

Abstract
Background
Transient receptor potential melastatin 3 (TRPM3) cation channels are ubiquitously expressed by multiple cells and have an important regulatory role in calcium-dependent cell signalling to help maintain cellular homeostasis. TRPM3 protein expression has yet to be determined on Natural Killer (NK) cells and B lymphocytes. Multiple single nucleotide polymorphisms have been reported in TRPM3 genes from isolated peripheral blood mononuclear cells, NK and B cells in Chronic fatigue syndrome/Myalgic encephalomyelitis (CFS/ME) patients and have been proposed to correlate with illness presentation. The object of the study was to assess TRPM3 surface expression on NK and B lymphocytes from healthy controls, followed by a comparative investigation examining TRPM3 surface expression, and cytoplasmic and mitochondrial calcium influx in CD19+ B cells, CD56bright and CD56dim cell populations from CFS/ME patients.
Results
TRPM3 cell surface expression was identified for NK and B lymphocytes in healthy controls (CD56bright TRPM3 35.72 % ± 7.37; CD56dim 5.74 % ± 2.00; B lymphocytes 2.05 % ± 0.19, respectively). There was a significant reduction of TRPM3 surface expression on CD19+ B cells (1.56 ± 0.191) and CD56bright NK cells (17.37 % ± 5.34) in CFS/ME compared with healthy controls. Anti-CD21 and anti-IgM conjugated biotin was cross-linked with streptavidin,and subsequently treatment with thapsigargin. This showed a significant reduction in cytoplasmic calcium ion concentration in CD19+ B lymphocytes. CD56bright NK cells also had a significant decrease in cytoplasmic calcium in the presence of 2-APB and thapsigargin in CFS/ME patients.
Conclusions
The results from this preliminary investigation identify, for the first time, TRPM3 surface expression on both NK and B lymphocytes in healthy controls. We also report for the first time, significant reduction in TRPM3 cell surface expression in NK and B lymphocytes, as well as decreased intracellular calcium within specific conditions in CFS/ME patients. This warrants further examination of these pathways to elucidate whether TRPM3 and impaired calcium mobilisation has a role in CFS/ME.
Electronic supplementary material
The online version of this article (doi:10.1186/s40659-016-0087-2) contains supplementary material, which is available to authorized users.
Keywords: Chronic fatigue syndrome, Transient receptor potential, Calcium signalling, Myalgic encephalomyelitis
Background
Mammalian Transient receptor potential ion channels (TRPs) are comprised of six main groups including the TRPA (ankyrin), TRPC (canonical), TRPM (melastatin), TRPML (mucolipin), TRPP (polycystin) and TRPV (vanilloid) [1]. These have been found to have different biological functions, activation mechanisms and differential expression on tissues throughout the body [2]. TRPs are extensively expressed on almost all cells and their dysregulation has been associated with pathological conditions and diseases such as cancer, glomerulosclerosis, pain syndrome, Olmsted syndrome, mucolipidoses, and polycystic kidney disease [3–5].
TRPs are activated following fluctuations or deviations in the cellular environment, including pathogens, temperature, pressure, chemicals, oxidation and reduction, toxins, osmolarity, and pH [6, 7]. TRPs are calcium (Ca2+) permeable cation channels that act as an excitatory signal when open to induce depolarisation of the cell and allow Ca2+ influx. Ca2+ plays a role in intracellular signalling pathways, contributing to a number of cellular processes, for example cell division, apoptosis and transcriptional events [8]. TRPM3 protein expression has been identified on pancreatic β cells, [9] as well as on cells in the central nervous system and the eye [2]. Chronic fatigue syndrome, also referred to as Myalgic encephalomyelitis (CFS/ME) (is a disorder marked by unexplained, debilitating fatigue accompanied by a range of symptoms relating to multiple physiologies [12]. CFS/ME patients may have reactions to a number of environmental and biological factors [13]. TRP channels may be targeted during inflammatory reactions as they are easily activated in the presence of irritants, inflammatory products, and xenobiotic toxins. Symptoms relating to sensitivity to toxins and irritants have also been associated with CFS/ME [14], however the causes of these sensitivities have not been fully investigated.
Immunological dysfunction is commonly reported in CFS/ME. A significant reduction in NK cytotoxic activity is a consistent finding in CFS/ME patients compared with healthy controls [15–26]. Furthermore, a significant increase in CD20+CD5+ B cells and CD19+IgM+ B cells has been reported in CFS/ME patients compare with healthy controls [27–31]. Atypical SNPs of TRP genes, particularly SNPs in TRPM3 from peripheral blood mononuclear cells, NK and B cells have been recently reported in CFS/ME groups compared with healthy controls [10, 11, 32]. Thus, the first aim of this preliminary investigation was to identify TRPM3 surface protein expression on NK and B lymphocytes from healthy controls, using flow cytometric protocols. To date, there has been no previous evidence of this expression. Furthermore, protocols used to identify TRP receptors on specific cell subsets are based on spectroscopy and crystallography methods. The second aim of this study was to conduct a comparative assessment between CFS/ME and healthy controls for TRPM3 surface protein expression and changes in intracellular calcium influx in NK and B lymphocytes. Perturbations of TRPM3 expression and consequent changes in intracellular calcium influx could provide further knowledge as to the possibility of TRPM3 contributing to the pathogenesis of CFS/ME.
Methods
Subjects
This study consisted of 17 CFS/ME patients that were aged matched with 19 healthy controls (Table 1). Of the 17 CFS/ME patients (age = 48.68 ± 1.06 years), 14 (82 %) were females and 3 (18 %) were males. The 19 healthy controls (age = 46.48 ± 1.22 years) comprised 13 (68 %) females and 6 (32 %) males. CFS/ME patients were defined according to the Fukuda criteria, which required persistent, ongoing fatigue for at least 6 months accompanied by at least 4 of the following: post-exertional malaise, impaired short term memory or concentration, unrefreshed sleep, headaches, sore throat, tender lymph nodes, muscle pain, and joint pain [12]. The average duration of illness of this group was 8.4 years and reported no alternative disease or illness that would explain the onset of their symptoms. Any patients on potential immune modulating medication were excluded from this study. Healthy controls did not meet any CFS/ME criteria. All participants in this study were of Caucasian decent and residents of Australia at the time of blood collection. Exclusion criteria for all participants involved in this study were those who were smokers, pregnant or breastfeeding.
80 ml of whole blood was collected from all participants into EDTA blood tubes. Written consent was obtained from all participants prior to sample collection. Pathology testing parameters (full blood count, electrolytes, high sensitive C reactive protein and erythrocyte sedimentation rate) were performed on all participants. No significant differences in these pathology testing parameters were identified between groups (data not shown). This investigation was conducted under the Griffith University Ethics reference number (HREC/15/QGC/63).
Peripheral blood mononuclear cells preparations
Peripheral blood mononuclear cells (PBMCs) were isolated from 40 ml EDTA blood after obtaining participants’ written consent. PBMCs were isolated using Ficoll density gradient centrifugation (Life technologies) as previously described [22]. PBMCs were counted using Trypan blue and concentration was adjusted to 1 × 107 cells in 100 μl in Dulbecco’s phosphate-buffered saline (DPBS) with Ca2+ and magnesium (Life technologies) for all assays.
TRPM3 immunophenotyping assay
Due to the constitutive active property of TRPM3 protein and readily available anti-TRPM3 antibody, this study utilised anti-TRPM3 antibody to measure protein expression as the antibody binds to the extracellular surface of the protein structure. Initially, PBMCs were incubated in 20 μl of FCR blocking reagent (Miltenyi Biotec) for 10 min at room temperature and washed with phosphate buffer saline (PBS) prior to centrifuge at 400g for 5 min. Supernatant was removed and incubated with primary fluorochrome labelled antibodies (CD19-BV421, CD3-PerCP, CD56-BV421 and CD16-APC Cy7, purchase from BD Bioscience) for 30 min at room temperature in the dark. Labelled cells were washed and incubated with 10 μg final concentration of goat anti-human TRPM3 antibody for 30 min. This was followed by a wash and resuspension in a final concentration of 5 % (v/v) of Bovine Serum Albumin (Sigma) for 30 min. Cells were washed again and incubated with 5 μg final concentration of donkey anti-goat IgG FITC (Santa Scruz) for 30 min. Cells were washed and resuspended in 200 μl of staining buffer (BD Bioscience) and acquired at 50,000 events using LSRFortessa X-20 (BD Bioscience).
LSR Fortessa X-20 flow cytometry analysis
Lymphocyte populations were identified using forward scatter and side scatter (FSC, SSC) dot plots. Exclusions were CD3+ cells and only CD3− lymphocytes were further used to characterised B lymphocytes and NK cell subset populations using CD19, CD56 and CD16 as previously described [22, 33]. Total B cells were identified as CD19+, whereas NK cell subsets were characterised using the expression of CD56BrightCD16Dim/− NK cells, CD56DimCD16Bright/+ NK cells and CD56−CD16+ NK cells (Fig. 1). TRPM3 expression was measured as percentage of parent cells (%) and mean fluorescence intensity (MFI) (Additional file 1: Table S1). LSRFotessa X-20 flow cytometry was utilised for sequential determination of cytoplasmic calcium [Ca2+]C and mitochondrial [Ca2+]M, to help compare cytoplasmic or mitochondrial Ca2+ influx kinetics in B lymphocytes and NK cells. Characterising kinetic measurements using median florescence of Fura-AM or Rhod-2 AM dye were used and smoothing curve method was applied to measure the area under the curve (AUC).
Fig. 1.
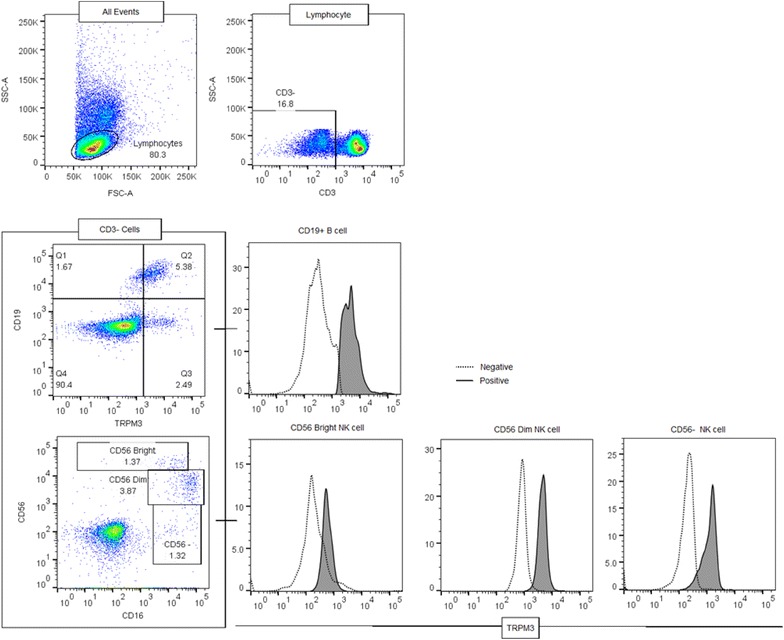
Immunophenotype of TRPM3 channel surface expression on B and NK cells by flow cytometry. Total B cells were identified using Anti-CD19. NK cells were characterised using the expression of CD56BrightCD16Dim/− NK cells, CD56DimCD16Bright/+ NK cells and CD56−CD16+ NK cells, followed by, identification of TRPM3 surface expression
Cytoplasmic calcium influx assay
Following phenotypic staining, the cells were incubated with 0.5 ml staining buffer that contained 0.02 % Pluronic® F-127 and 1 μM Fura-red AM or Rhod-2 AM for 30 min in the incubator at 37 °C. Stained cells were washed with DPBS without calcium and magnesium. Fura AM stained cells were stimulated after 30 s of flow cytometric acquisition in the presence of a final concentration of 1.4 μg streptavidin, 714 ng ionomycin, 50 μg 2-APB or 14 μg thapsigargin. Data was recorded over 4 min. Rhod-2 AM stain cells were incubated for a further 12 h, prior to acquisition. Unstimulated cells were used as a baseline when assessing the stimulation response’s AUC.
Thapsigargin is a potent inhibitor for Calcium-ATPase receptors and raises cytoplasmic calcium concentration by inhibiting the ability for the cells to pump calcium into the endoplasmic reticulum (ER) [34]. 50 ug 2-aminoethoxydiphenyl borate (2-APB) was used given its inhibition of ER and IP3R as previously described [35]. NK receptors (NG2DA and NKp46) were cross-linked [36] to ensure co-stimulation of human NK cells to measure calcium influx. CD19 and CD21 (complement receptor CR2) on B cells are responsible for activating the Immunoglobulin M (IgM) receptor and the three proteins together make the B cell receptor complex. This complex was then stabilised by streptavidin-mediated cross-linking to induce permanent calcium influx and enhance activation of CD19+ B cells [37].
Statistical data analyses
Statistical analyses were performed using IBM SPSS Statistics version 22 software (SPSS, Chicago, USA). Significance was tested by MANOVA and reported at the P < 0.05 level between CFS/ME and healthy control groups for parameters including TRPM3 protein expression, and calcium influx in B lymphocytes and NK cells. Data are given as mean ± SEM. Flowjo was employed to analyse FCS files extracted from FACSDiva 8 software (BD Bioscience). Post Hoc test was further performed to determine specifically where the significance was between healthy controls and CFS/ME. The Levene test was used to analyse homogeneity of variance between groups.
Results
Subject characteristics
There was no significant difference in age between healthy controls (n = 19, 48.32 ± 2.71) and CFS/ME patients (n = 17, 51.24 ± 2.14)
Identification of TRPM3 on Natural Killer cells and B lymphocytes
The fluorescence of fluorescein isothiocyanate conjugated (FTIC) secondary antibody was employed to quantitatively measure TRPM3 surface protein expression. Forward and side scatter dot plots were utilised to identified lymphocyte population prior to gating CD56Bright NK cells (CD3−CD56BrightCD16−), CD56Dim NK cells (CD3−CD56dimCD16+) and total B cells (CD3−CD19+) (Fig. 1). TRPM3+ NK cell subsets (Additional file 2: Figure 1) and TRPM3+ B cells (Additional file 3: Figure 2) were further identified for CFS/ME and healthy controls.
Comparative assessment of TRPM3 on Natural Killer cells and B lymphocytes between healthy controls and CFS/ME patients
CD56Bright NK cells showed significantly decreased TRPM3 expression (17.37 ± 5.34 %) in CFS/ME compared with healthy controls (35.72 ± 7.38 %) (Fig. 2a). Similarly, CD19+ cells demonstrated significantly decreased TRPM3 expression in CFS/ME (1.56 ± SEM 0.191 %) compared with healthy controls (2.06 ± SEM 0.16 %) (Fig. 2b). CD56Dim cells showed no significant difference between groups (Fig. 2a).
Fig. 2.
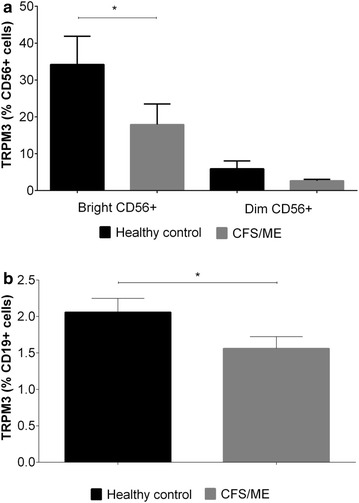
TRPM3 expression (%) on B lymphocytes and NK cells gated from HC and CFS/ME peripheral mononuclear cells. a NK cells subsets were charactered as CD56Bright NK cells and CD56Dim NK cells. Identification of TRPM3 surface expression on the NK cell subsets was analysed using indirect flow cytometry. b B cells were characterised as total B cells (CD3−CD19+) and indirect flow cytometry was employed to identify TRPM3 surface expression on B cells. Histograms report the means ± SEM. Asterisk denotes P < 0.05. HC healthy controls, CFS Chronic fatigue syndrome
Cytoplasmic and mitochondrial calcium influx
Fura-2 AM and Rhod-2 AM exhibit an increase in fluorescence upon binding to cytoplasmic and mitochondrial calcium ions. Changes in mitochondrial calcium concentration in the presence of stimulants showed no significance. CFS/ME patients showed significantly reduced cytoplasmic calcium ion concentration in CD19+ B cells during cross-linking of CD21 and IgM receptor (P < 0.01) (Fig. 3). CD56bright NK cells demonstrated a significantly decreased cytoplasmic calcium influx in the presence of 50 μg 2-APB and 14 μg thapsigargin (P < 0.05)(Fig. 4).
Fig. 3.
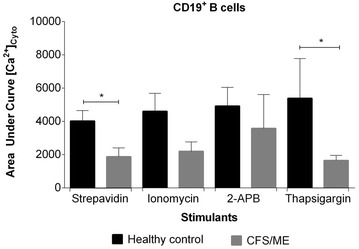
Fura-AM cytoplasmic calcium influx in CD19 + B cells. Calcium influx response curve reported as area under the curve was measure during Anti-IgM and anti-CD21 conjugated biotins were cross-linked with streptavidin or in the presence of ionomycin, 2-APB or thapsigargin. Histograms report the means ± SEM. Asterisk denotes statistically significance at P < 0.05
Fig. 4.
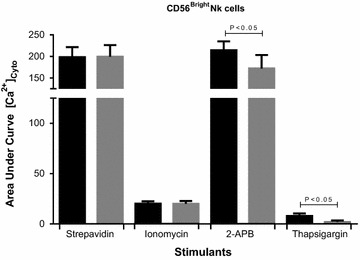
Fura-AM cytoplasmic calcium influx response during CD56Bright NK cell receptors, Anti-CD314 and anti-CD335 conjugated biotins were cross-linked with streptavidin or in the presence of ionomycin, 2-APB or thapsigargin. Histograms report the means ± SEM. Asterisk denotes statistically significance at P < 0.05
Discussion
The present paper reports, for the first time, the identification of TRPM3 surface expression on NK cells and B lymphocytes in CFS/ME patients as well as healthy controls. Further, diminished expression of TRPM3 surface expression was found on B lymphocytes and NK cells in CFS/ME patients compared with healthy controls. We also report, for the first time, a significant reduction in cytoplasmic calcium ion concentration in CD19+ B lymphocytes during cross-linking between anti-CD21 and anti-IgM following treatment with stepadividin or thapsigargin in CFS/ME patients. CD56bright NK cells also had a significant decrease in cytoplasmic calcium in the presence of 2-APB and thapsigargin in CFS/ME patients.
Investigation of TRP ion channels expression on lymphocytes has been very limited due to the difficulties of methodology as TRP channels are in relatively low abundance and there is limited availability of specific and high-affinity antibodies. Other methods of identification of TRP include molecular genetics by assessing TRP mRNA expression or isolation of protein from plasma membrane. Also, investigations of TRP ion channel functions and their roles in disease states have been reported through voltage clamping methodology or in vitro cell lines. Knock-out animal models have also been reported to study TRP ion channels (see review: [38]). TRPM3 has been reported to be expressed on a wide variety of tissues, such as kidneys, eyes, sensory neurons of the dorsal root ganglia and the β islet cells of the pancreases [2]. Only TRPM4, TRPM5, TRPM2 and TRPM7 surface expression have been reported on B cells, bone marrow cells, splenic cells, lymph node B cells and T and mast cells [39]. However, no previous investigation has reported TRPM3 on B lymphocytes or NK cells. Our novel findings suggest TRPM3 on B lymphocytes and NK cells may also be involved in calcium-mediated roles, such as those seen with other TRP family members. These calcium-mediated TRP functions contribute to a number of cellular processes and cellular functions. These processes include regulating enzymatic function and transcription factors, lymphocyte proliferation and differentiation of naïve cells into effector and memory cells as well as the production of cytokines and chemokines (see review [8]). TRPM3 alone or in conjunction with TRPC members, members of the TRPV1 family, together with muscarinic M3 receptors [40] and calcium release activated calcium (CRAC) channels [41] may need to be considered for calcium intracellular-mediated roles.
Our preliminary research findings report a significant reduction in TRPM3 surface expression for B lymphocytes and CD56Bright cells from CFS/ME patients compared with healthy controls. Our group has recently reported significant associations of SNPs, predominately in TRPM3 genes, from NK and B lymphocytes in CFS/ME patients compared with healthy controls [10, 11, 32]. The significant reduction in TRPM3 surface protein expression in conjunction with a significant reduction in Ca2+ influx from CD56bright NK cells suggests important functional implications. When Ca2+ influx is prevented or reduced an immunological synapse is not formed [42, 43]. A rise or pulsatile influx in intracellular Ca2+ is required in cells, such as Natural Killer cells and cytotoxic lymphocytes to initiate cell function, such as the cell lysis of a target infected cell, as well as formation of mitotic spindles for cytoskeleton movement to facilitate the secretory vesicles to fuse with the plasma membrane to ultimately form the immune synapse. Moreover, the production of lytic granules for targeting and killing infected cells is also dependent on Ca2+ [44].
Treatment with streptavidin cross-linked with NKD2A and NKp46, which are Ca2+ dependent, showed no significant reduction for cytoplasmic calcium in CD56Bright NK cells. In contrast, treatment with thapsigargin or 2-APB, suggests impairment of Ca2+ influx and storage. Treatment with 2-APB, which is both non-selective for TRPM3 [45] and an IP3R antagonist [35] showed a significant difference in intracellular Ca2+. Treatment with thapsigargin showed a significant reduction in Ca2+ in the CFS/ME group compared with the healthy control group, suggesting Ca2+ stores may be a consequence of reduced Ca2+ influx into the cell, ultimately lowering the function and intracellular signalling capability of the cell. Recent research has identified that CD56bright NK cells are very likely precursor cells of the CD56dim subset. CD56dim NK cells have been shown to have shorter telomeres than CD56bright NK cells from peripheral blood, suggesting CD56bright cells are not as mature as CD56dim [46]. Additionally, purified CD56bright CD16− NK cells have been shown to differentiate into CD56dim cells that have the characteristic phenotypic and functional features of peripheral blood CD56dim NK cells [47]. CD56bright NK cells constitutively express the high- and intermediate-affinity IL-2 receptors and expand in vitro and in vivo in response to low (picomolar) doses of IL-2 [48, 49] and after activation with IL-2 or IL-12, CD56bright cells exhibit similar or enhanced cytotoxicity against NK targets compared with CD56dim cells [50]. We and others have shown significant reductions in CD56bright NK cells in CFS/ME patients, significant reductions in lytic function of CD56bright NK cells as well as significantly reduced lytic granules [16, 18, 26, 51] in CFS/ME patients.
Importantly, cross linking anti-IgM and anti-CD21 initiates a calcium-dependent pathway through transmembrane proteins, most notably Igα (CD79a) and Igβ (CD79b). B cell linker protein (BLNK) binds to tyrosine of Igα via the intracellular mechanism of crosslinking Igα and Igβ cytoplasmic tails and phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAM) tyrosines which occurs by Src-family tyrosine kinases (SFTKs) and/or Syk, ultimately leading to calcium influx and protein kinase C (PKC) activation [52]. In the present study, B lymphocytes treated with streptavidin showed a significant reduction of intracellular Ca2+. In contrast, treatment with 2-APB showed no significant reduction in intracellular Ca2+. However, treatment with thapsigargin showed a significant difference in intracellular Ca2+. One rationale for this outcome may be due to calcium signalling by other TRP families identified, for example TRPM4, TRPM5 and TRPM2 that have been identified on B cells, bone marrow cells and lymph node B cells. It is feasible these other TRP subfamilies [39, 53], may play a role in intracellular Ca2+ influx and storage. Interestingly, muscarinic acetylcholine receptors have been found to inhibit TRPM3 via the action of phospholipase C [40]. Such interaction may provide additional complexity to Ca2+ cell signalling as we have recently reported a significant association of SNPs in genes of muscarinic acetylcholine receptors M3 from the same cohort of CFS/ME patients who had significant association of SNPs in their TRPM3 genes from peripheral blood mononuclear cells [54]. Future investigations comparing cell subsets that do not express TRPM3 need to be explored. Additionally, pathways that potentially influence calcium influx may discern whether reduced TRPM3 has a role in reduced intracellular calcium ion concentration.
Conclusions
Our novel discovery of TRPM3 on NK and B lymphocytes and their significantly reduced expression in CFS/ME patients, together with the preliminary discovery of impaired Ca2+ cytoplasmic concentration in these cells, suggests impaired Ca2+ metabolism may be implicated as a novel pathway for pathogenesis of CFS/ME. Further investigation may elucidate intracellular Ca2+ signalling pathways and the potential role this may play on transcriptional factors, such as NFκB and NFAT as both are activated by calcium through the calmodulin dependent protein kinases and phosphatases. Moreover, as TRPs are widely expressed on almost all human cells and tissues, this raises the question as to whether similar changes of TRPM3 expression or function of these receptors promotes more widespread disruption of intracellular signalling homeostasis in CFS/ME patients.
Authors’ contributions
SMG, DRS, PS, BN and TN designed and developed all experiments as well as analysis, revisions and final preparation of this article. SMG, DRS and TN were involved in the sample preparation and drafting manuscript. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by funding from the Stafford Fox Medical Research Foundation, Change for ME Charity, Alison Hunter Memorial Foundation, Mason Foundation and Queensland Co-Investment Program.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- 2-APB
2-aminoethoxydiphenyl borate
- AUC
area under the curve
- Ca2+
calcium
- CFS/ME
Chronic fatigue syndrome/Myalgic encephalomyelitis
- CNS
central nervous system
- CRAC
calcium release activated calcium
- EDTA
ethylenediaminetetraacetic acid
- ER
endoplasmic reticulum
- FTIC
fluorescein isothiocyanate conjugated
- ITAM
immunoreceptor tyrosine-based activation motif
- MANOVA
multivariate analysis of variance
- NK
Natural Killer cell
- PBMCs
peripheral blood mononuclear cells
- PBS
phosphate buffer saline
- SFTK
src-family tyrosine kinases
- SNP
single nucleotide polymorphisms
- TRPA
Transient receptor potential ankyrin
- TRPC
Transient receptor potential canonical
- TRPM
Transient receptor potential melastatin
- TRPM3
Transient receptor potential melastatin subfamily 3
- TRPML
Transient receptor potential mucolipin
- TRPP
Transient receptor potential polycystin
- TRPV
Transient receptor potential vanilloid
Additional files
10.1186/s40659-016-0087-2 The cell populations CD56 Bright, CD56 Dim and CD19+ cells expressing TRPM3 as percentage of parent cells (%) and Mean Fluorescence Intensity (MFI).
10.1186/s40659-016-0087-2 Dot plots gating strategies for TRPM3 expressed on NK cells. Identification of NK cell subsets and TRPM3 positive in healthy controls (a.) and CFS/ME patient (b.) that are based on the isotype controls (c.).
10.1186/s40659-016-0087-2 Dot plots gating strategies for TRPM3 expressed on B cells. Identification of B cell subsets and TRPM3 positive in healthy controls (a.) and CFS/ME patient (b.) that are based on the isotype controls (c.).
Contributor Information
T. Nguyen, Email: thao.nguyen2@griffithuni.edu.au
D. Staines, Email: d.staines@griffith.edu.au
B. Nilius, Email: Bernd.Nilius@med.kuleuven.be
P. Smith, Email: pksm@mac.com
S. Marshall-Gradisnik, Email: s.marshall-gradisnik@griffith.edu.au
References
- 1.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426(6966):517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 2.Oberwinkler J, Philipp SE. Trpm3. Handb Exp Pharmacol. 2014;222:427–459. doi: 10.1007/978-3-642-54215-2_17. [DOI] [PubMed] [Google Scholar]
- 3.Nilius B, Szallasi A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev. 2014;66(3):676–814. doi: 10.1124/pr.113.008268. [DOI] [PubMed] [Google Scholar]
- 4.Vennekens R, Menigoz A, Nilius B. TRPs in the brain. Rev Physiol Biochem Pharmacol. 2012;163:27–64. doi: 10.1007/112_2012_8. [DOI] [PubMed] [Google Scholar]
- 5.Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011;12(3):218. doi: 10.1186/gb-2011-12-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moran MM, et al. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10(8):601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- 7.Nieto-Posadas A, Jara-Oseguera A, Rosenbaum T. TRP channel gating physiology. Curr Top Med Chem. 2011;11(17):2131–2150. doi: 10.2174/156802611796904870. [DOI] [PubMed] [Google Scholar]
- 8.Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7(9):690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- 9.Colsoul B, Vennekens R, Nilius B. Transient receptor potential cation channels in pancreatic beta cells. Rev Physiol Biochem Pharmacol. 2011;161:87–110. doi: 10.1007/112_2011_2. [DOI] [PubMed] [Google Scholar]
- 10.Marshall-Gradisnik S. Natural killer cells and single nucleotide polymorphisms of specific ion channels and receptor genes in myalgic encephalomyelitis/chronic fatigue syndrome. Appl Clin Genet. 2016;9:39–47. doi: 10.2147/TACG.S99405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marshall-Gradisnik S, et al. Single nucleotide polymorphisms and genotypes in transient receptor potential ion channel and acetylcholine receptor genes from isolated B lymphocytes in myalgic ecephalomyelitis/chronic fatigue syndrome patients. J Int Med Res. 2016 (in review). [DOI] [PMC free article] [PubMed]
- 12.Fukuda K, et al. The chronic fatigue syndrome: a comprehensive approach to its definition and study. International chronic fatigue syndrome study group. Ann Intern Med. 1994;121(12):953–959. doi: 10.7326/0003-4819-121-12-199412150-00009. [DOI] [PubMed] [Google Scholar]
- 13.Brown MM, Jason LA. Functioning in individuals with chronic fatigue syndrome: increased impairment with co-occurring multiple chemical sensitivity and fibromyalgia. Dyn Med. 2007;6:6. doi: 10.1186/1476-5918-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carruthers BM, et al. Myalgic encephalomyelitis: international consensus criteria. J Intern Med. 2011;270(4):327–338. doi: 10.1111/j.1365-2796.2011.02428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenu EW, et al. Natural killer cells in patients with severe chronic fatigue syndrome. Auto Immun Highlights. 2013;4(3):69–80. doi: 10.1007/s13317-013-0051-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brenu EW, et al. Role of adaptive and innate immune cells in chronic fatigue syndrome/myalgic encephalomyelitis. Int Immunol. 2014;26(4):233–242. doi: 10.1093/intimm/dxt068. [DOI] [PubMed] [Google Scholar]
- 17.Caligiuri M, et al. Phenotypic and functional deficiency of natural killer cells in patients with chronic fatigue syndrome. J Immunol. 1987;139(10):3306–3313. [PubMed] [Google Scholar]
- 18.Maher KJ, Klimas NG, Fletcher MA. Chronic fatigue syndrome is associated with diminished intracellular perforin. Clin Exp Immunol. 2005;142(3):505–511. doi: 10.1111/j.1365-2249.2005.02935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ojo-Amaize EA, Conley EJ, Peter JB. Decreased natural killer cell activity is associated with severity of chronic fatigue immune dysfunction syndrome. Clin Infect Dis. 1994;18(Suppl 1):S157–S159. doi: 10.1093/clinids/18.Supplement_1.S157. [DOI] [PubMed] [Google Scholar]
- 20.Aoki T, et al. Low NK syndrome and its relationship to chronic fatigue syndrome. Clin Immunol Immunopathol. 1993;69(3):253–265. doi: 10.1006/clin.1993.1178. [DOI] [PubMed] [Google Scholar]
- 21.Brenu EW, et al. Longitudinal investigation of natural killer cells and cytokines in chronic fatigue syndrome/myalgic encephalomyelitis. J Transl Med. 2012;10:88. doi: 10.1186/1479-5876-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardcastle SL, et al. Characterisation of cell functions and receptors in Chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) BMC Immunol. 2015;16:35. doi: 10.1186/s12865-015-0101-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huth TK, et al. Pilot study of natural killer cells in chronic fatigue syndrome/myalgic encephalomyelitis and multiple sclerosis. Scand J Immunol. 2016;83(1):44–51. doi: 10.1111/sji.12388. [DOI] [PubMed] [Google Scholar]
- 24.Levine PH, et al. Dysfunction of natural killer activity in a family with chronic fatigue syndrome. Clin Immunol Immunopathol. 1998;88(1):96–104. doi: 10.1006/clin.1998.4554. [DOI] [PubMed] [Google Scholar]
- 25.Barker E, et al. Immunologic abnormalities associated with chronic fatigue syndrome. Clin Infect Dis. 1994;18(Suppl 1):S136–S141. doi: 10.1093/clinids/18.Supplement_1.S136. [DOI] [PubMed] [Google Scholar]
- 26.Klimas NG, et al. Immunologic abnormalities in chronic fatigue syndrome. J Clin Microbiol. 1990;28(6):1403–1410. doi: 10.1128/jcm.28.6.1403-1410.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradley AS, Ford B, Bansal AS. Altered functional B cell subset populations in patients with chronic fatigue syndrome compared to healthy controls. Clin Exp Immunol. 2013;172(1):73–80. doi: 10.1111/cei.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klimas NG, Koneru AO. Chronic fatigue syndrome: inflammation, immune function, and neuroendocrine interactions. Curr Rheumatol Rep. 2007;9(6):482–487. doi: 10.1007/s11926-007-0078-y. [DOI] [PubMed] [Google Scholar]
- 29.Natelson BH, Haghighi MH, Ponzio NM. Evidence for the presence of immune dysfunction in chronic fatigue syndrome. Clin Diagn Lab Immunol. 2002;9(4):747–752. doi: 10.1128/CDLI.9.4.747-752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robertson MJ, et al. Lymphocyte subset differences in patients with chronic fatigue syndrome, multiple sclerosis and major depression. Clin Exp Immunol. 2005;141(2):326–332. doi: 10.1111/j.1365-2249.2005.02833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramos S, et al. Characterisation of B cell subsets and receptors in chronic fatigue syndrome patients. J Clin Cell Immunol. 2015;16:35. doi: 10.1186/s12865-015-0101-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall-Gradisnik S, et al. Examination of single nucleotide polymorphisms (SNPs) in transient receptor potential (TRP) ion channels in chronic fatigue syndrome patients. Immunol Immunogenet Insights. 2015;7:1–6. doi: 10.4137/III.S25147. [DOI] [Google Scholar]
- 33.Ramos S, et al. Characterisation of B cell subsets and receptors in chronic fatigue syndrome patients. J Clin Cell Immunol. 2015;16(1):35. doi: 10.1186/s12865-015-0101-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding WX, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282(7):4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 35.Bootman MD, et al. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2 + release. FASEB J. 2002;16(10):1145–1150. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- 36.Bryceson YT, et al. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107(1):159–166. doi: 10.1182/blood-2005-04-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rossbacher J, Shlomchik MJ. The B cell receptor itself can activate complement to provide the complement receptor 1/2 ligand required to enhance B cell immune responses in vivo. J Exp Med. 2003;198(4):591–602. doi: 10.1084/jem.20022042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desai BN, Clapham DE. TRP channels and mice deficient in TRP channels. Pflugers Arch. 2005;451(1):11–18. doi: 10.1007/s00424-005-1429-z. [DOI] [PubMed] [Google Scholar]
- 39.Wu LJ, Sweet TB, Clapham DE. International union of basic and clinical pharmacology. LXXVI. current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010;62(3):381–404. doi: 10.1124/pr.110.002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badheka D, Borbiro I, Rohacs T. Transient receptor potential melastatin 3 is a phosphoinositide-dependent ion channel. J Gen Physiol. 2015;146(1):65–77. doi: 10.1085/jgp.201411336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parekh AB. Mitochondrial regulation of store-operated CRAC channels. Cell Calcium. 2008;44(1):6–13. doi: 10.1016/j.ceca.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 42.Delon J, et al. Imaging antigen recognition by naive CD4+ T cells: compulsory cytoskeletal alterations for the triggering of an intracellular calcium response. Eur J Immunol. 1998;28(2):716–729. doi: 10.1002/(SICI)1521-4141(199802)28:02<716::AID-IMMU716>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 43.Bhakta NR, Oh DY, Lewis RS. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat Immunol. 2005;6(2):143–151. doi: 10.1038/ni1161. [DOI] [PubMed] [Google Scholar]
- 44.Lyubchenko TA, Wurth GA, Zweifach A. Role of calcium influx in cytotoxic T lymphocyte lytic granule exocytosis during target cell killing. Immunity. 2001;15(5):847–859. doi: 10.1016/S1074-7613(01)00233-3. [DOI] [PubMed] [Google Scholar]
- 45.Xu SZ, et al. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: a differential, extracellular and voltage-dependent effect. Br J Pharmacol. 2005;145(4):405–414. doi: 10.1038/sj.bjp.0706197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouyang Q, et al. Telomere length in human natural killer cell subsets. Hematopoietic Stem Cells Vi. 2007;1106:240–252. doi: 10.1196/annals.1392.001. [DOI] [PubMed] [Google Scholar]
- 47.Chan A, et al. CD56(bright) human NK cells differentiate into CD56(dim) cells: role of contact with peripheral fibroblasts. J Immunol. 2007;179(1):89–94. doi: 10.4049/jimmunol.179.1.89. [DOI] [PubMed] [Google Scholar]
- 48.Nagler A, et al. Comparative studies of human FcRIII-positive and negative natural killer cells. J Immunol. 1989;143(10):3183–3191. [PubMed] [Google Scholar]
- 49.Jacobs R, et al. CD56brightNK cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol. 2001;31(10):3121–3126. doi: 10.1002/1521-4141(2001010)31:10<3121::AID-IMMU3121>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 50.Ellis TM, Fisher RI. Functional heterogeneity of Leu 19″bright” + and Leu 19″dim” + lymphokine-activated killer cells. J Immunol. 1989;142(8):2949–2954. [PubMed] [Google Scholar]
- 51.Fletcher MA, et al. Biomarkers in chronic fatigue syndrome: evaluation of natural killer cell function and dipeptidyl peptidase IV/CD26. PLoS ONE. 2010;5(5):e10817. doi: 10.1371/journal.pone.0010817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120(6):1175–1184. doi: 10.1182/blood-2012-02-362624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446(7133):284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- 54.Marshall-Gradisnik S, et al. Examination of single nucleotide polymorphisms in acetylcholine receptors in chronic fatigue syndrome patients. Immunol Immunogenet Insights. 2015;7:7–20. doi: 10.4137/III.S25105. [DOI] [Google Scholar]