

Abstract
Prion diseases or transmissible spongiform encephalopathies are fatal neurodegenerative diseases characterized by the aggregation and deposition of the misfolded prion protein in the brain. α-synuclein (α-syn)-associated multiple system atrophy has been recently shown to be caused by a bona fide α-syn prion strain. Several other misfolded native proteins such as β-amyloid, tau and TDP-43 share some aspects of prions although none of them is shown to be transmissible in nature or in experimental animals. However, these prion-like “prionoids” are causal to a variety of neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. The remarkable recent discovery of at least two new α-syn prion strains and their transmissibility in transgenic mice and in vitro cell models raises a distinct question as to whether some specific strain of other prionoids could have the capability of disease transmission in a manner similar to prions. In this overview, we briefly describe human and other mammalian prion diseases and comment on certain similarities between prion and prionoid and the possibility of prion-like transmissibility of some prionoid strains.
Keywords: Neurodegeneration, prion, prionoid, protein aggregate
Looking back, a half a century ago Carlton Gajdusek et al. first reported the successful transmission of kuru to an experimental animal in 1966.[1] Subsequently, Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), and familial fatal insomnia (FFI) were transmitted to nonhuman primates or transgenic (Tg) mice. Kuru, CJD, GSS, and FFI were eventually found to be caused by PrPsc prions that were initially discovered in hamsters with experimental scrapie.
Milton Shy and Glenn Drager in 1960 described two male patients suffering from autonomic insufficiency, along with a movement disorder that resembled Parkinson's disease (PD).[2] Nine years later, Graham and Oppenheimer suggested, based on overlapping clinical features that Shy-Drager syndrome should be combined with striatonigral degeneration and olivopontocerebellar atrophy and that these three entities be called multiple system atrophy (MSA).[3] Subsequently, the brains of MSA patients as well as Lewy bodies from the brains of PD patients were shown to harbor silver-positive glial (MSA) and neuronal (PD) intracytoplasmic inclusions, which exhibited positive immunostaining for α-synuclein (α-syn).[4] Parallel molecular genetic investigations discovered a genetic linkage between the A53T point mutation in asnl gene that encodes α-syn and inherited PD.[5] Most recently, Stanley Prusiner et al. have demonstrated the successful transmissibility of MSA by a unique α-syn strain to TgM83+/- mice expressing human asnl (A53G) mutation,[6] thus demonstrating α-syn as a new human prion and MSA as a new prion disease.[6]
Similar to sporadic CJD and MSA, late-age neurodegenerative diseases, including Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and PD, are characterized by the coalescence of misfolded protein oligomers into highly ordered protein aggregates in the affected brain. Just like prion, aggregates of misfolded proteins progressively spread to the structurally- or functionally-related neuronal network, resulting in progressive neuronal loss as the disease progresses, a characteristic hallmark of all neurodegenerative diseases.[7,8] Additionally, it is becoming clear that the chemical identity and topography of each of these misfolded proteins is unique and that the principle governing protein aggregation and deposition into the affected brain is strikingly stereotypical in each of the prion and prionoid misfolding diseases (PMDs).[9] It is also becoming obvious that these misfolded protein aggregates can spread from cell to cell,[7,8,9,10,11,12] eventually affecting the whole system of neuronal network similar to prion diseases. For these reasons, these misfolded proteins are collectively called prion-like “prionoids.”[9,12] However, they are not found to transmit between individuals or to experimental models so far and thus, they are not strictly infectious in nature. They are rather prion-like misfolded proteins and further research will determine if some specific strain of prionoids can behave as true transmissible prions. This overview focuses on the recent advances in understanding the mechanism of prion and prionoid formation and their spread and disease causation in PMDs.
Misfolded Protein Monomers, Oligomers, and Higher Order Aggregates
Prion diseases or transmissible spongiform encephalopathies (TSEs) are the best characterized PMDs. Prion diseases or TSEs can be genetic, acquired, or sporadic. Prion diseases are caused by the conversion of the normal cellular prion protein (PrPc) into a pathogenic β-sheet-enriched prion isoform designated as PrPsc, which is able to self-propagate by recruiting native PrPc molecules.[13] This conformational change confers PrPsc with an increased tendency to aggregate, high resistance to heat and chemical denaturation, insolubility in nonionic detergents, and resistance to protease digestion.[9,13,14,15]
The term “prion” (protein infectious particle) was first articulated by Prusiner in the “prion only hypothesis” of TSEs in 1982.[15] Subsequently, a number of studies have supported this concept, including the successful induction of neurodegenerative disease in experimental animals just from the recombinant prion forms of proteins.[16] Whether misfolded PrPsc is itself an infectious agent or it requires further specific posttranslational changes or other cofactors for infectivity remains unresolved. Nevertheless, prion diseases are entirely dependent on the expression of endogenous PrPc (for template induction), as evidenced by the fact that prnp gene knockout mice are totally resistant to experimental prion disease induction.[17] The mechanism by which PrPc converts into PrPsc and adopts the capability to self-template and self-propagate is not well-understood. However, PrPc can fold into a variety of heat-resistant conformers whose mixture in a relative proportion may result in more than one prion strain.[7,11,15] Every conformer may not be pathogenic but each pathological prion strain displays a specific disease phenotype including incubation times, clinical signs, brain PrPsc deposition-patterns, and histopathological lesions, all of which is faithfully recapitulated upon serial passages within the same Tg host genotype. Thus, existence of a variety of prion strains explains the spectrum of disease phenotypes such as CJD, GSS, FFI, and variant CJD (vCJD).[9,13,14,18,19,20] However, the molecular mechanisms by which one or more PrPsc conformer is/are produced and selected has not been yet clearly elucidated.
Nonprion PMDs, including AD, FTD, PD, and ALS, resemble prion diseases in that the alteration in the secondary structure of proteins leads to the formation of abnormally folded conformers and their deposition and propagation in the neuronal network is the pathological substrate of these disease phenotypes.[7,8,9,10,11,12] However, apart from an important exception of α-syn in MSA,[6] all nonprion PMDs differ from putative prion diseases in that the disease transmission in the former has not yet been demonstrated.
Most existing evidence indicates a common model of disease progression in all PMDs, including in putative prion diseases [Figure 1]. Folded or partially unfolded conformer(s) of a disease-linked protein can template the native protein monomers and they interact with each other to form cross-beta sheets.[9,13,14,18,19,20] Two related models of prion self-templation and propagation and disease causation are proposed. In prion diseases, abnormally the nuclear polymerization model proposed by Jarrett and Lansbury (1993), the PrPsc and PrPc are in a reversible thermodynamic equilibrium and in the presence of stable oligomeric PrPsc aggregates, the change from PrPc to PrPsc is favored.[21] In this model, PrPsc aggregates (seeds) are indispensable for prion spread. In Prusiner's template-assisted model (1991), a high energy barrier prevents spontaneous conversion of PrPc into PrPsc and specifically, PrPsc recruits and converts PrPc to form nascent PrPsc.[22] Homodimers of PrPsc-PrPsc then dissociate to allow the formation of a new heterodimer (PrPsc-PrPc) and the generation of further PrPsc molecules. In Prusiner's model, the PrPsc aggregate seeds are not considered essential for the prion conversion and disease propagation process. Debate still exists as to whether it is the accumulation of PrPsc or possibly an intermediate form of the prion during the conversion of PrPc into PrPsc that is responsible for neurotoxicity and neurodegeneration.
Figure 1.
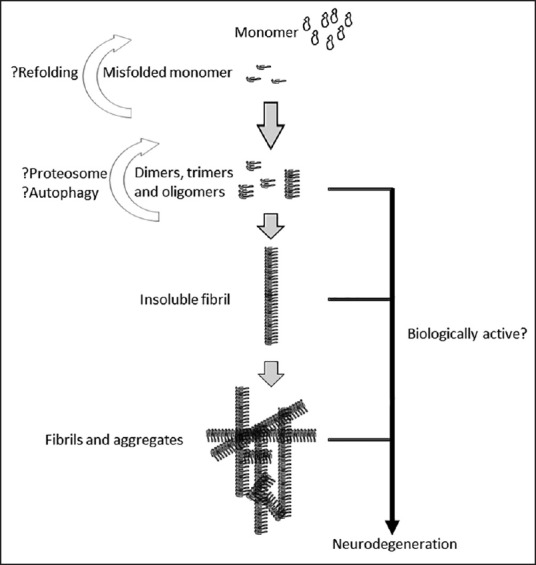
The life cycle of protein monomers, oligomers, and aggregates in prions and other protein misfolding diseases (PMDs)
Since PrPsc accretion typically occurs along a single axis, its product is not a three-dimensional (3D) crystal but rather filamentous structures sometimes called fibrillary filaments. Multiple fibrillary filaments can interact with each other and can form higher order fibrillary aggregates [Figure 1], which following appropriate staining can be visualized by light microscopy. It is unclear whether misfolded oligomers or fibrillary fragments liberated from larger aggregates form additional seeds that possess templating and therefore, self-perpetuating activity of their own. Of note, this model of cyclical amplification and spread does not entirely explain the toxicity associated with the deposition of protein aggregates. It appears likely that the PrPsc oligomers and seeds or the larger aggregates or both cause neuronal toxicity and loss. However, the exponential cascade of prion production and spread explains the rapid progression of CJD with typical disease duration of a few months from onset to death.[14] In principle, substances that reduce or reverse protein misfolding and aggregate formation could possess therapeutic potential.
Prion Diseases in Humans and Other Mammals
Prion diseases or TSEs are fatal neurodegenerative diseases that affect a diversity of mammalian species including CJD, GSS, FFI, vCJD, variably protease-sensitive prionopathy,[23] and prion disease-associated with diarrhea and autonomic neuropathy[24] in humans, scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle, feline spongiform encephalopathy (FSE) in cats, transmissible mink encephalopathy in mink, and chronic wasting disease in deer and elk.[14,19,20,25] PrPc encoded by prnp gene is universally conserved throughout evolution in mammals. PrPc is mostly expressed in the central nervous system but also in the lymphoreticular tissue, skeletal muscle, heart, kidney, digestive tract, skin, mammary gland, and endothelia.[19,20] Despite its ubiquitous expression and distribution, the function of PrPc is not yet clear.
Each of the prion diseases can occur as an inherited disorder (vertical transmission), arise spontaneously, or be acquired by infection (horizontal transmission). Transmissions within the same animal species and occasionally also between different species have been reported for some prion diseases and at least one of them, the BSE, is considered a zoonosis (vCJD) now.
Scrapie is a naturally occurring TSE affecting sheep, goats, and mouflons and it is endemic in many countries worldwide. After a long incubation period (2–5 years), scrapie heralds with behavioral changes, trembling, gait incoordination, weight loss, emaciation, visual impairment, and pruritus, and wool loss.[19,20] The animal usually dies in 1-6 months after the disease onset. Although vertical transmission in scrapie is known, the most prevalent route of prion infection is contact transmission between ewes and her lambs around the time of birth.[20] Additionally, the high resistance of scrapie prion agent against denaturing factors contributes to its persistence in the environment, i.e., in soil, and consequently favors a horizontal transmission within the sheep and goat herds.[25] Rapid loss of sheep and goat herds and the attendant economic loss have prompted their contact isolation as disease control and eradication measure. Additionally, genotype-resistant animal breeding programs have emerged as a tool to control and eradicate scrapie in sheep and goat herds.[26]
BSE, commonly known as “mad cow disease,” was first reported in the UK in 1986 and soon spread worldwide, becoming endemic in many countries. BSE-infected cattle display more than 2 years of incubation period, progressive neurodegeneration affecting the cerebellum and cerebrum, and death in a few months.[27] The practice of feeding cattle with meat and bone meal contaminated with infectious prions was proposed as the most likely mechanism for the BSE epidemic. BSE agent has demonstrated a high capacity to cross species' barriers.[19,28] During the BSE epidemic of the 1980s, not only did it spread to humans with the emergence of the now well-known vCJD but also to cats and a variety of zoo animals, probably including FSE in cats. BSE has been experimentally transmitted to mice, hamsters, sheep, goats, pigs, minks, and nonhuman primates with a high degree of efficiency. Interestingly, upon serial passage in sheep, neurotropic BSE becomes more lymphotropic and becomes transmissible by the injection of lymphoid-blood tissue to human-prnp (PrPc) Tg mice.[19,28] This has led to regulatory restrictions prohibiting the use of processed animal protein in feed to all livestock.
Prionoids and Other Protein Misfolding Diseases
The ability to misfold and self-propagate is not exclusive to prion proteins. Most later-age neurodegenerative disorders are associated with the accumulation of misfolded abnormal forms of specific proteins in the nervous system of humans as well as animals. This heterogeneous group of diseases, prionoid diseases, is caused by the conformational change of physiologically soluble proteins into a β-sheet-enriched forms.[9] Similar to prions, such abnormal conformational change triggers insolubility, aggregation propensity, and resistance of protein aggregates to physical denaturants, favoring their deposition in the nervous system and disruption of neuronal/brain function.
To date, over 20 different misfolding proteins have been reported in humans and animals, including α-syn in in PD and MSA, amyloid precursor protein (APP), and tau in AD, TDP-43 in ALS an FTD, huntingtin in Huntington disease, and amyloid polypeptide in systemic amyloid disorders.[9] The exact mechanism by which these misfoldings and aggregations occur remains unknown but is reminiscent of prion replication. Further, evidence supporting prion-like infectious properties of α-syn has been recently reported.[6] However, in contrast to prions, infectious transmission between individuals has not yet been documented with α-syn or any other prionoid so far.
α-synuclein: PD and MSA are neurodegenerative disorders characterized by the accumulation of protein aggregates called Lewy bodies in the dopaminergic neurons of the substantia nigra (PD) and intracytoplasmic inclusions in the cytoplasm of glial (mainly oligodendroglia) cells in the brain (MSA).[4] Overtime, this accumulation of abnormal aggregated proteins is linked to toxicity and neurodegeneration, leading to characteristic neurological syndromes. Similar to intracytoplasmic inclusions in MSAs, the Lewy bodies in PD principally comprise α-syn.[4] Mutations in asln gene and even overexpression of wild type asln can cause PD.[5] In an important experiment, aggregated α-syn was found to induce PD-like disease in wild type mice and rats.[29] Cytoplasmic levels of α-syn appear to increase with age[30] but the factors that trigger PD in specific cohorts of individuals are unknown.
In a landmark recent experiment, intracerebral injection of α-syn aggregates from the brain of the patients with MSA successfully transmitted the disease in transgenic mice harboring human asln mutation (TgM83+/−) and in transfected human embryonic kidney cells (α-syn140*A53T).[6] It remains to be seen if some strain of α-syn propagates in nature as a truly infectious disease.
β-amyloid and tau: AD, the most prevalent neurodegenerative disease, is characterized by the deposition of extracellular Aβ-containing amyloid plaques and intraneuronal aggregates of hyperphosphorylated tau protein in the form of neurofibrillary tangles (NFTs).[31] Aβ is a proteolytic product of amyloid precursor protein (APP), which is processed into amyloidogenic fragments by the β and γ secretases. Many different peptides are generated in vivo, of which Ab1-42 has the highest propensity to aggregate. Mice expressing an APP transgene that bears the “Swedish” mutation (K670N/M67L) produce elevated levels of Ab1-42 peptide and show cognitive impairment.[32] Similarly, mutations in the tau gene can cause neurodegeneration with NFTs,[33] thereby providing genetic evidence for the causal role of tau aggregation in disease. As with prions, it is unclear whether small oligomeric species or larger aggregates of Aβ play a major pathogenic role in AD.
Although early studies of AD transmission to primates were inconclusive, injection of Aβ accelerates the deposition of endogenous Aβ in transgenic mice overexpressing APP.[34] But these phenomena are different from those occurring in prion transmission since they occur only in recipients that overexpress Aβ and are therefore, predisposed to the disease process. Overexpression of Aβ is shown to form strains, which may correspond to distinct conformational states of aggregates and they are preserved even after serial transmission to recipient Tg animals.[35] Whether any of these Aβ strains in the inoculum can transmit the AD phenotype is currently an area of intense research.
TDP-43: TDP-43 is a soluble, nuclear protein that undergoes cytoplasmic redistribution and aggregation in a majority of the cases of sporadic and familial ALS and FTD.[36,37] Missense mutations in TDP-43 gene are linked to familial ALS,[38] strongly supporting the idea that TDP-43 proteinopathy is indeed central to the pathogenesis of these diseases. Furthermore, in ALS and FTD, the disease spreads over time and the underlying pathologic substrate in these diseases indicates a focal beginning and a regional spread as the disease progresses, leading eventually to diffuse neuronal loss.[12,37] Such disease transmission form cell to cell likely involves a donor cell-releasing prionoid aggregate and the acceptor cell that is competent for maintaining propagation. Transmission of TDP-43 pathology by protein aggregate seeds in experimental animals has been unsuccessful so far and thus, it remains unclear whether it can be propagated as a truly transmissible disease.
In the studies cited here, apart from the lack of transmissibility of prionoids, another key question remains about whether misfolded protein oligomers or higher order aggregates or both are the toxic drivers in prion diseases and other PMDs. Some researchers have questioned the neuronal toxicity of all aggregates or plaques.[39] It is possible that fibril aggregates break spontaneously once they reach a critical length, producing an equilibrium between the growth and fragmented byproducts or seeds [Figure 1]. Studies in yeast have identified Hsp104, HSP70, and HSP40 disaggregases, a sophisticated machinery that can dissolve cytosolic aggregates, including the yeast Ψ prions.[40] As expected, overexpression of Hsp-104 can cure yeast from Ψ prions but curiously, yeast cells lacking Hsp-104 display a phenotype, which is resistant to prion infection,[41] suggesting that aggregation/disaggregation is a dynamic process and probably disaggregation (oligomeric seeds) is essential for prion replication. The existence of the mammalian cellular disaggregase system remains largely unexplored. Nonetheless, the identification of the cellular machinery that enables aggregation and disaggregation of misfolded proteins is likely to offer inroads into the therapy of PMDs, including prion diseases.
Prion diseases, as also other PMDs, are currently incurable, though investigators in several research centers are searching for treatments to prevent the formation or reverse the preformed PrPsc into PrPc or block the action of PrPsc (see review[42]). Diverse approaches including drug therapy, antibodies, gene silencing through RNA interference (RNAi) and antisense oligonucleotides (ASOs), vaccines, and stem cell therapy remain areas of current active research.[42,43,44,45] Drug treatments such as quinacrine, and the antibiotics tetracycline and doxycline initially offered some promise but ultimately failed in placebo-controlled trials.[43] Drugs (tacrolimus and astemizole) that reduced PrPsc levels in prion-infected mice and prolonged their survival have been recommended for treatment in humans and for prophylaxis in familial prion cases.[44] Until disease mechanism-based therapy is discovered, the therapy in PMDs remains symptomatic and palliative.
Conclusion
As the average life expectancy of the world's population is rising and age remains the strongest risk factor for neurodegenerative diseases, the overall burden of PMDs is expected to reach to epidemic proportions in the coming decades. While many important phenomena remain unexplained and efficacious therapies for most PMDs are still lacking, rapidly accelerating prion basic research is a strong reason for optimism. Accumulating knowledge in prion diseases may notably help researchers in deciphering the nature of more common PMDs, offering opportunities for the control and prevention of neurodegenerative disorders. Without doubt, we will see important theoretical advances in PMDs in the near future, from which effective therapeutic approaches will eventually emerge.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Gajdusek DC, Gibbs CJ, Alpers M. Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature. 1966;209:794–6. doi: 10.1038/209794a0. [DOI] [PubMed] [Google Scholar]
- 2.Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension: A clinical pathologic study. Arch Neurol. 1960;2:511–27. doi: 10.1001/archneur.1960.03840110025004. [DOI] [PubMed] [Google Scholar]
- 3.Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry. 1969;32:28–34. doi: 10.1136/jnnp.32.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 5.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 6.Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015;112:E5308–17. doi: 10.1073/pnas.1514475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costanzo M, Zurzolo C. The cell biology of prion-like spread of protein aggregates: Mechanisms and implication in neurodegeneration. Biochem J. 2013;452:1–17. doi: 10.1042/BJ20121898. [DOI] [PubMed] [Google Scholar]
- 8.Goedert M, Falcon B, Clavaguera F, Tolnay M. Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies. Curr Neurol Neurosci Rep. 2014;14:495. doi: 10.1007/s11910-014-0495-z. [DOI] [PubMed] [Google Scholar]
- 9.Aguzzi A, Lakkaraju AK. Cell biology of prions and prionoids: A status report. Trends Cell Biol. 2016;26:40–51. doi: 10.1016/j.tcb.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Tyson T, Steiner JA, Brundin P. Sorting out release, uptake and processing of alpha-synuclein during prion-like spread of pathology. J Neurochem. 2015 doi: 10.1111/jnc.13449. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis J, Dickson DW. Propagation of tau pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131:27–48. doi: 10.1007/s00401-015-1507-z. [DOI] [PubMed] [Google Scholar]
- 12.Verma A. Protein aggregates and regional disease spread in ALS is reminiscent of prion-like pathogenesis. Neurol India. 2013;61:107–10. doi: 10.4103/0028-3886.111109. [DOI] [PubMed] [Google Scholar]
- 13.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadowski M, Verma A, Wisniewski T. Infections of the nervous system. Chapter 59G. Prion diseases. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, editors. Neurology in Clinical Practice. 5th ed. Newton, MA: Butterworth-Heinmann; 2008. pp. 1566–82. [Google Scholar]
- 15.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 16.Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science. 2004;305:673–6. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 17.Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, et al. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A. 1993;90:10608–12. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haïk S, Brandel JP. Infectious prion diseases in humans: Cannibalism, iatrogenicity and zoonoses. Infect Genet Evol. 2014;26:303–12. doi: 10.1016/j.meegid.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Aguilar-Calvo P, García C, Espinosa JC, Andreoletti O, Torres JM. Prion and prion-like diseases in animals. Virus Res. 2015;207:82–93. doi: 10.1016/j.virusres.2014.11.026. [DOI] [PubMed] [Google Scholar]
- 20.Dickinson AG. Scrapie in sheep and goats. Front Biol. 1976;44:209–41. [PubMed] [Google Scholar]
- 21.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 22.Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–22. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 23.Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Zou WQ, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mead S, Gandhi S, Beck J, Caine D, Gajulapalli D, Carswell C, et al. A novel prion disease associated with diarrhea and autonomic neuropathy. New Engl J of Med. 2013;269:1904–14. doi: 10.1056/NEJMoa1214747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saunders SE, Bartz JC, Bartelt-Hunt SL. Soil-mediated prion transmission: Is local soil-type a key determinant of prion disease incidence? Chemosphere. 2012;87:661–7. doi: 10.1016/j.chemosphere.2011.12.076. [DOI] [PubMed] [Google Scholar]
- 26.Dobly A, Van der Heyden S, Roels S. Trends in genotype frequency resulting from breeding for resistance to classical scrapie in Belgium (2006-2011) J Vet Sci. 2013;14:45–51. doi: 10.4142/jvs.2013.14.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wells GA, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M, et al. A novel progressive spongiform encephalopathy in cattle. Vet Rec. 1987;121:419–20. doi: 10.1136/vr.121.18.419. [DOI] [PubMed] [Google Scholar]
- 28.Priem J, Langeveld JP, van Keulen LJ, van Zijderveld FG, Andreoletti O, Bossers A. Enhanced virulence of sheep-passaged bovine spongiform encephalopathy agent is revealed by decreased polymorphism barriers in prion protein conversion studies. J Virol. 2014;88:2903–12. doi: 10.1128/JVI.02446-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–4. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- 30.Jiang P, Gan M, Ebrahim AS, Castanedes-Casey M, Dickson DW, Yen SH. Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of α-synuclein oligomers and decrease of neurites. Neurobiol Aging. 2013;34:1504–15. doi: 10.1016/j.neurobiolaging.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cowan CM, Quraishe S, Mudher A. What is the pathological significance of tau oligomers? Biochem Soc Trans. 2012;40:693–7. doi: 10.1042/BST20120135. [DOI] [PubMed] [Google Scholar]
- 32.Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–67. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crimins JL, Pooler A, Polydoro M, Luebke JI, Spires-Jones TL. The intersection of amyloid β and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer's disease. Ageing Res Rev. 2013;3:757–63. doi: 10.1016/j.arr.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heilbronner G, Eisele YS, Langer F, Kaeser SA, Novotny R, Nagarathinam A, et al. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013;14:1017–22. doi: 10.1038/embor.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watts JC, Condello C, Stöhr J, Oehler A, Lee J, DeArmond SJ, et al. Serial propagation of distinct strains of Aβ prions from Alzheimer's disease patients. Proc Natl Acad Sci U S A. 2014;111:10323–8. doi: 10.1073/pnas.1408900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 37.Verma A. Tale of two diseases: Amyotrophic lateral sclerosis and frontotemporal dementia. Neurol India. 2014;62:347–51. doi: 10.4103/0028-3886.141174. [DOI] [PubMed] [Google Scholar]
- 38.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zetterberg H, Blennow K. Biomarker evidence for uncoupling of amyloid build-up and toxicity in Alzheimer's disease. Alzheimers Dement. 2013;9:459–62. doi: 10.1016/j.jalz.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 41.Tuite MF. Genetics. Psi no more for yeast prions. Nature. 1994;370:327–8. doi: 10.1038/370327a0. [DOI] [PubMed] [Google Scholar]
- 42.Bolognesi ML, Legname G. Approaches for discovering anti-prion compounds: Lessons learned and challenges ahead. Expert Opin Drug Discov. 2015;10:389–97. doi: 10.1517/17460441.2015.1016498. [DOI] [PubMed] [Google Scholar]
- 43.Haïk S, Marcon G, Mallet A, Tettamanti M, Welaratne A, Giaccone G, et al. Doxycycline in Creutzfeldt-Jakob disease: A phase 2, randomized, double-blind, placebo controlled trial. Lancet Neurol. 2014;13:150–8. doi: 10.1016/S1474-4422(13)70307-7. [DOI] [PubMed] [Google Scholar]
- 44.Karapetyan YE, Sferrazza GF, Zhou M, Ottenberg G, Spicer T, Chase P, et al. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc Natl Acad Sci U S A. 2013;110:7044–9. doi: 10.1073/pnas.1303510110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohsawa N, Song CH, Suzuki A, Furuoka H, Hasebe R, Horiuchi M. Therapeutic effect of peripheral administration of an anti-prion protein antibody on mice infected with prions. Microbiol Immunol. 2013;57:288–97. doi: 10.1111/1348-0421.12037. [DOI] [PubMed] [Google Scholar]