regulation of gene expression during embryonic development. Transgenic Res. 2001;10:247–258. doi: 10.1023/a:1016632110931. [DOI] [PubMed] [Google Scholar]
- 22.Jamieson BD, Zack JA. Murine models for HIV disease. AIDS. 1999;13(Suppl. A):S5–S11. [PubMed] [Google Scholar]
- 23.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 24.Agha-Mohammadi S, Lotze MT. Regulatable systems: applications in gene therapy and replicating viruses. J Clin Invest. 2000;105:1177–1183. doi: 10.1172/JCI10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sauer B, Henderson N. Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol. 1990;2:441–449. [PubMed] [Google Scholar]
- 26.Zhu Z, Ma B, Homer RJ, Zheng T, Elias JA. Use of the tetracycline-controlled transcriptional silencer (tTS) to eliminate transgene leak in inducible overexpression transgenic mice. J Biol Chem. 2001;276:25222–25229. doi: 10.1074/jbc.M101512200. [DOI] [PubMed] [Google Scholar]
- 27.Efrat S, Fusco-DeMane D, Lemberg H, al EO, Wang X. Conditional transformation of a pancreatic beta-cell line derived from transgenic mice expressing a tetracycline-regulated oncogene. Proc Natl Acad Sci USA. 1995;92:3576–3580. doi: 10.1073/pnas.92.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiota G, Rhoads DB, Wang TC, Nakamura T, Schmidt EV. Hepatocyte growth factor inhibits growth of hepatocellular carcinoma cells. Proc Natl Acad Sci USA. 1992;89:373–377. doi: 10.1073/pnas.89.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandgren EP, Quaife CJ, Pinkert CA, Palmiter RD, Brinster RL. Oncogene-induced liver neoplasia in transgenic mice. Oncogene. 1989;4:715–724. [PubMed] [Google Scholar]
- 30.Manickan E, Satoi J, Wang TC, Liang TJ. Conditional liver-specific expression of simian virus 40 T antigen leads to regulatable development of hepatic neoplasm in transgenic mice. J Biol Chem. 2001;276:13989–13994. doi: 10.1074/jbc.M009770200. [DOI] [PubMed] [Google Scholar]
- 31.Held WA, Mullins JJ, Kuhn NJ, Gallagher JF, Gu GD, Gross KW. T antigen expression and tumorigenesis in transgenic mice containing a mouse major urinary protein / SV40 T antigen hybrid gene. EMBO J. 1989;8:183–191. doi: 10.1002/j.1460-2075.1989.tb03363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.May T, Hauser H, Wirth D. Transcriptional control of SV40 T-antigen expression allows a complete reversion of immortalization. Nucleic Acids Res. 2004;32:5529–5538. doi: 10.1093/nar/gkh887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seinsoth S, Uhlmann-Schiffler H, Stahl H. Bidirectional DNA unwinding by a ternary complex of T antigen, nucleolin and topoisomerase I. EMBO Rep. 2003;4:263–268. doi: 10.1038/sj.embor.embor770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slack A, Cervoni N, Pinard M, Szyf M. DNA methyltransferase is a downstream effector of cellular transformation triggered by simian virus 40 large T antigen. J Biol Chem. 1999;274:10105–10112. doi: 10.1074/jbc.274.15.10105. [DOI] [PubMed] [Google Scholar]
- 35.Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, Freire R, Entwistle A, Golemis EA, Roberts TM, Jat PS, Gjoerup OV. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci USA. 2004;101:947–952. doi: 10.1073/pnas.0308006100. [DOI] [PMC free article] [PubMed] [Google Scholar]