

In this study, Tsabar et al. investigated how the DNA damage checkpoint is extinguished and found that dissociation of histone H3 from Asf1, a histone chaperone, is required for efficient recovery. They also show that Asf1 is required for complete dephosphorylation of Rad53 when the upstream DNA damage checkpoint signaling is turned off, providing new insights into the mechanisms regulating the response to DNA damage.
Keywords: Asf1, Rad53, Rtt101, Rtt109, DNA damage checkpoint, DNA double-stranded breaks
Abstract
To allow for sufficient time to repair DNA double-stranded breaks (DSBs), eukaryotic cells activate the DNA damage checkpoint. In budding yeast, Rad53 (mammalian Chk2) phosphorylation parallels the persistence of the unrepaired DSB and is extinguished when repair is complete in a process termed recovery or when the cells adapt to the DNA damage checkpoint. A strain containing a slowly repaired DSB does not require the histone chaperone Asf1 to resume cell cycle progression after DSB repair. When a second, rapidly repairable DSB is added to this strain, Asf1 becomes required for recovery. Recovery from two repairable DSBs also depends on the histone acetyltransferase Rtt109 and the cullin subunit Rtt101, both of which modify histone H3 that is associated with Asf1. We show that dissociation of histone H3 from Asf1 is required for efficient recovery and that Asf1 is required for complete dephosphorylation of Rad53 when the upstream DNA damage checkpoint signaling is turned off. Our data suggest that the requirements for recovery from the DNA damage checkpoint become more stringent with increased levels of damage and that Asf1 plays a histone chaperone-independent role in facilitating complete Rad53 dephosphorylation following repair.
Cells use several pathways to repair double-stranded breaks (DSBs). G1-arrested cells repair DSBs mainly by nonhomologous end-joining (NHEJ) (Lewis and Resnick 2000; Ferreira and Cooper 2004), but, after CDK1 activation and the initiation of 5′-to-3′ resection of DSB ends, the repair pathway choice shifts to homologous recombination (HR) (Aylon et al. 2004; Ira et al. 2004). HR requires that the cell harbor a homologous sequence that can be used as a template to repair the DSB. When the donor sequence is homologous to both sides of the DSB, cells will primarily repair using gene conversion (GC), a largely error-free process (Krogh and Symington 2004; Haber 2013). When the DSB is created in a region flanked by homologous sequences, the DSB can be repaired by single-strand annealing (SSA) (Krogh and Symington 2004). SSA depends on extensive 5′-to-3′ resection followed by the annealing of complementary single strands and results in a deletion of the sequences between the homologies.
To allow for sufficient time to repair, cells activate the DNA damage checkpoint (Harrison and Haber 2006; Ciccia and Elledge 2010). In budding yeast, the two PI3K-like kinases Mec1 and Tel1 (homologs of mammalian ATR and ATM, respectively) phosphorylate a cascade of downstream effectors to enforce G2/M arrest (Harrison and Haber 2006; Gobbini et al. 2013). Tel1 is recruited to the DSB ends through its interaction with the MRX complex, a complex required for the initiation of resection (Nakada et al. 2003). Mec1 forms a complex with the essential protein Ddc2, the yeast homolog of the mammalian ATRIP (Paciotti et al. 2000). After resection takes place, RPA rapidly coats the ssDNA, and the RPA-coated ssDNA recruits Ddc2 to the sites of damage; this recruitment is essential for checkpoint signaling by Mec1 (Rouse and Jackson 2000; Cortez et al. 2001; Melo et al. 2001; Wakayama et al. 2001; Zou and Elledge 2003).
Chk1 and Rad53, the budding yeast homologs of mammalian Chk1 and Chk2, are phosphorylated in response to damage by Mec1 and Tel1 (Sanchez et al. 1996, 1999; Sun et al. 1996; Gardner et al. 1999), although, in budding yeast, Mec1 is the dominant kinase for this modification. Another target of Mec1 and Tel1 phosphorylation, Rad9 (yeast ortholog of 53BP1), serves as a scaffold for Rad53 binding after their phosphorylation (Sun et al. 1998; Gilbert et al. 2001; Schwartz et al. 2002; Sweeney et al. 2005). Upon association, Rad53 undergoes autophosphorylation and is released from Rad9 to promote arrest (Gilbert et al. 2001). Rad53 hyperphosphorylation is correlated to the duration of repair. Repair events that are resolved rapidly do not trigger Rad53 hyperphosphorylation and do not cause a noticeable cell cycle delay, whereas DSBs that are slow to repair trigger Rad53 hyperphosphorylation (Pellicioli et al. 2001; Vaze et al. 2002; Kim and Haber 2009).
Following repair of a DSB, cells turn off the DNA damage checkpoint and resume cell cycle progression. This process, termed recovery, requires dephosphorylation of Rad53 by the PP2C family phosphatases Ptc2 and Ptc3 (Leroy et al. 2003). The H3–H4 histone chaperones Asf1 and CAF-1 also play a redundant role in recovery, although their role remains unclear (Chen et al. 2008; Kim and Haber 2009; Tsabar and Haber 2013). In mammalian cells, Asf1 and CAF-1 function synergistically to re-establish nucleosomes following nucleotide excision repair (Mello et al. 2002). Cells that fail to repair the DSB can also turn off the DNA damage checkpoint in a process termed adaptation (Sandell and Zakian 1993; Toczyski et al. 1997; Lee et al. 1998). In this process, cells arrest for 12–15 h and then resume cell cycle progression. Cells that suffer more than one unrepaired DSB fail to adapt (Lee et al. 1998), perhaps because Rad53 is hyperphosphorylated to a greater extent. Indeed, the level of hyperphosphorylation is well correlated to the number of DSBs that the cells suffer (Mantiero et al. 2007). These findings demonstrate that Rad53 acts as an indicator for both the persistence and the extent of DNA damage.
Although multiple irreparable DSBs elicit a stronger checkpoint response, it is unclear whether multiple repairable DSBs trigger a stronger checkpoint response as well. In this study, we asked whether cells can monitor the number of repairable DSBs, even if one of these DSBs is repaired so rapidly that it does not itself elicit Rad53 hyperphosphorylation. We show that an addition of a second, repairable DSB does not cause a recovery defect in wild-type cells. However, unlike our finding for a single DSB where both Asf1 and CAF-1 participate in recovery, when the cell is challenged by two repairable DSBs, deletion of ASF1 alone is sufficient to cause a recovery defect, suggesting that the requirements for recovery from a single DSB and multiple DSBs are different. This two-DSB system provides us with a tool to study the requirements for recovery from more than one DSB.
We also explored how proteins that genetically or physically interact with Asf1 affect recovery. After binding to Asf1, histone H3 undergoes acetylation on Lys56 by the histone acetyltransferase Rtt109 (Collins et al. 2007; Driscoll et al. 2007; Han et al. 2007; Tsubota et al. 2007; Fillingham et al. 2008). Rtt101, a Cul4 subunit of the Roc1-dependent E3 ubiquitin ligase, ubiquitylates histone H3 on Lys121, Lys122, and Lys125, with a preference for histone H3 that has been acetylated on Lys56 (Han et al. 2013). Rtt101-mediated ubiquitylation of H3 promotes the handoff of the histone H3–H4 heterodimer from Asf1 to CAF-1 (Han et al. 2013). We found that RTT101 and RTT109 are epistatic to ASF1, suggesting that the handoff of the histone H3–H4 heterodimer from Asf1 to CAF-1 is required for recovery from the DNA damage checkpoint.
The modifications of histone H3 also affect the association of Asf1 with Rad53. Histone H3 and Rad53 share the same Asf1-binding site and therefore compete for Asf1 binding (Emili et al. 2001; Jiao et al. 2012). In budding yeast, Asf1 binds most of the free and unmodified Rad53 in the cell (Emili et al. 2001; Hu et al. 2001). Phosphorylation of Rad53 following DNA damage prevents this interaction and frees Rad53 to undergo autophosphorylation and allows Asf1 to bind histone H3K56ac (Emili et al. 2001; Hu et al. 2001). We show that impairing the interaction between histone H3 and Asf1 rescues the recovery defects of rtt101Δ and rtt109Δ but not asf1Δ, demonstrating that the alleviation of the interaction between Asf1 and histone H3 is required for recovery. We propose that the dissociation of histone H3 from Asf1 frees Asf1 to promote recovery by binding and sequestering dephosphorylated Rad53.
Results
Addition of a rapidly repairable DSB to a slowly repaired DSB does not exacerbate the DNA damage checkpoint activation in wild-type cells
Our first goal was to see whether adding a rapidly repaired DSB to a cell in which a DSB was slowly repaired by ectopic GC would lead to more robust checkpoint activation. A second DSB, even rapidly repaired, might lead to reduced viability compared with a strain with only the slowly repaired break, analogous to the fact that two DSBs prevent adaptation.
We began with strain YJK17, in which an HO endonuclease-induced DSB within the MATα locus on chromosome 3 (Chr 3) is repaired by GC, using a donor consisting of a cloned segment of MATa-inc carrying a mutation that prevents HO cleavage, inserted at the ARG5,6 locus on the right arm of Chr 5 (Kim and Haber 2009). In this strain, the two normal homologous donors to repair a DSB at MAT (HML and HMR) have been deleted. Inducing a DSB in this strain activates the DNA damage checkpoint and results in GC repair over a 9-h time span, with ∼70% viability (Kim and Haber 2009).
To see whether adding a rapidly repairable DSB would create a greater checkpoint “load,” we integrated plasmid pNS102 at the ura3-52 locus to produce an SSA substrate in which the flanking 1-kb URA3 and ura3-52 homologous sequences are each separated by 2 kb from an HO endonuclease cleavage site (Fig. 1A; Sugawara and Haber 1992). SSA repair was complete in 3–5 h (Sugawara and Haber 1992). Addition of a rapidly repaired DSB (strain YFA01) did not lead to decreased viability in the wild-type background (Fig. 1B), indicating that both recovery and repair are proficient when two repairable DSBs are present.
Figure 1.

The two repairable DSB system. (A) Schematic of the two-DSB system. The top panel describes the GC assay (slower to repair), while the bottom panel describes the SSA repair construct (faster to repair). (B) Viability of the wild type and the cac1Δ, asf1Δ, and asf1Δ cac1Δ mutants in the ectopic GC (YJK17) and ectopic GC+SSA (YFA01) backgrounds. n ≥ 3. (**) P < 0.005, calculated relative to wild type. (C) Schematic of YMT170. After HO induction, repair requires a 25-kb resection and is executed through SSA, as in strain YMV080 (Vaze et al. 2002). Strain YMT170 is similar to YMV080, but the NAT cassette has been switched with HPH. (D) Viability of wild type (YMT170) and asf1Δ (YMT169).
Asf1 is not required for repair of two DSBs
Previously, we published that deletion of the H3–H4 histone chaperone ASF1 does not impede recovery in the YJK17 ectopic GC system, but, in conjunction with deletion of CAC1 (the largest subunit of CAF-1), recovery is reduced (Kim and Haber 2009). However, another study suggested that deletion of ASF1 alone was sufficient to impede recovery in a single-DSB system (Chen et al. 2008). To address this discrepancy, we tested the effect of asf1Δ or cac1Δ on viability of the one and two repairable DSB strains.
We first repeated the viability experiments for the previously studied systems: YJK17 (Kim and Haber 2009) and YMV80 (Vaze et al. 2002; Chen et al. 2008). As we previously reported, in strain YJK17, deletion of either CAC1 or ASF1 did not cause a reduction in viability, whereas viability in a cac1Δ asf1Δ double-mutant strain was reduced to 35% (Fig. 1B). In YMV80, repair occurs by SSA, but one of the flanking repeats is 25-kb away from the HO cleavage site and requires 25 kb of resection to expose both regions of homology; consequently, this repair event does not begin until ∼6 h and leads to strong checkpoint activation (Vaze et al. 2002). We used a derivative of YMV080 in which the NAT cassette had been replaced with HPH (YMT170) (Fig. 1C). Deleting ASF1 did not prevent recovery in this system (Fig. 1D), further supporting our previous findings that deletion of ASF1 in a single-DSB system does not impair recovery when the cell needs to repair a single DSB.
We next tested the effect of deleting ASF1 and CAC1 in the two-DSB system YFA01. As with the single DSB, the viability of cac1Δ was comparable with wild type, but deletion of ASF1 alone was sufficient to reduce viability in the two-DSB system from 70% to 40% (Fig. 1B). The viability of the cac1Δ asf1Δ double mutant was not further reduced as compared with a single DSB (35%).
We next tested the effect of ASF1 deletion on repair. We monitored GC and SSA separately by Southern blot. In wild-type cells, GC was 90% completed by 9 h (Fig. 2A,E). Repair of this DSB in the two-DSB system was similar in outcome and kinetics to those previously reported when only the ectopic GC was present (Kim and Haber 2009). Repair of the SSA DSB was 100% completed by 3–5 h (Fig. 2A,E), comparable with the kinetics and outcome previously reported in the system that contained only this SSA event (Sugawara and Haber 1992). Repair in asf1Δ is comparable with wild type for both the ectopic GC and SSA (Fig. 2B,E). The observation that deletion of ASF1 led to a reduction in viability without impeding repair suggests that deletion of ASF1 causes a recovery defect when the cells experience two repairable DSBs.
Figure 2.

Repair kinetics in the two-DSB system. Southern blot monitoring repair of the GC DSB (top panel) and the SSA DSB (bottom panel) in wild-type (YFA01) (A), asf1Δ (YFA02) (B), rtt109Δ (YFA06) (C), and rtt101Δ (YLK03) (D) cells. (E) Quantification of repair of both GC and SSA DSBs. GC product was normalized to the uncut restriction fragment at 0 h. n = 2. SSA was normalized to product at 24 h. n = 1. Error bars in GC are ranges.
Asf1 is required for recovery after repair of two DSBs
Checkpoint activation can be monitored by a Western blot in which Rad53 phosphorylation is seen as more slowly migrating bands. In strain YJK17, Rad53 was phosphorylated by 3 h after HO induction and was dephosphorylated by 9 h (Kim and Haber 2009). Repair of the SSA event alone (Fig. 1A) did not elicit an observable Rad53 hyperphosphorylation (Fig. 3A). When a DSB was induced in strain JKM179 (Lee et al. 1998) lacking donors and with a single HO cleavage site, Rad53 hyperphosphorylation resulted in a mobility shift that was detectable 3 h after induction and was lost 12 h after induction when the cells adapted to the DNA damage (Fig. 3B). In strain YJK17 (1-DSB wild type), Rad53 hyperphosphorylation was detectable 3 h after HO induction and was lost ∼9 h following induction of HO (Fig. 3B). A similar pattern of Rad53 activation was observed in YFA01 (2-DSB wild type) (Fig. 3B), but dephosphorylation was delayed in this strain compared with YJK17. Rad53 hyperphosphorylation in the YFA01 was only completely lost 12 h after induction. After Rad53 activation, three distinct bands appeared, the lowest ran as unphosphorylated Rad53, and the two higher bands indicated apparently increasingly hyperphosphorylated Rad53. In both YJK17 and YFA01, the two higher-migrating bands were most observable 7 h after HO induction. By 9 h, the two upper phosphorylated bands were still observable, but the lowest-migrating band was much more prominent, indicating the regeneration of unphosphorylated Rad53. By 12 h, the slower-migrating bands were completely lost. These kinetics of phosphorylation and dephosphorylation correlate with GC repair kinetics.
Figure 3.
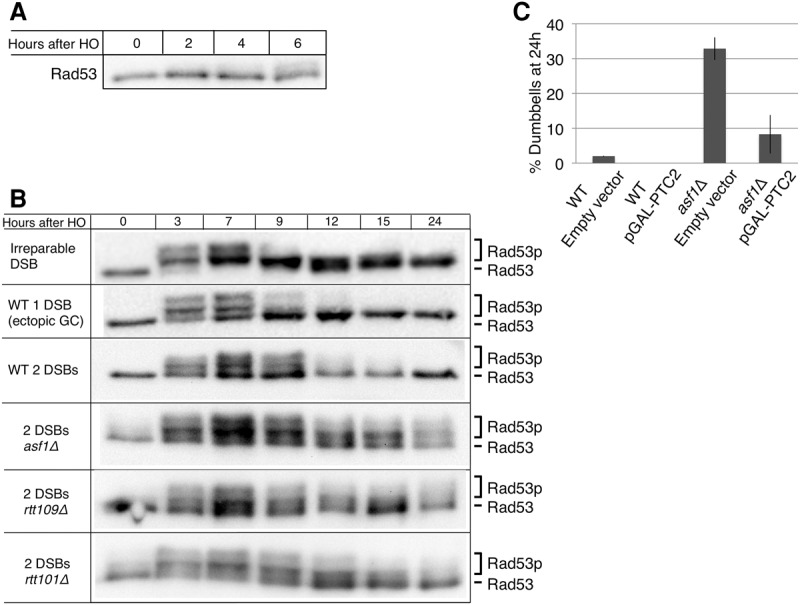
The effect of ASF1, RTT109, and RTT101 on Rad53 phosphorylation during repair in the two-DSB system. (A) Rad53 Western blot in a strain that carries only the rapidly repairable SSA (tNS024). Samples were run on a 6% SDS-PAGE gel. (B) Rad53 Western blot after induction of an irreparable DSB (JKM179), one ectopic GC (YJK17), and two repairable HO-cleaved DSBs (2-DSB wild type [YFA01], 2-DSB asf1Δ [YFA02], 2-DSB rtt109Δ [YFA06], and 2-DSB rtt109Δ [YLK03]). Rad53 hyperphosphorylation can be seen as higher-migrating forms. Samples were run on a 6% SDS-PAGE gel. (C) PTC2 overexpression rescues the checkpoint arrest in asf1Δ after induction of two DSBs. Single cells were plated on galactose plates, and G2–M-arrested cells (dumbbells) were counted 24 h after induction. n = 50.
Similar to wild type, in the two-DSB asf1Δ cells, HO induction led to Rad53 hyperphosphorylation by 3 h (Fig. 3B). In asf1Δ, however, phosphorylation of Rad53 was more extensive than in wild type, resulting in more prominent phosphorylated bands. Strikingly, despite similar repair kinetics and outcomes in wild type and asf1Δ, Rad53 in asf1Δ remains hyperphosphorylated up to 24 h, long after repair has been completed. In contrast, when only the ectopic GC is present, Rad53 phosphorylation in asf1Δ cells is similar to wild type (Kim and Haber 2009). This result supports the conclusion reached from the viability analysis above that ASF1 is required for recovery when cells suffer two DSBs.
If failure to turn off the DNA damage checkpoint following repair is indeed responsible for the lower viability in asf1Δ in the two-DSB system, then alleviating the checkpoint should rescue this defect. Overexpression of PTC2 is sufficient to dephosphorylate Rad53 (Leroy et al. 2003). Although overexpression of PTC2 results in lethality, recovery of the cells can be monitored microscopically on a galactose plate by observing the ability of single cells to grow beyond the dumbbell (G2/M-arrested) state. Overexpression of PTC2 rescues the arrest of asf1Δ cac1Δ cells, allowing them to escape G2/M arrest 24 h after HO induction when a single DSB had to be repaired by ectopic GC (Kim and Haber 2009). We found a similar result when two repairable DSBs are induced; overexpressing PTC2 had no significant effect on wild-type cells at 24 h but reduced the proportion of G2/M-arrested asf1Δ cells from 35% to 5% (Fig. 3C).
Rtt109 and Rtt101 are required for recovery after repairing two DSBs
Asf1 associates with the histone H3–H4 heterodimer (English et al. 2006) and promotes nucleosome deposition during replication (Tyler et al. 1999). It is possible that depositing H3–H4 dimers is required for recovery following repair of multiple DSBs. We hypothesized that we would see results similar those of to asf1Δ if we deleted Rtt109. We measured the viability of rtt109Δ in both the one- and two-DSB systems. Indeed, rtt109Δ behaved similarly to asf1Δ, both with one DSB and when two DSB were induced (Fig. 4A). The asf1Δ rtt109Δ double mutant behaved similarly to each single mutant (Fig. 4A), indicating that asf1Δ and rtt109Δ are epistatic. Furthermore, the viability of rtt109Δ cac1Δ was similar to asf1Δ cac1Δ in both one- and two-DSB systems (Fig. 4A), further suggesting that asf1Δ and rtt109Δ act in the same pathway.
Figure 4.
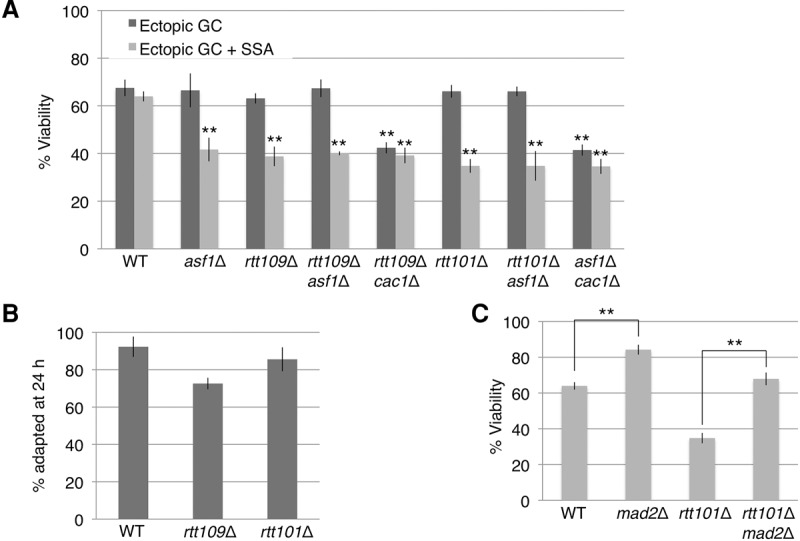
The effect of the histone H3 handoff pathway on recovery when the DNA damage checkpoint is activated by two DSBs. (A) Epistasis analysis of YLK03 (rtt101Δ) and YFA06 (rtt109Δ) with asf1Δ and cac1Δ. n ≥ 3. (**) P < 0.005, calculated relative to wild type. (B) Adaptation assay in rtt109Δ and rtt101Δ. Adapted cells were counted at 24 h. Error bars indicate ranges. n = 2. (C) The viability of wild type, mad2Δ, rtt101Δ, and rtt101Δ mad2Δ. n ≥ 3. (**) P < 0.005.
We noted that deletion of RTT109 led to a delay in repair of the GC DSB, whereas asf1Δ did not (Fig. 2C,E). There was no delay in repair of the SSA DSB (Fig. 2C,E). Despite the slower repair kinetics of the GC DSB, repair was almost 80% complete by 12 h. Slower repair in rtt109Δ might account for some of the lower viability seen in this mutant when two DSBs have to be repaired. These data suggest that, although rtt109Δ and asf1Δ share many roles, repair by GC may require RTT109 acting independently of ASF1. Indeed, previous reports show that, although rtt109Δ and asf1Δ show epistasis in many cases, they differ in a few key genetic interactions (Haber et al. 2013). We therefore wished to know whether rtt109Δ and asf1Δ had a similar adaptation phenotype. asf1Δ cells are proficient for adaptation to a single DSB (Kim and Haber 2009). To test adaptation in rtt109Δ, we deleted RTT109 in strain JKM179 and plated these cells on galactose plates. Like asf1Δ, we found that rtt109Δ cells are adaptation-proficient, with >75% of the cells adapted 24 h after a single irreparable DSB was induced (Fig. 4B). Adaptation-defective cells typically allow <30% of cells to progress (Harrison and Haber 2006).
Recently, Rtt101-mediated ubiquitylation of histone H3 has been shown to promote the handoff of histone H3 from Asf1 to CAF-1 (Han et al. 2013). Because Rtt101-mediated ubiquitylation of histone H3 occurs preferentially on histones containing the H3K56ac modification, we tested the effect of rtt101Δ on viability and recovery. Viability of rtt101Δ was significantly reduced when two DSBs activated the checkpoint (Fig. 4A), similar to rtt109Δ and asf1Δ. The viability of asf1Δ rtt101Δ was not lower than either single mutant when two DSBs are induced (Fig. 4A). Like asf1Δ and rtt109Δ, viability of rtt101Δ was similar to wild type when only one DSB was induced (Fig. 4A). Moreover, similar to asf1Δ and rtt109Δ, rtt101Δ cells were adaptation-proficient (Fig. 4B). These results suggest that RTT101 promotes recovery from the DNA damage checkpoint through the same pathway as ASF1.
Unlike rtt109Δ, deletion of RTT101 did not reduce repair efficiency or impair repair kinetics of either the GC or SSA DSB (Fig. 2D,E). This result suggests that the reduced viability in rtt101Δ in the two-DSB systems is caused by impaired recovery from the DNA damage checkpoint.
Finally, we examined Rad53 phosphorylation in both rtt109Δ and rtt101Δ. Similar to asf1Δ and wild type, Rad53 was hyperphosphorylated by 3 h after damage (Fig. 3B). Unlike wild type, phosphorylation persisted in both rtt109Δ and rtt101Δ at least up to 15 h. By 24 h, however, Rad53 in both rtt109Δ and rtt101Δ was mostly dephosphorylated, whereas, in asf1Δ, Rad53 hyperphosphorylation was clearly observable at that time point (Fig. 3B). The observation that rtt109Δ and rtt101Δ fail to properly recover despite eventually dephosphorylating Rad53 suggests that cells that fail to turn off the DNA damage checkpoint by 12 h are unable to recover even if Rad53 is dephosphorylated at a later time.
Many adaptation-defective mutations can be suppressed by deleting the spindle assembly checkpoint (SAC) gene MAD2, which suggests that there may be a handoff from the DNA damage checkpoint to the SAC to sustain arrest (Garber and Rine 2002; Dotiwala et al. 2010). To address the possibility that the SAC is required to sustain permanent arrest in the two-DSB system, we deleted MAD2 in the wild-type and rtt101 two-DSB strains. Deletion of MAD2 resulted in an increase in viability from 64% in two-DSB wild type to 84% in two-DSB mad2Δ (Fig. 4C). In rtt101Δ, deletion of MAD2 caused an increase in viability from 35% to 68% (Fig. 4C). These results indicate that the SAC partakes in sustaining arrest after the DNA damage checkpoint is turned off.
Rad53 dissociation from Asf1 is dependent on the number of DSBs
In budding yeast, Asf1 associates with Rad53 in cycling cells (Emili et al. 2001; Hu et al. 2001). After treatment with HU or MMS, this association is lost. This loss depends on Mec1-dependent phosphorylation of Rad53, as Asf1 does not bind phosphorylated Rad53. We hypothesized that this dissociation would be exacerbated when more than one DSB is induced.
First, we tested whether a single irreparable DSB is sufficient to disturb Asf1–Rad53 interaction as viewed by coimmunoprecipitation. We tagged Asf1 with HA in strain JKM179 (Lee et al. 1998). We then immunoprecipitated Asf1 using anti-HA-conjugated agarose beads and blotted using either anti-HA or anti-Rad53 antibodies. To detect both Asf1 and-HA and Rad53, we ran the samples on a 10% SDS-PAGE gel. This percentage gel does not allow a clear separation of phosphosrylated forms of Rad53 (cf. Figs. 5A, 3B). Rad53 was associated with Asf1-HA before induction of a DSB (Fig. 5A), as previously published (Emili et al. 2001; Hu et al. 2001; Jiao et al. 2012). This association gradually decreased after HO induction. By 6 h following HO induction, the Rad53 signal in the immunoprecipitation samples was reduced to 40% of the signal at 0 h. This correlated to the pattern of Rad53 phosphorylation (Fig. 3B). Cells that suffer a single irreparable HO DSB adapt by 15 h after HO induction. Accordingly, Rad53 is dephosphorylated in these cells by 15 h (Fig. 3B), and Rad53 association with Asf1 is partially re-established (80% compared with 0 h) (Fig. 5A,D).
Figure 5.

Coimmunoprecipitation of Asf1-HA and Rad53. Samples were run on 10% SDS-PAGE gels. Immunoprecipitation performed with anti-HA-conjugated agarose beads and blotted with anti-HA (Asf1-HA) or anti-Rad53. (A) Coimmunoprecipitation of Asf1-HA and Rad53 in an irreparable DSB system (JKM179). (B) Coimmunoprecipitation of Asf1-HA and Rad53 in a system with one repairable ectopic GC DSB (YJK17) and two repairable DSBs (YFA01). (C) Coimmunoprecipitation of Asf1-HA and Rad53 in a system with two repairable DSBs in rtt101Δ (left panel) or rtt109Δ (right panel). (D) Quantification of Rad53 association with Asf1. Rad53 immunoprecipitation signal was normalized to Asf1 immunoprecipitation signal and 0 h. Error bars indicate ranges. n = 2. (E) Fold increase of Rad53 levels. Rad53 levels were normalized to 0 h. Protein loading was quantified using a Bradford assay. Error bars indicate ranges. n = 2.
Next, we tested whether a single repairable ectopic GC event also leads to dissociation of Rad53 from Asf1. As in the irreparable DSB, Rad53 hyperphosphorylation was detectable by 3 h following HO induction but was lost by 12 h following HO induction (Fig. 3B). Unexpectedly, after HO induction, the Rad53 signal in the immunoprecipitated fraction increased (Fig. 5B,D), indicating that, after suffering a single repairable DSB, more Rad53 is bound to Asf1. Rad53 levels doubled by 6 h following induction of a single DSB (Fig. 5E); we hypothesize that the increase in association between Rad53 and Asf1 may be the result of an increase in the total pool of Rad53 which does not result in more hyperphosphorylated Rad53.
We then tested the association of Rad53 with Asf1 in the two-DSB strain. Unlike the single GC DSB, two DSBs caused a significant Rad53 dissociation from Asf1 6 h after HO induction (40% association compared with 0 h), comparable with the results with the irreparable DSB (Fig. 5B,D). By 12 h after HO induction, the levels of Rad53 in the immunoprecipitated fraction increased to 80% compared with 0 h, paralleling Rad53 dephosphorylation following repair (Fig. 3B). Rad53 levels in the two-DSB strain increased fourfold compared with twofold enrichment when one DSB is induced (Fig. 5E). These results suggest that both dissociation of Rad53 from Asf1 after DNA damage and Rad53 protein levels are correlated with the amount of DNA damage suffered by the cells.
Finally, we tested whether deletion of RTT101 or RTT109 affects the association between Asf1 and Rad53. Rad53 signal decreased in both mutants after induction of HO and did not increase even 12 h after HO induction, when repair was complete (Fig. 5C,D). This result indicates that ablation of Rtt101 or Rtt109 leads to a reduced ability of Asf1 to reassociate with Rad53 following repair.
Degradation of Rad53 rescues the recovery defect of asf1Δ
The coimmunoprecipitation results demonstrate that the association between Asf1 and Rad53 is re-established after the DNA damage checkpoint has been turned off. We hypothesized that, in asf1Δ, Rad53 could be reactivated even when repair has occurred. This hypothesis predicts that elimination of Rad53 following repair would rescue the recovery defect of asf1Δ in our two-DSB system. Deletion of Rad53 significantly reduces the cell cycle arrest after damage (Gardner et al. 1999; Sanchez et al. 1999), and this may impair the ability of the cells to properly repair the two DSBs prior to dividing (Kaye et al. 2004). We therefore integrated an auxin-inducible degron (AID) (Morawska and Ulrich 2013) into the C terminus of Rad53 in strains YFA01 (two-DSB wild-type) and YFA02 (two-DSB asf1Δ). Rad53 levels, as monitored by Western blot, were significantly reduced 1 h after treatment with auxin (Supplemental Fig. S1A). Equal numbers of YEP-lactose-grown cells were plated on galactose plates (where HO is induced) and dextrose plates (0 h). We then induced HO in the liquid cultures for 8 h. By that time, repair is almost complete (Fig. 2B,E). We then split the liquid cultures, and half were treated with auxin for 1 h, after which an equal number of auxin-treated and untreated cells were plated on galactose plates. Reduction of Rad53-AID levels by 1 h of auxin treatment was sufficient to significantly increase viability from 40% to 70% in the asf1Δ strains (Fig. 6A). This result demonstrates that the reduction in viability seen in asf1Δ cells suffering two DSBs can be attributed in its entirety to a defect in checkpoint deactivation in the absence of ASF1. Furthermore, this result supports the role of Rad53 in this recovery defect, as transient reduction of Rad53 levels in asf1Δ cells experiencing two DSBs led to viability comparable with that of wild-type cells suffering two DSBs (Figs. 1B, 6A).
Figure 6.

Rescue of the recovery defect in asf1Δ, rtt109Δ, and rtt101Δ. (A) Reducing Rad53 levels for 1 h rescues the recovery defect in two-DSB asf1Δ. Two-DSB asf1Δ cells containing Rad53-AID (YMT222) were allowed to repair the DSB for 8 h prior to treatment with 500 µM auxin. One hour after treatment with auxin, asf1Δ cells were plated on galactose plates. Colony numbers were normalized to 0 h of YEPD. The viability of wild-type cells with Rad53-AID is shown as a reference. n = 3. Error bars indicate standard errors. (B) Histone H3-R129E rescues the recovery defect of rtt101Δ and rtt109Δ but not asf1Δ. n = 3. Error bars indicate standard errors. (*) P < 0.05. (C) Overexpression of Asf1 rescues rtt109Δ. Wild type (YFA01) and rtt109Δ (YFA06) were transformed with either an empty vector or pASF1. n ≥ 3. (**) P < 0.005. (D) Asf1 is required for complete dephosphorylation of Rad53 after Ddc2 degradation. A strain containing a single irreparable DSB with Ddc2-AID and either wild-type (left four lanes) or asf1Δ (right four lanes). Six hours after HO induction, the culture was split and half was treated with 500 µM auxin for 2 h (8 h after HO induction).
Impairing Asf1–H3 association rescues the recovery defect of rtt101Δ and rtt109Δ
The DNA damage-dependent dissociation of Rad53 from Asf1 allows Asf1 to bind more H3 after damage (Emili et al. 2001). We hypothesized that the acetylation of H3K56 by Rtt109 and the subsequent ubiquitylation by Rtt101 serve to weaken the interaction of Asf1 with H3, allowing Rad53 to outcompete histone H3 for Asf1 binding and thus promote recovery.
To test our hypothesis, we sought to weaken Asf1–H3 interaction without interfering with the Asf1–Rad53 interactions. The histone H3-R129E mutation weakens Asf1–H3 interaction without affecting the interaction between Asf1 and Rad53 (Agez et al. 2007; Jiao et al. 2012). In budding yeast, transcription of HHT2 contributes ∼85% of total H3 mRNA (Cross and Smith 1988; Liang et al. 2012). We therefore introduced the HHT2-R129E mutation into our two-DSB system in the wild-type, asf1Δ, rtt101Δ, and rtt109Δ backgrounds.
The HHT2-R129E mutation did not significantly reduce the viability of wild-type cells (Fig. 6B). Likewise, HHT2-R129E did not change the viability of asf1Δ cells. These results demonstrate that the HHT2-R129E mutation is neither toxic nor sufficient to promote recovery from checkpoint-mediated arrest in asf1Δ. Strikingly, HHT2-R129E rescued the recovery defect of both rtt101Δ and rtt109Δ cells to about the same extent as degrading Rad53 rescued asf1Δ (Fig. 6, cf. B and A). These results suggest that, while the presence of Asf1 is required to promote recovery when there are two repairable DSBs, the activities of Rtt101 and Rtt109 are dispensable if the interaction between Asf1 and histone H3 is reduced and raises the possibility that, in the absence of Rtt109 or Rtt101, the interaction between Asf1 and histone H3 limits the amount of Asf1 available to promote recovery. We therefore tested whether overexpression of Asf1 would rescue rtt109Δ in a strain that suffers two repairable DSBs. Plasmid pPK196 is a centromeric plasmid containing a single copy of Asf1 with its endogenous promoter. Overexpression of Asf1 did not change wild-type viability (Fig. 6C). The viability of rtt109Δ, however, was rescued to wild-type levels by expression of a single additional copy of Asf1 (Fig. 6C). We propose that the role of Asf1 in recovery is separate from its histone chaperone activity and that Rtt101 and Rtt109 promote recovery by modifying histone H3 and weakening the interaction between histone H3 and Asf1.
Asf1 is required for complete dephosphorylation of Rad53 following checkpoint deactivation
Irreparable DSBs in budding yeast lead to Rad53 phosphorylation, and this modification is lost after cells adapt or after Mec1 is degraded using a temperature degron (Pellicioli et al. 2001). We asked whether Asf1 is required for complete dephosphorylation of Rad53 following inhibition of the Mec1 signaling pathway. We integrated an AID at the C terminus of Ddc2 in a strain containing one irreparable DSB. Six hours after induction of HO endonuclease in a wild-type strain, there was robust Rad53 phosphorylation with essentially all of the kinase in hyperphosphorylated forms (Fig. 6D). At this point, the culture was divided, and half was treated with 500 µM auxin to degrade Ddc2. Addition of auxin led to depletion of Ddc2 30 min following treatment (Supplemental Fig. S1B). Two hours later, Rad53 phosphorylation was still maintained in the untreated culture, whereas, in the auxin-treated cells, Rad53 phosphorylation was significantly reduced (Fig. 6D), confirming that Mec1 signaling is continuously required to maintain the phosphorylation state of Rad53 (Pellicioli et al. 2001). Checkpoint activation was then monitored in asf1Δ in the Ddc2-AID strain. As in wild-type, strong Rad53 phosphorylation was evident 6 h after DSB induction. Interestingly, 2 h following auxin treatment, Rad53 phosphorylation was still detected (Fig. 6D). While the highest migrating forms of Rad53 were lost, indicating some dephosphorylation, Rad53 persisted in a middle migrating band and failed to return to the lowest migrating form observed prior to DSB induction (Fig. 6D). These results suggest that although Asf1 does not itself dephosphorylate Rad53, it is required to promote complete dephosphorylation of Rad53 following the deactivation of the upstream Ddc2–Mec1 signaling. Given that Rad53 amplifies its Mec1-dependent phosphorylation by autophosphorylation, these results might imply that Asf1 plays a role in either preventing autophosphorylation or promoting turnover of phosphorylated forms of the protein.
Discussion
Budding yeast cells can sense the number of DSBs that they have suffered (Lee et al. 1998; Mantiero et al. 2007). This sensitivity is apparently achieved by regulating the level of Rad53 phosphorylation in response to more DSBs (Mantiero et al. 2007). Maintenance of γ-H2AX in the 50-kb region around each of two DSBs is regulated independently so that it is removed around a repaired DSB even as it persists around an unrepaired DSB (Tsabar et al. 2015). Here we provide the first evidence that cells can sense the number of repairable DSBs even if one of these DSBs repairs rapidly enough so that it does not by itself elicit detectable Rad53 phosphorylation. In addition, we show that the genetic requirements for recovery are different when there are two DSBs versus one. We propose that Asf1 participates in a dynamic DNA damage-sensing mechanism. When the cells sense DSBs, Mec1 phosphorylates Rad53, leading to its dissociation from Asf1 and hyperactivation by autophosphorylation. When Rad53 is dephosphorylated by Ptc2 and Ptc3, it can reassociate with Asf1. However, so long as the damage persists, Mec1 phosphorylation of Rad53 will lead to its dissociation from Asf1 and its activation by autophosphorylation, but, when the DSB has been repaired, Asf1 can rapidly sequester Rad53. Asf1 can thus prevent Rad53 phosphorylation (which occurs in trans) (Pellicioli et al. 1999; Gilbert et al. 2001) by Rad53 that has not yet been dephosphorylated (Fig. 7).
Figure 7.
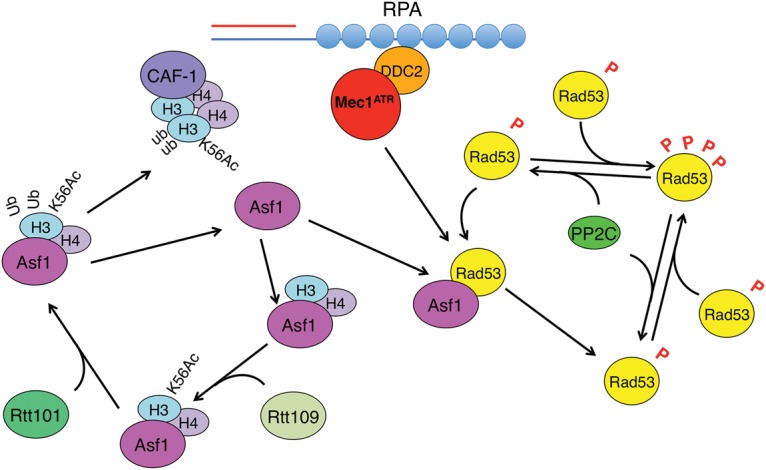
A model for the role of Asf1 in the dynamic sensing of DNA damage. After a Ddc2–Mec1 recognizes a DSB, Mec1 phosphorylates Rad53, leading to Rad53 dissociation from Asf1. Rad53 then undergoes hyperphosphorylation. PP2C dephosphorylates Rad53, and the dephosphorylated Rad53 reassociates with Asf1. If the damage persists, Mec1 can rephosphorylate Rad53 and lead to dissociation from Asf1 and reactivation. In addition, activated Rad53 can counter the dephosphorylation by autophosphorylation. In parallel, Asf1-bound histone H3 undergoes modifications by Rtt109 and Rtt101, which facilitate the handoff of the H3–H4 histone dimer to CAF-1, freeing Asf1 to reinteract with Rad53.
Chromatin remodeling plays extensive roles in the DNA damage response. While chromatin remodelers such as Fun30, Ino80, the RSC complex, and Rad54 facilitate different HR pathways (Kent et al. 2007; Shim et al. 2007; Hicks et al. 2011; Chen et al. 2012; Costelloe et al. 2012; Eapen et al. 2012; Tsabar and Haber 2013), other chromatin-modifying factors, including Asf1, CAF-1 Rdh54, Ino80, and Swr1, participate in regulating the DNA damage checkpoint (Emili et al. 2001; Hu et al. 2001; Lee et al. 2001; Papamichos-Chronakis et al. 2006; Kim and Haber 2009; Tsabar and Haber 2013). Here we show that Asf1 regulates the DNA damage checkpoint by a mechanism separate from its histone chaperone activity.
In replication, Asf1 interacts with histone H3 and, together with the histone acetyltransferase Rtt109, acetylates H3K56 (Collins et al. 2007; Driscoll et al. 2007; Han et al. 2007; Tsubota et al. 2007). H3K56ac was suggested to facilitate chromatin re-establishment following repair and signal for recovery (Chen et al. 2008), although a more recent study suggested that H3K56ac is required for repair of specific types of DNA damage induced by MMS (Wurtele et al. 2012). We found that deletion of RTT09 only slightly impaired repair by GC and did not affect SSA repair. Moreover, both asf1Δ and rtt109Δ exhibit a recovery defect only when more than one DSB has to be repaired. Neither asf1Δ nor rtt109Δ affect adaptation to a single DSB; it is not possible to see whether these mutants respond differently to two unrepaired DSBs because two irreparable DSBs already confer an adaptation defect (Lee et al. 1998). Last, we show that the histone H3-R129E mutation rescues the recovery defect of rtt109Δ cells in our two-DSB system. All of these data strongly suggest that H3K56ac does not act as a recovery signal.
Rtt101 ubiquitylates histone H3 in vitro, with a preference for histone H3 that has been acetylated on Lys56 (Han et al. 2013). This ubiquitylation is suggested to facilitate the handoff of histone H3–H4 heterodimers from Asf1 to CAF-1 and Rtt106. In vivo, Rtt101 functions in the same genetic pathway as H3K56ac to confer resistance to MMS damage (Wurtele et al. 2012). Like deletion of RTT109 and ASF1, deletion of RTT101 does not confer an adaptation defect or a repair defect. Similarly to rtt109Δ, the recovery defect of rtt101Δ in the two-DSB system is rescued by weakening the interaction of Asf1 with H3. Together with our genetic data, this finding suggests that Rtt101 and Rtt109 act on the same pathway to promote recovery and that their role in recovery is to facilitate the removal of histone H3 from Asf1.
Rad53 phosphorylation in asf1Δ cells suffering two repairable DSBs persisted 24 h after induction of the DSBs, long after repair is completed. This suggests that the DNA damage checkpoint responds to the number of DSBs that it experienced even if one of these DSBs itself does not trigger a checkpoint activation. Rad53 phosphorylation in rtt109Δ and rtt101Δ persisted 15 h after HO induction but did not persist up to 24 h. It is noteworthy that adaptation occurs ∼12–15 h following induction of a single irreparable DSB. It is possible that the 12- to 15-h time frame following DNA damage checkpoint activation represents a time window by which the DNA damage checkpoint must be resolved by a productive pathway leading to recovery. One possibility raised by our findings is that failure to turn off the DNA damage checkpoint by 12 h results in a handoff of the arrest to the SAC, which prevents productive recovery. Accordingly, deletion of MAD2 had a more significant effect on rtt101Δ cells exhibiting a longer checkpoint than on wild-type cells.
The association of Asf1 with Rad53 is disrupted by exposure to the genotoxic agents MMS and HU, and this disruption is dependent on DNA damage checkpoint signaling (Emili et al. 2001; Hu et al. 2001). Interestingly, Rad53 phosphorylation in response to HU is delayed in asf1Δ (Hu et al. 2001). As Asf1 also binds histone H3 and CAF-1, this may indicate that the role of Asf1 is to concentrate Rad53 close to the replication forks, which are very susceptible to DNA damage (Hu et al. 2001). Here we show that a single irreparable DSB is sufficient to disrupt Asf1's association with Rad53 significantly. Moreover, we show that adaptation and recovery temporally correlate with the re-establishment of the association between Asf1 and Rad53. This, together with the observation that ablation of Rad53 rescues the viability of asf1Δ cells in a system with two repairable DSBs and the prolonged Rad53 phosphorylation in asf1Δ, may suggest that re-establishment of the Asf1–Rad53 interaction plays a significant role in regulating the DNA damage checkpoint during recovery and adaptation. Supporting this model, our observation that Rad53 phosphorylation persists in asf1Δ cells even when Ddc2 is depleted demonstrates that Asf1 is required for the complete turning off of the DNA damage checkpoint.
The role of Asf1 in recovery manifests itself only when the cells are exposed to multiple DSBs. The reason for the requirement for Asf1 when the cell is exposed to multiple DSBs remains unclear. One clue may come from studying the association of Rad53 and Asf1. A single repairable DSB did not cause dissociation between Asf1 and Rad53 that was seen with two DSBs. This difference may reflect how one or two DSBs alter total Rad53 protein levels and its hyperphosphorylation. The observation that total Rad53 protein levels increase in a DNA damage-dependent manner (Smolka et al. 2005) and that the magnitude of the increase and phosphorylation of Rad53 are correlated to the number of DSBs suggests that, in response to two DSBs, there is a larger pool of free Rad53, and this may lead to more active Rad53 in response to two DSBs. In this case, when only one DSB triggers the DNA damage checkpoint, Ptc2 and Ptc3 dephosphorylation of Rad53 is sufficient to deactivate the checkpoint; however, when more than one DSB is induced, Rad53 levels increase further, and there may be more autophosphorylation. In this event, Asf1 might be required to sequester Rad53 and prevent it from being reactivated.
In mammalian cells, the DNA damage checkpoint is highly sensitive to the amount of damage inflicted on the cells (Buscemi et al. 2004; Deckbar et al. 2007; Loewer et al. 2013). These studies suggest that studying recovery from more than one DSB in budding yeast may be more physiologically similar to the condition in mammalian cells. Nevertheless, despite the high level of conservation of Asf1 and Rad53 (mammalian Chk2) between yeast and mammals, the interaction between Asf1 and Chk2 was not detected in mammalian cells (Groth et al. 2005). This suggests that, in mammalian cells, a large fraction of Chk2 is free from interaction with Asf1. Another possibility is that the interaction between Asf1 and Rad53 is cell cycle-regulated. If this is the case, then we are able to capture the interaction between Asf1 and Rad53 because an asynchronous population of budding yeast contains cells in all stages of the cell cycle, and this interaction might be missed in mammalian cells that are predominantly in G0/G1. Another possibility is that the absence of this interaction may be a result of fundamental differences between budding yeast and mammalian cells in the mechanism of DNA damage checkpoint regulation. However, Asf1 has been shown to participate in nucleosome deposition following repair (Mello et al. 2002) and interact with tousled-like kinases (TLKs), a family of kinases specific to metazoans that plays an extensive role in the regulation of replication, transcription, and repair (Groth et al. 2003, 2005; Mousson et al. 2007; De Benedetti 2012). These observations implicate Asf1 with regulation of the DNA damage checkpoint in mammalian cells as well. The findings presented in our study demonstrate that the DNA damage checkpoint is regulated by both the duration of damage persistence and the amount of damage, link chromatin assembly to recovery from the DNA damage checkpoint, and contribute to our understanding of mechanisms by which cells sense the completion of repair.
Materials and methods
Strains
Strains were based on strain YJK17 (Kim and Haber 2009), which contains a glactose-inducible HO gene and is deleted for the normal HML and HMR donors to repair a DSB at MAT. This MATα strain carries a 4-kb MATa-inc sequence, which cannot be cleaved by HO, inserted at the arg5,6 locus on Chr 5. In some strains, an additional HO cut site was inserted at the ura3-52 locus on the opposite arm of Chr 5 such that it was flanked by ∼1 kb of flanking homology (Sugawara and Haber 1992).
To construct the histone H3-R129E mutation, the region containing HHF2 and HHT2 was amplified using quantitative PCR (qPCR) and introduced to plasmid pRS405 at its HpaI site to create plasmid pMT05. The R129E mutation was introduced by site-directed mutagenesis using the QuikChange II kit (Agilent) to create plasmid pMT06. HHT2-R129E and the adjacent LEU2 were amplified from this plasmid and integrated at the endogenous HHT2 in strains YFA01, YFA02, YLK03, and YFA06 so that LEU2 was downstream from HHT2-R129E. Integration of LEU2 and the coinserted R129E mutation was verified by PCR followed by DNA sequencing.
A full list of strains is in Supplemental Table S1. All strains and sequences of oligos for PCR are available on request.
Plating assays
For viability experiments, cells were grown overnight in 5 mL of YEPD, washed twice with 5 mL of YEP-lactose, and kept for at least 6 h in lactose. Next, cells were counted, and ∼100 cells were plated on YEP galactose plates (where HO is induced) and on YEPD plates as a control. Colonies were counted 3–5 d after plating.
Adaptation assays were conducted by inoculating cells overnight in 5 mL of YEP-lactose and then plating them on YEP-galactose plates. Single cells were manipulated and monitored for 24 and 48 h following plating. A PTC2 overexpression assay was performed similarly to the adaptation assay, using strains containing plasmid pJH2447 with PTC2 under the control a galactose promoter.
Southern blots
Southern blots were performed as described in Kim and Haber (2009). To monitor the ectopic GC, genomic DNA extracted by phenol extraction from a time course was digested overnight with EcoRI. A radiolabeled probe for the Z1Z2 region at MAT was used (Kim and Haber 2009). The product was normalized to the donor signal and 0 h. To monitor SSA repair at ura3, DNA was digested with BglII. A radiolabeled probe for the 3′ end of the URA3 gene was used. The signal was normalized to the total signal and the signal from a cell that had completed SSA.
Western blots
Western blots were done as described (Pellicioli et al. 2001). Proteins were extracted using TCA preparation. An antibody against Rad53 (Abcam, ab166859) was used to detect Rad53. Ddc2-AID-Myc degradation was monitored by an antibody against Myc.
Degradation of proteins using AID
Proteins were degraded using the AID system from H. Ullrich (Morawska and Ulrich 2013). Rad53-AID cells were grown overnight in YEPD and washed twice in YEP-lactose for 6 h, and then a larger culture was inoculated and grown until cells reached log-phase growth (5 × 106 cells per milliliter). Cells were then counted, and ∼100 colony-forming units (CFUs) were plated on YEPD and YEP-galactose plates to monitor viability. The liquid culture was treated with 2% galactose to induce the HO endonuclease. Eight hours after HO induction, the culture was split in two, and one of the two cultures was treated with 500 µM auxin. One hour after auxin treatment, cells were counted, and 100 CFUs were plated on galactose plates. Western blots were performed to confirm loss of the protein (Fig. 5E).
Ddc2-AID-Myc cells were grown as described above. A sample was collected prior to galactose induction, and then 2% galactose was added to the medium. Six hours after galactose induction, a sample was collected, the culture was split, and one half was treated with 500 µM auxin for 2 h. A final sample was collected 8 h after galactose induction (2 h after auxin induction). Western blots were used to confirm loss of Ddc2.
Coimmunoprecipitation
Coimmunoprecipitation experiments were conducted as previously published (Hu et al. 2001) with a modified lysis buffer (50 mM Tris at pH 7.5, 50 mM NaCl, 5 mM EDTA, 0.5% Triton X-100, proteinase inhibitors).
Supplementary Material
Acknowledgments
We thank Helle Ulrich for plasmids to generate the AIDs. We also thank Paul Kaufman for the plasmid pPK196. This work was supported by National Institutes of Health grants GM20056 and GM61766 to J.E.H. D.P.W. is supported by National Institutes of Health Genetics training grant T32GM007122.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.280685.116.
References
- Agez M, Chen J, Guerois R, van Heijenoort C, Thuret JY, Mann C, Ochsenbein F. 2007. Structure of the histone chaperone ASF1 bound to the histone H3 C-terminal helix and functional insights. Structure 15: 191–199. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, Kupiec M. 2004. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 23: 4868–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscemi G, Perego P, Carenini N, Nakanishi M, Chessa L, Chen J, Khanna K, Delia D. 2004. Activation of ATM and Chk2 kinases in relation to the amount of DNA strand breaks. Oncogene 23: 7691–7700. [DOI] [PubMed] [Google Scholar]
- Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. 2008. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134: 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, Ira G. 2012. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature 489: 576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, et al. 2007. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446: 806–810. [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. 2001. ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716. [DOI] [PubMed] [Google Scholar]
- Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, et al. 2012. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489: 581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SL, Smith MM. 1988. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol 8: 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Benedetti A. 2012. The tousled-like kinases as guardians of genome integrity. ISRN Mol Biol 2012: 627596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckbar D, Birraux J, Krempler A, Tchouandong L, Beucher A, Walker S, Stiff T, Jeggo P, Lobrich M. 2007. Chromosome breakage after G2 checkpoint release. J Cell Biol 176: 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotiwala F, Harrison JC, Jain S, Sugawara N, Haber JE. 2010. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr Biol 20: 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll R, Hudson A, Jackson SP. 2007. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 315: 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE. 2012. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol 32: 4727–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emili A, Schieltz DM, Yates JR III, Hartwell LH. 2001. Dynamic interaction of DNA damage checkpoint protein Rad53 with chromatin assembly factor Asf1. Mol Cell 7: 13–20. [DOI] [PubMed] [Google Scholar]
- English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. 2006. Structural basis for the histone chaperone activity of Asf1. Cell 127: 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MG, Cooper JP. 2004. Two modes of DNA double-strand break repair are reciprocally regulated through the fission yeast cell cycle. Genes Dev 18: 2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillingham J, Recht J, Silva AC, Suter B, Emili A, Stagljar I, Krogan NJ, Allis CD, Keogh MC, Greenblatt JF. 2008. Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol Cell Biol 28: 4342–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber PM, Rine J. 2002. Overlapping roles of the spindle assembly and DNA damage checkpoints in the cell-cycle response to altered chromosomes in Saccharomyces cerevisiae. Genetics 161: 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R, Putnam CW, Weinert T. 1999. RAD53, DUN1 and PDS1 define two parallel G2/M checkpoint pathways in budding yeast. EMBO J 18: 3173–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert CS, Green CM, Lowndes NF. 2001. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol Cell 8: 129–136. [DOI] [PubMed] [Google Scholar]
- Gobbini E, Cesena D, Galbiati A, Lockhart A, Longhese MP. 2013. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair (Amst) 12: 791–799. [DOI] [PubMed] [Google Scholar]
- Groth A, Lukas J, Nigg EA, Sillje HH, Wernstedt C, Bartek J, Hansen K. 2003. Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint. EMBO J 22: 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G. 2005. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell 17: 301–311. [DOI] [PubMed] [Google Scholar]
- Haber JE. 2013. Genome stability: DNA repair and recombination. Garland Science, New York. [Google Scholar]
- Haber JE, Braberg H, Wu Q, Alexander R, Haase J, Ryan C, Lipkin-Moore Z, Franks-Skiba KE, Johnson T, Shales M, et al. 2013. Systematic triple-mutant analysis uncovers functional connectivity between pathways involved in chromosome regulation. Cell Rep 3: 2168–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. 2007. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315: 653–655. [DOI] [PubMed] [Google Scholar]
- Han J, Zhang H, Wang Z, Zhou H, Zhang Z. 2013. A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 155: 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JC, Haber JE. 2006. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet 40: 209–235. [DOI] [PubMed] [Google Scholar]
- Hicks WM, Yamaguchi M, Haber JE. 2011. Real-time analysis of double-strand DNA break repair by homologous recombination. Proc Natl Acad Sci 108: 3108–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Alcasabas AA, Elledge SJ. 2001. Asf1 links Rad53 to control of chromatin assembly. Genes Dev 15: 1061–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. 2004. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431: 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Seeger K, Lautrette A, Gaubert A, Mousson F, Guerois R, Mann C, Ochsenbein F. 2012. Surprising complexity of the Asf1 histone chaperone-Rad53 kinase interaction. Proc Natl Acad Sci 109: 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, Toczyski DP. 2004. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol 14: 2096–2106. [DOI] [PubMed] [Google Scholar]
- Kent NA, Chambers AL, Downs JA. 2007. Dual chromatin remodeling roles for RSC during DNA double strand break induction and repair at the yeast MAT locus. J Biol Chem 282: 27693–27701. [DOI] [PubMed] [Google Scholar]
- Kim JA, Haber JE. 2009. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc Natl Acad Sci 106: 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh BO, Symington LS. 2004. Recombination proteins in yeast. Annu Rev Genet 38: 233–271. [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. 1998. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409. [DOI] [PubMed] [Google Scholar]
- Lee SE, Pellicioli A, Malkova A, Foiani M, Haber JE. 2001. The Saccharomyces recombination protein Tid1p is required for adaptation from G2/M arrest induced by a double-strand break. Curr Biol 11: 1053–1057. [DOI] [PubMed] [Google Scholar]
- Leroy C, Lee SE, Vaze MB, Ochsenbein F, Guerois R, Haber JE, Marsolier-Kergoat MC. 2003. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol Cell 11: 827–835. [DOI] [PubMed] [Google Scholar]
- Lewis LK, Resnick MA. 2000. Tying up loose ends: nonhomologous end-joining in Saccharomyces cerevisiae. Mutat Res 451: 71–89. [DOI] [PubMed] [Google Scholar]
- Liang D, Burkhart SL, Singh RK, Kabbaj MH, Gunjan A. 2012. Histone dosage regulates DNA damage sensitivity in a checkpoint-independent manner by the homologous recombination pathway. Nucleic Acids Res 40: 9604–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer A, Karanam K, Mock C, Lahav G. 2013. The p53 response in single cells is linearly correlated to the number of DNA breaks without a distinct threshold. BMC Biol 11: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Clerici M, Lucchini G, Longhese MP. 2007. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep 8: 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello JA, Sillje HH, Roche DM, Kirschner DB, Nigg EA, Almouzni G. 2002. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep 3: 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JA, Cohen J, Toczyski DP. 2001. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 15: 2809–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawska M, Ulrich HD. 2013. An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast 30: 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousson F, Ochsenbein F, Mann C. 2007. The histone chaperone Asf1 at the crossroads of chromatin and DNA checkpoint pathways. Chromosoma 116: 79–93. [DOI] [PubMed] [Google Scholar]
- Nakada D, Matsumoto K, Sugimoto K. 2003. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev 17: 1957–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciotti V, Clerici M, Lucchini G, Longhese MP. 2000. The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev 14: 2046–2059. [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Krebs JE, Peterson CL. 2006. Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev 20: 2437–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M. 1999. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18: 6561–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. 2001. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell 7: 293–300. [DOI] [PubMed] [Google Scholar]
- Rouse J, Jackson SP. 2000. LCD1: an essential gene involved in checkpoint control and regulation of the MEC1 signalling pathway in Saccharomyces cerevisiae. EMBO J 19: 5801–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. 1996. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271: 357–360. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. 1999. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286: 1166–1171. [DOI] [PubMed] [Google Scholar]
- Sandell LL, Zakian VA. 1993. Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell 75: 729–739. [DOI] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. 2002. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell 9: 1055–1065. [DOI] [PubMed] [Google Scholar]
- Shim EY, Hong SJ, Oum JH, Yanez Y, Zhang Y, Lee SE. 2007. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol 27: 1602–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka MB, Albuquerque CP, Chen SH, Schmidt KH, Wei XX, Kolodner RD, Zhou H. 2005. Dynamic changes in protein-protein interaction and protein phosphorylation probed with amine-reactive isotope tag. Mol Cell Proteomics 4: 1358–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Haber JE. 1992. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol 12: 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF. 1996. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev 10: 395–406. [DOI] [PubMed] [Google Scholar]
- Sun Z, Hsiao J, Fay DS, Stern DF. 1998. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281: 272–274. [DOI] [PubMed] [Google Scholar]
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. 2005. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol 15: 1364–1375. [DOI] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, Hartwell LH. 1997. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90: 1097–1106. [DOI] [PubMed] [Google Scholar]
- Tsabar M, Haber JE. 2013. Chromatin modifications and chromatin remodeling during DNA repair in budding yeast. Curr Opin Genet Dev 23: 166–173. [DOI] [PubMed] [Google Scholar]
- Tsabar M, Eapen VV, Mason JM, Memisoglu G, Waterman DP, Long MJ, Bishop DK, Haber JE. 2015. Caffeine impairs resection during DNA break repair by reducing the levels of nucleases Sae2 and Dna2. Nucleic Acids Res 43: 6889–6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, Denu JM, Kaufman PD. 2007. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell 25: 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler JK, Adams CR, Chen SR, Kobayashi R, Kamakaka RT, Kadonaga JT. 1999. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402: 555–560. [DOI] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber JE. 2002. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10: 373–385. [DOI] [PubMed] [Google Scholar]
- Wakayama T, Kondo T, Ando S, Matsumoto K, Sugimoto K. 2001. Pie1, a protein interacting with Mec1, controls cell growth and checkpoint responses in Saccharomyces cerevisiae. Mol Cell Biol 21: 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtele H, Kaiser GS, Bacal J, St-Hilaire E, Lee EH, Tsao S, Dorn J, Maddox P, Lisby M, Pasero P, et al. 2012. Histone H3 lysine 56 acetylation and the response to DNA replication fork damage. Mol Cell Biol 32: 154–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. 2003. Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.