

Abstract
Background:
Few papers address the comprehensive prognosis in infantile spasms and look into the seizure profile and psychomotor outcome.
Objective:
We aimed to follow up children with infantile spasms to study: a) the etiology, demographics, semiology, electroencephalogram (EEG), and radiological pattern; b) seizure control, psychomotor development, and EEG resolution with treatment; c) the effects of various factors on the control of spasms, resolution of EEG changes, and psychomotor development at 3-year follow-up.
Materials and Methods:
Fifty newly diagnosed cases with a 1-12 month age of onset and who had hypsarrhythmia in their EEG were recruited and 43 were followed up for 3 years.
Results:
Of the children followed up, 51% were seizure-free and 37% had a normal EEG at the 3-year follow-up. Autistic features were seen in 74% of the children. Only 22.7% among the seizure-free (11.6% of the total) children had normal vision and hearing, speech with narration, writing skills, gross and fine motor development, and no autism or hyperactivity. On multivariate analysis, two factors could predict bad seizure outcome — the occurrence of other seizures in addition to infantile spasms and no response to 28 days of adrenocorticotropic hormone (ACTH). No predictor could be identified for abnormal psychomotor development.
Discussion and Conclusion:
In our study, we could demonstrate two factors that predict seizure freedom. The cognitive outcome and seizure control in this group of children are comparable to the existing literature. However, the cognitive outcome revealed by our study and the survey of the literature are discouraging.
Keywords: Development, infantile spasms, cognitive outcome, prognosis, psychomotor development, seizure freedom, outcome, West syndrome
Introduction
It is known that seizures and electrical epileptogenic activity result in cognitive decline.[1] Infantile spasms (West syndrome) is an epileptic encephalopathy characterized by spasms and hypsarrhythmia on the electroencephalogram (EEG).[2]
Despite treatment, the prognosis of West syndrome is poor with the majority (88-96%) having psychomotor retardation at follow-up.[3,4,5] The pathophysiology is still unknown; two-third patients have a demonstrable underlying cause and there is no consensus on the treatment. Applying logic, the etiology is different in the developing countries with a greater burden of birth-related problems. As the most accepted treatment modality [adrenocorticotropic hormone (ACTH)] involves immunosuppression, the treatment in the same region has to be tailored to the needs of the population (exposed to increased risk of infectious diseases). Earlier studies observed that in the subgroup of cryptogenic patients, a delay in the initiation of treatment may be associated with a worse outcome.[3,6] However, different groups have described different prognostic factors for outcome in infantile spasms.
Objectives
To describe the semiology and the demographical, etiological, EEG and radiological patterns in children with infantile spasms.
To describe seizure control, psychomotor development (using standardized psychometric assessments), and EEG resolution with treatment in children with infantile spasms.
To evaluate the effects of various factors on the control of spasms, resolution of EEG changes, and psychomotor development at 3 years after initiation of treatment.
Materials and Methods
We prospectively recruited 50 consecutive patients in this longitudinal follow-up study conducted in a tertiary care pediatric neurology center in South India from August 2009 to February 2012.
Inclusion criteria
Newly diagnosed children fulfilling the International League Against Epilepsy (ILAE) criteria for infantile spasms;[7]
An age of onset between 1 month and 12 months;
Presence of hypsarrhythmia or modified hypsarrhythmia on EEG
Exclusion criteria
The present study excluded nonconsenting patients and those previously treated with ACTH, steroids, or vigabatrin.
Ethical considerations
The study protocol was approved by the Ethical Review Committee of our institution. Informed consent was obtained from caretakers of every patient enrolled.
Definitions[8]
A child was considered seizure-free at 28 days of age when he was free of seizures for a minimum period of 7 days. At a 3-year follow-up, a child was considered seizure-free if he had no seizures for at least the past 3 months.
The term symptomatic infantile spasms was used for children who had a clearly defined underlying cause and/or significant developmental delay prior to the onset of spasms.[9] Each child was classified as symptomatic or cryptogenic at the end of the study, sometimes after a long-lasting diagnostic workup. The cryptogenic cases included those in whom no underlying cause could be detected.
Parents recorded the type and number of seizures using daily record cards. Each patient had at least a monthly visit. At each visit, the investigator confirmed the seizure assessment and screened for presence of psychomotor retardation using the Trivandrum Developmental Screening Chart (TDSC).[10] Socioeconomic status was assessed using modified Kuppuswamy Scale 2003.[11]
Tools and techniques
A semi-structured pretested questionnaire was used to collect demographic details and the clinical and treatment history. Chromosomal analysis and metabolic workup were done in cases where it was clinically deemed necessary. Magnetic resonance imaging (MRI) scan of the head using a 1.5 Tesla machine and if necessary an additional computed tomography (CT) scan of the brain in cases of suspected calcification were performed.
At 1-year age, the Developmental Assessment Scale for Indian Infants (DASII) was administered by trained developmental therapists at the Child Developmental Centre, Thiruvananthapuram, Kerala, India and each child's mental and motor developmental quotients were obtained.[12] At 3 years follow-up, each child had the Childhood Autism Rating Scale (CARS) administered at the Child Developmental Centre, Thiruvananthapuram, Kerala, India and the children were classified as nonautistic, having mild to moderate autism, and having severe autism.[13] The presence of hyperactivity was judged based on the DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD). Visual acuity testing was performed in the cognitively normal child by picture card matching and letter recognition if the child was old enough. In the cognitively abnormal child, fixation of eyes on colored objects and light and following of light or object was used to assess vision. Behavioral observation audiometry was performed in the majority of cases to assess hearing at the 3-year follow-up. Patients who could not cooperate were assessed using auditory brain stem-evoked response. Gross and fine motor skills were assessed using TDSC. Writing skills of the child was assessed depending on the age of the child at the 3-year follow-up. The minimum standard was set at being able to copy a circle. An informal picture story narration test was used to assess narrative skills.
After clinical evaluation and laboratory tests, every patient had initially 28 days of ACTH. Synthetic ACTH was used at a dose of 20 IU subcutaneously (s/c) per day for all infants more than 5 kg weight and 10 IU s/c per day for infants who weighed less than 5 kg. As a low dose was used, a daily regimen was given instead of the alternate day regimen. Other antiepileptics were tapered and stopped initially. No standardized treatment protocol was implemented after the initial 28 days for this follow-up study. Patients who were controlled with ACTH were continued on the oral medications; if they showed a partial response or no response, another 28 days of ACTH was given. Later other oral antiepileptics including vigabatrin, lamotrigine, topiramate, and benzodiazepines were given as the situation warranted, depending on the response and emergence of adverse effects. Vigabatrin was withdrawn after 6 weeks if there was no response. In patients who responded, vigabatrin was continued for 6-8 months and tapered. Patients who remained seizure-free were continued on sodium valproate monotherapy wherever possible or drug combinations for 2 total years of seizure freedom and EEG normalization (a normal EEG). Potential worsening by phenobarbitone, phenytoin, and carbamazepine was kept in mind and these drugs were avoided.
At each monthly or more frequent follow-up visits, treatment emergent adverse effects were recorded. Patients on vigabatrin were seen by an ophthalmologist every 3 months and retinal changes were assessed as in this age group, visual fields could not be examined and a large number of patients already had cortical blindness due to the initial cerebral insult.
Statistical analysis
The data were coded, entered, and analyzed using Windows Statistical Package for the Social Sciences (SPSS) version 11. Descriptive statistics was used to describe the social and demographic profiles, the seizure semiology, and the EEG. Bivariable analysis was performed using t-test and chi-square as appropriate for the variable. A p value <0.05 was considered to be significant. For multivariate analysis, binary logistic regression was used. Statistical analysis was performed by the second author (an epidemiologist).
Observations and Results
Fifty consecutive cases of infantile spasms with hypsarrhythmia in their EEG were recruited over a 2.5-year period. The eligible study candidates were 55. The caretakers of two children refused to give consent to join the study. Three cases were excluded as the initial EEG was normal.
Baseline characteristics
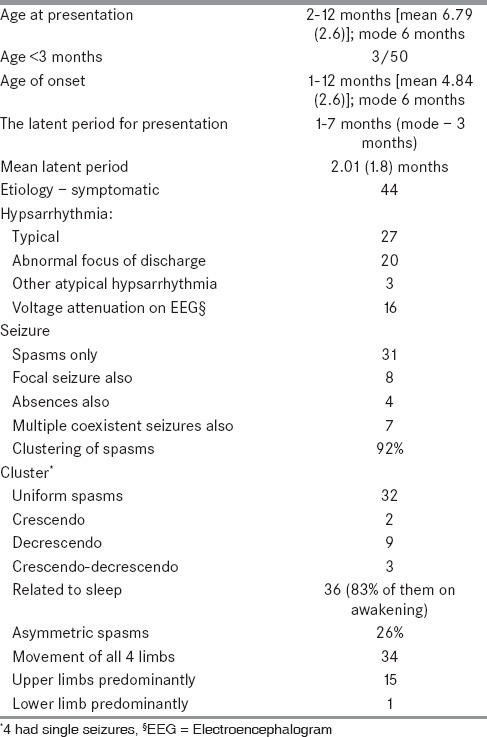
Table 1 shows the baseline characteristics of the 50 children. Among the children with family history of epilepsy, only one had family history of infantile spasms. The vast majority of our patients belonged to the middle income group — 36/50. Table 2 shows the patient details and characteristics of the seizures of the group.
Table 1.
Baseline characteristics of the study population
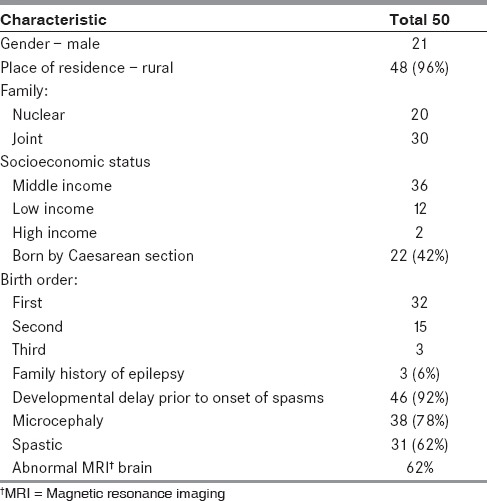
Table 2.
Seizure, patient, and EEG characteristics
Etiology
Six out of the total 50 were cryptogenic. The symptomatic group included 20 patients with perinatal problems including hypoxic ischemic encephalopathy (HIE), neonatal jaundice (NNJ), and hypoglycemic brain injury (HBI), 10 patients with cerebral malformation, 5 patients with intrauterine infections, 4 patients with inborn errors of metabolism, 1 patient with neonatal meningitis, 1 patient with postneonatal ventilation induced anoxic brain damage, and 3 patients with tuberous sclerosis. For analysis, the symptomatic group was reclassified into two groups:
Perinatal; including HIE, NNJ, HBI, neonatal meningitis, and postneonatal ventilation anoxic damage, and
A structural metabolic group, which included brain malformations, the inborn errors of metabolism, intrauterine infections, and tuberous sclerosis.
Outcome
Early outcome
Seizure status
After 14 days of ACTH, 20% only were seizure-free. After 28 days of ACTH, 40% had full control of seizures, 28 (56%) had partial control, one had aggravation, and one died. Of the 20 children who were seizure-free at 28 days, 15 relapsed at some point to be controlled later; only 5 continued to be seizure-free without any relapse.
EEG at 28 days after adrenocorticotropic hormone
After 28 days of ACTH, EEG showed normalization in only 2 (4%) patients; 66% of the patients showed no change in their hypsarrhythmic pattern; 28% had some change for the better with more normal sleep patterns emerging and less of spiking, and one child died before the EEG on the 28th day.
Psychomotor development at one year age
At 1 year of age, 88% of the children were available for follow up, four were lost to follow-up, one child died at 2 weeks after enrolment, and one child died at 1 year. DASII was administered to 86% of the children at 1 year as one child who subsequently died at 14 months was too sick. Only 4/43 (9.3%) of the group showed normal motor and mental developments.
Associations with seizure freedom at 28 days of adrenocorticotropic hormone
On bivariable analysis, no factor had an association with seizure-free status at 28 days after initiation of ACTH.
Associations with psychomotor development at 1-year age
On bivariate analysis, no factor had an association with the psychomotor development of the children at 1 year of age.
Late outcome
Only 43 children were available for follow-up at 3 years as three died and four were lost to follow-up.
Mortality
One child who had severe pneumonia at the time of death, died at 2 weeks after enrolling into the study while on ACTH (had hyperammonemia but the metabolic defect could not be further characterized). The other two who died were not on ACTH at the time of death. Both had gross cerebral malformation — one had holoprosencephaly and cleft lip and palate and the other had lissencephaly.
Seizure status
At 3-year follow-up, 22 were seizure-free, 13 continued to have infantile spasms alone, 7 had other seizure types also, and 1 had Lennox Gastaut syndrome. However, among those who were seizure-free, 2 were off drugs and 10 each were on one drug and multiple drugs. Only 16/22 seizure-free children had a normal EEG, 5 had an interictal EEG, and 1 had persisting hypsarrhythmia. Though 22 (51.2%) of the 43 who could be followed up to 3 years were seizure-free for 3 months, only 16/43 (37.2%) had a normal EEG. Sixteen (37.2%) had an interictal EEG, 8 (18.6%) had persisting hypsarrhythmia, and 3 (6.9%) had a Lennox-Gastaut syndrome (LGS) type EEG (one of them had the semiology of LGS and the others continued to have multifocal myoclonic jerks only).
Though the number of children who attained seizure freedom at 28 days after treatment initiation and at 3-year follow-up were nearly the same — 20/49 (40.8%) — (one died at 2 weeks after recruitment) and 22/43 (51.2%), EEG normalization was markedly more at 3 years — 4% (2/49) at 28 days versus 37.2% (16/43) at 3-year follow-up.
Comprehensive outcome at 3-year follow-up
At 3-year follow-up (n = 43), visual impairment was seen in 30% children, hearing impairment in 16% children, normal fine and gross motor skills in 14% and 16% children, respectively, normal speech skills in 16% children and no autism in 25.5% children. Only 11.6% were seizure-free with no motor or cognitive sequelae (no autism or ADHD and normal learning, writing, and speech) and normal vision and hearing. However, only 7% children had the above attributes with a normal EEG as well. Of the children, 4.6% were seizure-free with a normal EEG and normal motor and cognitive development and were off drugs for at least 3 months. Only 5/22 (22.7%) who were totally seizure-free had normal gross motor and fine motor developments, no autism or hyperactivity, normal speech with narration, normal writing skills, and normal vision and hearing.
There was no statistically significant relation between age and latent period for presentation with seizure freedom at 3-year follow-up. To attempt to find the prognostic factors for seizure freedom at 3 years, bivariable analysis was performed using various factors.
Result of bivariable analysis of factors related to seizure freedom at 3-year follow-up [Table 3]
Table 3.
Result of bivariate analysis of factors associated with seizure control at 3-year follow-up
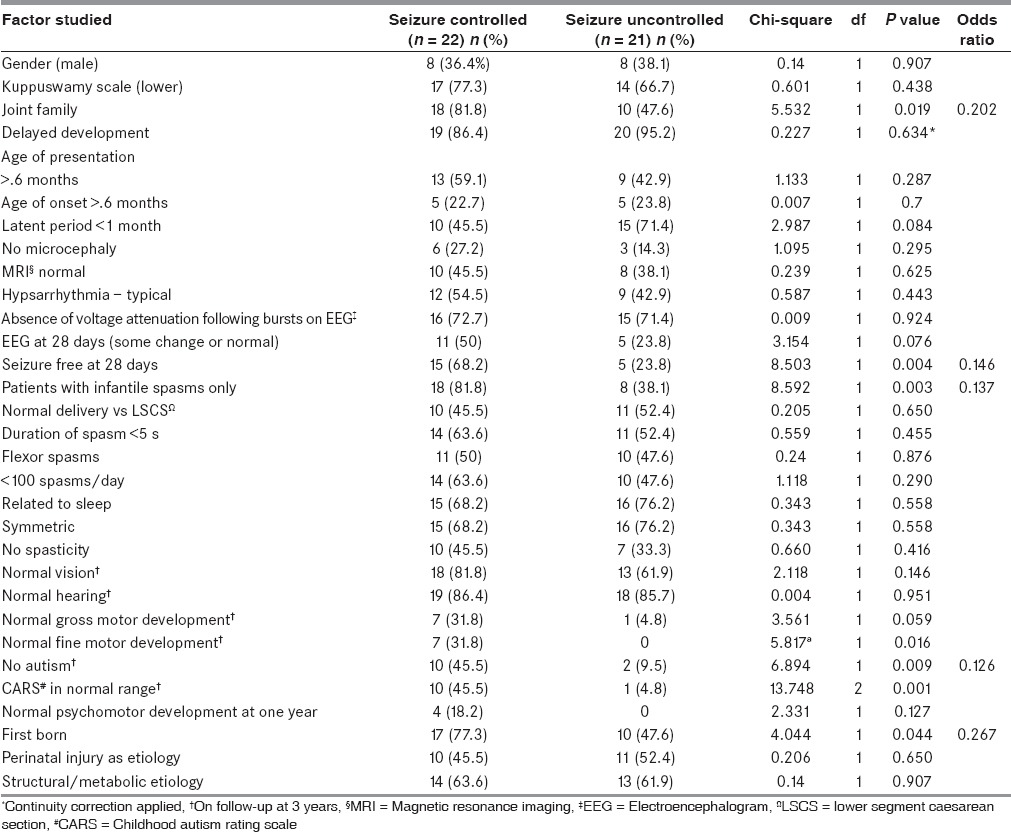
Seizure control at 28 days after the initiation of ACTH, occurrence of infantile spasms only versus occurrence of other seizure types along with the infantile spasms, no autism at 3-year follow-up, being part of a joint family, and being the first born had a significant positive association with seizure freedom at 3-year follow-up.
Result of multivariate analysis of factors to predict persistence of seizures at three years
On multivariate analysis, two factors were significant — the occurrence of other seizure types in addition to the infantile spasms p = 0.007; adjusted odds ratio (OR) [95% confidence interval (CI)] 11.105 (1.939-63.612) and no response to 28 days of ACTH p = 0.007 and adjusted OR (95% CI) 10.350 (1.896-56.491).
Association of etiology with electroencephalogram at 3-year follow-up and autism
The etiology when classified as cryptogenic, perinatal insult and structural metabolic, showed no association with the EEG at 3 years (p = 0.67) and to the development of autism as assessed with the CARS score at 3 years (p = 0.30).
Predictors of abnormal psychomotor development and abnormal electroencephalogram at 3 years
On bivariable analysis, no factor could be statistically significantly associated with abnormal psychomotor development or abnormal EEG at 3-year follow-up.
Treatment emergent adverse events
Out of the 50 children administered ACTH, 12 each had significant weight gain and severe infection, 15 had hypertension, and 1 had hypocalcemia with features of rickets. Of the 12 patients who developed severe infection while on ACTH, one died (pneumonia) and the rest survived. Among the survivors of severe infection, one each had chicken pox, severe conjunctivitis, orbital cellulitis, staphylococcal scalded skin syndrome, and measles. The rest had severe respiratory infection. Three children who were on ACTH developed oral candidiasis, two complained of high-pitched cry, five developed hyperpigmentation, and one had increased irritability and grunting. One patient on topiramate (n = 24) had loss of appetite at a 6 mg/kg dose. None of the patients on Vigabatrin (n = 17) had new visual deficits or macular changes (monitored by an ophthalmologist at three monthly intervals). Two children developed allergic skin reaction and consequently had to discontinue vigabatrin. One child each on valproate (n = 50) and lamotrigine (n = 7) had allergic skin reaction and had to discontinue the same. Three children who were on valproate showed a dose-dependent increase in liver enzymes. We did not look for brain atrophy as a side effect of ACTH or vigabatrin as described in the literature due to the cost involved.
The group with metabolic diseases
The metabolic diseases identified in this group included a child with Gaucher (enzyme proved) a case of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency (tandem mass spectroscopy), a case of hyperammonemia not further characterized and a case of biotidinase deficiency. Two of them continued to have seizures at 3 years and were autistic, the child with MCAD deficiency was seizure-free but was severely autistic and the child with hyperammonemia was lost to follow-up.
The children with intrauterine infection
There were two cases of rubella and three with Cytomegalovirus (CMV) infection. The two with CMV were lost to follow-up. The third was seizure-free but autistic at 3-year follow-up. Both the children with rubella had seizures and were autistic at 3-year follow-up.
Magnetic resonance imaging abnormality
The MRI was abnormal in 31/50; (25/43 who came for follow-up). Nine children had cerebral malformation (three hemimegalencephaly, two schizencephaly, one lissencephaly, one holoprosencephaly, one corpus callosal agenesis — as part of Aicardi syndrome and one with altered gyral morphology). One child had hydrocephalus, nine had cerebral atrophy, three had old infarcts, two had bilateral cystic encephalomalacia, two had extensive calcification (one frontal and the other basal ganglia), one each had bilateral thalamic hyperintensities and nonspecific white matter hyperintensities, and the three with tuberous sclerosis had multiple cortical tubers. Out of the 25 children with MRI abnormality, only 12 children were seizure-free at 3-year follow-up and only six children were nonautistic.
Discussion
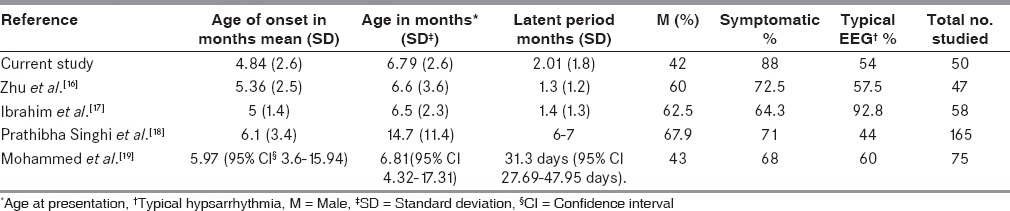
There are dificulties in comparing the results of a clinical series of infantile spasms from the literature because of their nonhomogeneity and different definitions of response rates. Table 4 shows the comparison of age, gender, etiology, and EEG with other studies. The other studies shown were all conducted a few years earlier and had fewer symptomatic cases. It is a known fact that patients diagnosed more recently are often subject to “more sensitive investigative techniques” that will lead to identified diagnoses. Surprisingly, our study showed more females than males in the group as observed in a few studies only.[17,19] None of the factors in our study correlated significantly with the etiology as observed by Mohammed et al.[19] The dose of ACTH used in our study was the same as that recommended by the American Academy of Neurology in its evidence-based guideline update 2013.[20]
Table 4.
Comparison of age, gender, etiology, and EEG with other studies
Developmental outcome
Though 51.2% of the children who came for follow-up in our study were seizure-free, it is noteworthy that only 11.3% were seizure-free and developmentally normal. However, the available literature also supports this observation. A review of 67 published studies with an average follow-up period of 31 months found that only 16% of the patients with infantile spasms had normal development.[21] Of the symptomatic patients, 80% and in the idiopathic group, 50% remained developmentally delayed and 60% only of the total remained seizure-free at a median follow-up of 2.38 years in a retrospective long-term follow-up study by Lagae et al.[22] In one population-based survey by Trevathan, 80% of 10-year-old children with a diagnosis of infantile spasms had some form of mental retardation.[15] In the series by Basheer, 87.5% had abnormal neurodevelopmental outcome.[19] Most studies have shown the best outcomes in the cryptogenic group, with normal development seen in 30-70% of these infants, especially when treated early.[3,14] In the series by Ibrahim, only 4/56 children (7.2%) showed normal developmental milestones after follow-up for 6 months.[17]
Only few studies have addressed autism as sequelae in children with infantile spasms. In a study by Saemundsen, only 6/17 (35.3%) showed autism spectrum disorder while we had 74.4% with autism, possibly reflecting the small number studied by Saemundsen.[23]
Predictors of abnormal developmental outcome and autism
On bivariable analysis, we could not find any predictor for an abnormal developmental outcome or autism. Poor prognostic factors for mental development in the 1981 study by Matsumoto were delayed development before the onset of spasms, neurological abnormalities, prenatal and perinatal etiologies, relapse after initial ACTH therapy, laughing attacks, and evolution into other types of fits.[6] Only in the cryptogenic cases in the available literature, was there a significant correlation between the delay in treatment and the long-term prognosis for mental development.
Long term seizure control
Among randomized controlled trials, the response rate (cessation of clinical spasms and normalization of the hypsarrhythmic EEG) to natural or synthetic ACTH ranged from 42-87% of the patients and up to 100% in the cryptogenic group.[2,24,25]
Baram recorded 87% spasm freedom with EEG normalization when he used the highest dose of 150 units/m2 twice daily of natural ACTH for 14 days followed by taper.[25] Lux used 40 units synthetic ACTH on alternate days and got 76% seizure freedom without EEG normalization.[2] Hrachovy et al. compared high-dose and low-dose natural ACTH in a class I randomized control trial. Primary outcome measurement was cessation of clinical spasms and the abolition of hypsarrhythmia on EEG. There were no differences in efficacy for each dosing group, with 50% responders in the high-dose group and 58% response in the low-dose group.[26] The adverse effect profile was similar except for a higher rate of hypertension in the high-dose group. The seizure control with high dose ACTH is contradictory in many well-quoted studies with Kivity and Yanagaki showing 91.89% and 100% suppression of hypsarrhythmia and total cessation of seizure with high dose ACTH, respectively.[14,24,25] In the study by Mohammed BP, 77.5% continued to have seizures.[19]
Predictors of seizure control on long term follow up
The factors that we could discern on bivariable analysis included being part of a joint family (possibly with the grandparents involved in bringing up the child and hence, a better drug compliance) and being first born (and possibly consequently having a younger mother and with less time allotted to other siblings). One other factor seen to be significantly associated with better seizure control was no autism at 3-year follow-up, which could be a consequence of the seizure control. On multivariate analysis, two of the five factors remained significant (seizure control at 28 days after the initiation of treatment and occurrence of infantile spasms alone with no other seizure type). These two factors could be an indicator of less severe initial pathology. Table 5 compares the factors predicting seizure control in other studies with the factors we have identified. No delay in the initiation of treatment has been endorsed in the studies by Lombroso and in an Indian study to be related to good outcome.[5,27] Singhi and independently Basheer, like us, did not find that correlation.[18,19] In the North Indian paper by Singhi and Ray, no factor other than developmental improvement could predict seizure control.[18] Matsumoto et al. as in the present study, have identified the presence of other seizures in addition to infantile spasms, a predictor of continuing seizures.[6] As observed in our study, Ibrahim et al. and Basheer et al. have identified early seizure control as a factor predicting seizure freedom later.[17,19] As in our study, Basheer also observed that etiology could not predict seizure control.[19]
Table 5.
Predictors for seizure control in various studies irrespective of drug combinations and doses used

Mortality
The mortality rate associated with infantile spasms has been estimated to be between 5% and 30%.[15,25] Our study had a mortality of 6.97% (3/43) and all died of infection as reported in other series.[28] Riikonen and Glaze and Hrachovy independently reported a mortality of 5%.[3,4] Death due to septicemia while on ACTH was reported in two out of 56 patients by Ibrahim.[17]
Side effect profile
Our study showed that ACTH has not negligible side effects in a large number of children. With regard to the side-effect profile, Ibrahim found that vigabatrin was better than ACTH with only 2/38 patients experiencing the adverse effects, as opposed to ACTH, where 6/18 patients reported ACTH induced hypertension.[17] In our study, 30% developed hypertension on ACTH. In other studies, hypertension has been reported in 0-37%.[24,26] Riikonen reported death in 5%, hypocalcemia in two patients, and hypertension in 11/162 children.[29] Irritability has been reported in 37-100%.[24,26] Only one each of our patients had hypocalcemia and severe irritability and grunting.
Conclusions
The two factors identified in our study to predict a good seizure outcome — the presence of infantile spasms alone versus with other seizure types and seizure control at 28 days after commencement of treatment could help us to predict seizure control in children with infantile spasms early in the disease course. The fact that we could demonstrate a seizure-free state lasting for at least 3 months at 3-year follow-up with low dose of ACTH in 51% should encourage us to persist in treating children with infantile spasms. However, it is alarming to note that only 11.6% were seizure free with no neurodevelopmental sequelae in spite of 51% being seizure free and that no factor in our study could predict the occurrence of these sequelae.
Though the number of patients with seizure freedom at 28 days after the commencement of treatment and 3-year follow-up were comparable, EEG normalization was of no comparison, pointing to the necessity for the continuation of antiepileptics until EEG normalization even if spasms are controlled.
Study Limitations and Strengths
The study was prospective and a formal cognitive assessment could be done at 1 year of age and at 3-year follow-up. Studies with larger numbers of patients should be undertaken to assess the developmental and cognitive outcomes of treated children with infantile spasms with and without EEG normalization.
Financial support and sponsorship
The study was funded by the Sree Avittom Thirunal (SAT) Endowment fund of the State Government of Kerala. The sponsoring agent had no role in the design and conduct of the study.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
The study was funded by the Sree Avittom Thirunal (SAT) Endowment fund of the State Government of Kerala. We would also like to acknowledge the work done by Prasanna GL, Rajee Krishnan R, Resmi VR, and Sunitha RM, developmental therapists and Lekshmi MA and Letha S, preschool teachers of the Child Development Centre, Thiruvananthapuram, Kerala, India.
References
- 1.Holmes GL. Effects of early seizures on later behavior and epileptogenicity. Ment Retard Dev Disabil Res Rev. 2004;10:101–5. doi: 10.1002/mrdd.20019. [DOI] [PubMed] [Google Scholar]
- 2.Lux AL, Osborne JP. A proposal for case definitions and outcome measures in studies of infantile spasms and west syndrome: Consensus statement of the West Delphi Group. Epilepsia. 2004;45:1416–28. doi: 10.1111/j.0013-9580.2004.02404.x. [DOI] [PubMed] [Google Scholar]
- 3.Riikonen R. A long-term follow-up study of 214 children with the syndrome of infantile spasms. Neuropediatrics. 1982;13:14–23. doi: 10.1055/s-2008-1059590. [DOI] [PubMed] [Google Scholar]
- 4.Glaze DG, Hrachovy RA, Frost JD, Jr, Kellaway P, Zion TE. Prospective study of outcome of infants with infantile spasms treated during controlled studies of ACTH and prednisone. J Pediatr. 1988;112:389–96. doi: 10.1016/s0022-3476(88)80318-4. [DOI] [PubMed] [Google Scholar]
- 5.Lombroso CT. A prospective study of infantile spasms: Clinical and therapeutic correlates. Epilepsia. 1983;24:135–58. doi: 10.1111/j.1528-1157.1983.tb04874.x. [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto A, Watanabe K, Negoro T, Sugiura M, Iwase K, Hara K, et al. Long-term prognosis after infantile spasms: A statistical study of prognostic factors in 200 cases. Dev Med Child Neurol. 1981;23:51–65. doi: 10.1111/j.1469-8749.1981.tb08446.x. [DOI] [PubMed] [Google Scholar]
- 7.Blume WT, Lüders HO, Mizrahi E, Tassinari C, van Emde Boas W, Engel J., Jr Glossary of descriptive terminology for ictal semiology: Report of the ILAE task force on classification and terminology. Epilepsia. 2001;42:1212–8. doi: 10.1046/j.1528-1157.2001.22001.x. [DOI] [PubMed] [Google Scholar]
- 8.Panayiotopoulos CP. Epileptic Encephalopathies in Infancy and Early Childhood in Which the Epileptiform Abnormalities May Contribute to Progressive Dysfunction. Oxfordshire (UK): Bladon Medical Publishing; 2005. The Epilepsies: Seizures, Syndromes and Management. Chapter 7. [PubMed] [Google Scholar]
- 9.Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, et al. Infantile spasms: A U. S. consensus report. Epilepsia. 2010;51:2175–89. doi: 10.1111/j.1528-1167.2010.02657.x. [DOI] [PubMed] [Google Scholar]
- 10.Nair MK, George B, Philip E, Lekshmi MA, Haran JC, Sathy N. Trivandrum developmental screening chart. Indian Pediatr. 1991;28:869–72. [PubMed] [Google Scholar]
- 11.Kumar N, Shekhar C, Kumar P, Kundu AS. Kuppuswamy's socioeconomic status scale-updating for 2007. Indian J Pediatr. 2007;74:1131–2. [PubMed] [Google Scholar]
- 12.Phatak P, Misra N. Developmental Assessment Scales for Indian Infants (DASII)1-30 months – Revision of Baroda norms with indigenous material. Psychol Stud. 1996;41:55–6. [Google Scholar]
- 13.Schopler E, Reichler RJ, DeVellis RF, Daly K. Toward objective classification of childhood autism: Childhood Autism Rating Scale (CARS) J Autism Dev Disord. 1980;10:91–103. doi: 10.1007/BF02408436. [DOI] [PubMed] [Google Scholar]
- 14.Kivity S, Lerman P, Ariel R, Danziger Y, Mimouni M, Shinnar S. Long-term cognitive outcomes of a cohort of children with cryptogenic infantile spasms treated with high-dose adrenocorticotropic hormone. Epilepsia. 2004;45:255–62. doi: 10.1111/j.0013-9580.2004.30503.x. [DOI] [PubMed] [Google Scholar]
- 15.Trevathan E, Murphy CC, Yeargin-Allsopp M. The descriptive epidemiology of infantile spasms among Atlanta children. Epilepsia. 1999;40:748–51. doi: 10.1111/j.1528-1157.1999.tb00773.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhu X, Chen O, Zhang D, Jin R, Li F, Wang Y, et al. A prospective study on the treatment of infantile spasms with first-line topiramate followed by low-dose ACTH. Epilepsy Res. 2011;93:149–54. doi: 10.1016/j.eplepsyres.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Ibrahim S, Gulab S, Ishaque S, Saleem T. Clinical profile and treatment of infantile spasms using vigabatrin and ACTH--a developing country perspective. BMC Pediatr. 2010;10:1. doi: 10.1186/1471-2431-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singhi P, Ray M. Profile of west syndrome in North Indian children. Brain Dev. 2005;27:135–40. doi: 10.1016/j.braindev.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Mohamed BP, Scott RC, Desai N, Gutta P, Patil S. Seizure outcome in infantile spasms-a retrospective study. Epilepsia. 2011;52:746–52. doi: 10.1111/j.1528-1167.2010.02963.x. [DOI] [PubMed] [Google Scholar]
- 20.Go CY, Mackay MT, Weiss SK, Stephens D, Adams-Webber T, Ashwal S, et al. Child Neurology Society; American Academy of Neurology. Evidence-based guideline update: Medical treatment of infantile spasms Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2012;78:1974–80. doi: 10.1212/WNL.0b013e318259e2cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hrachovy RA, Frost JD., Jr Infantile epileptic encephalopathy with hypsarrhythmia (infantile spasms/West syndrome) J Clin Neurophysiol. 2003;20:408–25. doi: 10.1097/00004691-200311000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Lagae L, Verhelst H, Ceulemans B, De Meirleir L, Nassogne MC, De Borchgrave V, et al. Treatment and long term outcome in West syndrome: The clinical reality. A multicentre follow up study. Seizure. 2010;19:159–64. doi: 10.1016/j.seizure.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 23.Saemundsen E, Ludvigsson P, Rafnsson V. Autism spectrum disorders in children with a history of infantile spasms: A population-based study. J Child Neurol. 2007;22:1102–7. doi: 10.1177/0883073807306251. [DOI] [PubMed] [Google Scholar]
- 24.Yanagaki S, Oguni H, Hayashi K, Imai K, Funatuka M, Tanaka T, et al. A comparative study of high-dose and low-dose ACTH therapy for West syndrome. Brain Dev. 1999;21:461–7. doi: 10.1016/s0387-7604(99)00053-4. [DOI] [PubMed] [Google Scholar]
- 25.Baram TZ, Mitchell WG, Tournay A, Snead OC, Hanson RA, Horton EJ. High-dose corticotropin (ACTH) versus prednisone for infantile spasms: A prospective, randomized, blinded study. Pediatrics. 1996;97:375–9. [PMC free article] [PubMed] [Google Scholar]
- 26.Hrachovy RA, Frost JD, Jr, Glaze DG. High-dose, long-duration versus low-dose, short-duration corticotropin therapy for infantile spasms. J Pediatr. 1994;124:803–6. doi: 10.1016/s0022-3476(05)81379-4. [DOI] [PubMed] [Google Scholar]
- 27.Sharma NL, Viswanathan V. Outcome in west syndrome. Indian Pediatr. 2008;45:559–63. [PubMed] [Google Scholar]
- 28.Snead OC, 3rd, Benton JW, Myers GJ. ACTH and prednisone in childhood seizure disorders. Neurology. 1983;33:966–70. doi: 10.1212/wnl.33.8.966. [DOI] [PubMed] [Google Scholar]
- 29.Riikonen R, Donner M. ACTH therapy in infantile spasms: Side effects. Arch Dis Child. 1980;55:664–72. doi: 10.1136/adc.55.9.664. [DOI] [PMC free article] [PubMed] [Google Scholar]