

Abstract
Despite continuing debate about the amyloid β‐protein (or Aβ hypothesis, new lines of evidence from laboratories and clinics worldwide support the concept that an imbalance between production and clearance of Aβ42 and related Aβ peptides is a very early, often initiating factor in Alzheimer's disease (AD). Confirmation that presenilin is the catalytic site of γ‐secretase has provided a linchpin: all dominant mutations causing early‐onset AD occur either in the substrate (amyloid precursor protein, APP) or the protease (presenilin) of the reaction that generates Aβ. Duplication of the wild‐type APP gene in Down's syndrome leads to Aβ deposits in the teens, followed by microgliosis, astrocytosis, and neurofibrillary tangles typical of AD. Apolipoprotein E4, which predisposes to AD in > 40% of cases, has been found to impair Aβ clearance from the brain. Soluble oligomers of Aβ42 isolated from AD patients' brains can decrease synapse number, inhibit long‐term potentiation, and enhance long‐term synaptic depression in rodent hippocampus, and injecting them into healthy rats impairs memory. The human oligomers also induce hyperphosphorylation of tau at AD‐relevant epitopes and cause neuritic dystrophy in cultured neurons. Crossing human APP with human tau transgenic mice enhances tau‐positive neurotoxicity. In humans, new studies show that low cerebrospinal fluid (CSF) Aβ42 and amyloid‐PET positivity precede other AD manifestations by many years. Most importantly, recent trials of three different Aβ antibodies (solanezumab, crenezumab, and aducanumab) have suggested a slowing of cognitive decline in post hoc analyses of mild AD subjects. Although many factors contribute to AD pathogenesis, Aβ dyshomeostasis has emerged as the most extensively validated and compelling therapeutic target.
Keywords: Aβ, Alzheimer, genetics, cell biology, treatment
Subject Categories: Neuroscience
Glossary
- Microgliosis
early non‐specific proliferation and migration of microglial cells, macrophage‐like cells in the central nervous system, as the first response to brain damage.
- Astrocytosis
final response to brain damage and injury with proliferation of astrocytes, a type of glial cell responsible for maintaining extracellular ion and neurotransmitter concentrations, modulating synapse function, and forming the blood–brain barrier.
- Neurofibrillary tangles
accumulation of hyperphosphorylated tau protein, commonly found in Alzheimer's disease, that aggregates inside nerve cell bodies, also known as dystrophic neurites.
- Plaque deposition
aggregates of amyloid fibrils that are deposited outside neurons in dense formations, also known as senile plaques or neuritic plaques.
- FAD
familial AD caused by inherited mutations in APP and presenilin (typically early‐onset) by opposition to “sporadic” or late‐onset AD
Introduction
Few problems in modern biomedicine have garnered as much scientific interest and public concern as has Alzheimer's disease. Virtually unknown to the general public four decades ago, AD has risen in prevalence to an estimated 40 million patients worldwide. The true number must be much higher, given the increasing recognition that the disease begins in the brain at least 2–3 decades before one first forgets the name of a grandchild or where one has parked one's car. Since molecular studies of AD began in earnest in the early 1980s, thousands of scientists and healthcare professionals have delved into all aspects of this complex, multifactorial syndrome, hoping to help patients now and prevent others from developing it in the future.
Although the progressive buildup of amyloids of diverse protein composition in various systemic organs has been known to cause devastating diseases for more than a century, the idea put forward by George Glenner (Glenner & Wong, 1984) that the particular amyloidogenic protein accumulating in AD (Aβ) could be causative has met with considerable skepticism over the ensuing years. Precisely why this idea has been so controversial is not clear (Selkoe, 2011), but the steady accrual of data from many preclinical and clinical studies has increasingly supported it. The amyloid (or Aβ) hypothesis (Beyreuther & Masters, 1991; Hardy & Allsop, 1991; Selkoe, 1991; Hardy & Higgins, 1992) has become the dominant model of AD pathogenesis and is guiding the development of potential treatments.
We reviewed the evidence for this hypothesis (Fig 1) a dozen years ago (Hardy & Selkoe, 2002). Space precludes a full examination here of the enormous literature on Aβ since that review; a monograph on AD pathobiology contains many details (Selkoe et al, 2012). But in the context of continuing concern about the concept and yet the recent emergence of apparently positive clinical trial data, a critical analysis of the latest developments in laboratory and clinic is warranted and timely. We review here numerous new developments since our prior review of this hypothesis, on which ever‐increasing scientific effort is being expended. We also summarize the salient findings over three decades that undergird the amyloid hypothesis (Box 1), and we discuss several alternative concepts or concerns that have been counterposed to it (Table 1).
Figure 1. The sequence of major pathogenic events leading to AD proposed by the amyloid cascade hypothesis.

The curved blue arrow indicates that Aβ oligomers may directly injure the synapses and neurites of brain neurons, in addition to activating microglia and astrocytes.
Box 1: Evidence supporting a key role for Aβ dyshomeostasis in initiating AD.
All AD patients undergo progressive Aβ deposition followed by surrounding neuritic and glial cytopathology in brain regions serving memory and cognition.
Mutations within and immediately flanking the Aβ region of APP cause aggressive forms of FAD.
Humans with trisomy 21 (Down's syndrome) harbor 3 copies of APP and invariably develop neuropathologically typical AD. Those who die in their early‐to‐mid teens (from other causes) show abundant diffuse Aβ plaques without neuritic dystrophy, microgliosis, astrocytosis, and tangle formation, all of which accrue gradually in such subjects in the late teens and beyond.
Inheritance of a missense mutation in APP that decreases the production and aggregation of Aβ lifelong protects against AD and age‐related cognitive decline.
Missense mutations in presenilin 1 or 2 are the most common cause of early‐onset AD, and presenilin is the catalytic subunit of γ‐secretase. The mutations result in relative increases in the production of Aβ42/43 peptides. These hydrophobic species self‐aggregate, leading to profound Aβ deposition in mid‐life.
ApoE4 carriers were once included in typical late‐onset AD. This allele was found to markedly increase AD risk and decrease brain clearance of Aβ, leading to excess Aβ aggregation and typical downstream AD neuropathology.
Aβ42 oligomers isolated from typical (late‐onset) AD brains decrease synapse density, inhibit LTP, and enhance long‐term synaptic depression in rodent hippocampus, and their intraventricular injection impairs memory in healthy adult rats.
Human Aβ42 oligomers induce tau hyperphosphorylation at AD‐relevant epitopes and cause neuritic dystrophy in cultured rat neurons; co‐administering Aβ antibodies fully prevents this.
Aβ oligomers occur in a penumbra around many neuritic plaques. Accordingly, synapse decreases occur in a centrifugal gradient: less abnormality at longer distances from the plaque edge.
Based on many human biomarker studies, low CSF Aβ42 and positive amyloid‐PET scans precede other AD‐related changes (increased CSF tau, decreased cerebral glucose metabolism, brain atrophy, clinical dementia) by years.
Trials of 3 different Aβ monoclonal antibodies (solanezumab, crenezumab, and aducanumab) have suggested slowing of cognitive decline in post hoc analyses of mild (but not moderate) AD patients.
Other amyloidogenic proteins have been proven to cause progressive human organ failure, and therapeutic lowering of the amyloid or its precursor protein yields therapeutic benefits in patients.
Table 1.
Findings that appear to undercut the amyloid hypothesis of AD and counterarguments that could explain these discrepancies
| Findings | Counterarguments |
|---|---|
| Amyloid plaque burden correlates much less well with degree of cognitive impairment than do neurofibrillary tangle counts | Aβ deposits appear to be a very early and widespread event that is distant to the clinical dementia and can lead to many downstream cellular and molecular changes (e.g., microgliosis, neuritic dystrophy, tangles, etc.) that are more proximate to and causative of neuronal dysfunction |
| Many humans show sometimes abundant Aβ deposits at death but were not noticeably demented | Some or many of these deposits are diffuse plaques (not rich in abnormal neurites and glia); the patients were often not tested rigorously before death; and Aβ oligomer levels per plaque are much lower than in AD brains (Esparza et al, 2013), suggesting that plaques can effectively sequester oligomers in a non‐diffusible, less neurotoxic state, at least up to a point |
| Some human neuropathological studies suggest tangles may precede amyloid plaques | Such studies may not have searched systematically for diffuse plaques or soluble Aβ oligomers in the brain. Human genetics proves that Aβ‐elevating APP mutations lead to downstream alteration and aggregation of wild‐type tau, whereas tau mutations do not lead to Aβ deposition and amyloid‐related dementia |
| A hypothesis that AD is fundamentally due to loss of presenilin function has been put forward | AD‐causing presenilin mutations may indeed act through partial loss of function of this protease, but these heterozygous mutations do not produce clinically detectable loss of presenilin function (e.g., Notch phenotypes), and organismal development and function are normal until the carriers develop typical AD symptoms in mid‐life, heralded by elevated Aβ42/43 to Aβ40 ratios. Moreover, 99.9% of all AD patients have wild‐type presenilins |
| Numerous clinical trials of anti‐amyloid agents have not met their pre‐specified endpoints | Several of these agents had inadequate preclinical data, poor brain penetration, little human biomarker change, and/or low therapeutic indexes (e.g., tramiprosate; R‐flurbiprofen; semagacestat). Most such failed trials enrolled many patients in the late‐mild and moderate stages of AD, whereas other trials conducted in very mild or mild AD produced suggestive evidence of clinical benefit. AD trials done prior to obligatory amyloid‐PET imaging turned out to have up to ~25% of subjects that were amyloid‐negative (i.e., did not have AD) |
New insights from AD genetics and APP homeostasis
The fact that AD‐causing mutations in APP and in presenilins 1 and 2 alter APP proteolytic processing in a way that elevates the relative levels of the Aβ42 or Aβ43 peptides has long been known (Scheuner et al, 1996; NB: Those mutations in APP that lie within the Aβ sequence increase the self‐aggregation of the resultant peptides, not their production). A key mechanistic explanation was the discovery that the presenilin genes encode the active site of the intramembrane‐cleaving γ‐secretase enzyme (De Strooper et al, 1998; Wolfe et al, 1999). Subsequent studies have begun to illuminate how presenilin mediates intramembrane proteolysis (Qi‐Takahara et al, 2005; Takami et al, 2009; Chavez‐Gutierrez et al, 2012; Okochi et al, 2013; Fernandez et al, 2014): an initial endopeptidase cleavage of APP near the transmembrane/cytoplasmic interface of APP (the ε‐cleavage) is followed by multiple carboxypeptidase cleavages that each sequentially removes 3 or 4 C‐terminal amino acids (i.e., approximately one turn of the intramembrane helix) (Fig 2). This process yields two product lines that start with either the Aβ48/49 or the Aβ49/50 ε‐cleavage. Although the precise molecular effects of different presenilin mutations differ somewhat, in all cases the mutations appear to decrease this C‐ to N‐terminal cleavage “processivity” and thus increase the relative production of longer (more hydrophobic and self‐aggregating) Aβ peptides. This elegant model provides a biochemical explanation for earlier work showing that pathogenic presenilin mutations often increase the Aβ42/Aβ40 ratio in humans. γ‐Secretase reactions conducted directly in presenilin‐mutant AD brain tissue showed that all presenilin mutations studied decreased this carboxypeptidase‐like activity, and assays in a few “sporadic” AD brains suggested that a similar decrease in processivity might occur in some non‐presenilin‐mutant cases (Szaruga et al, 2015). Aβ42, Aβ43, and longer Aβ peptides are highly self‐aggregating, whereas Aβ40 may actually be anti‐amyloidogenic (Kim et al, 2007).
Figure 2. Progressive cleavages of the APP transmembrane domain by the Presenilin/γ‐secretase complex.

One group has emphasized that the aforementioned mechanism represents a loss of function of presenilin and have proposed that the neural phenotype of AD patients is fundamentally due to a loss of presenilin function, independent of effects on Aβ production (Shen & Kelleher, 2007; Xia et al, 2015). They have studied presenilin‐1 mutations that generally lower Aβ and hardly raise relative Aβ42 levels, but this work may overlook an elevation of the Aβ43 and other longer species, which are highly amyloidogenic (Saito et al, 2011). Although AD‐causing presenilin mutations can indeed be interpreted as partial loss of function from a genetics perspective, pinpointing the function of presenilin as an aspartyl endopeptidase allows one instead to speak in biochemical terms of a functional shift of the principal proteolytic cleavages to more C‐terminal bonds in the substrate (Kretner et al, 2016). Humans with pathogenic presenilin mutations are heterozygotes and experience no loss of function of Notch cleavage; rather, they have accelerated Aβ42 and Aβ43 accumulation that long precedes their AD‐typical memory syndrome. Most importantly, > 99% of all AD patients (including all other forms of familial disease) express wild‐type presenilin, so loss of presenilin function cannot be a general mechanism of AD pathogenesis.
The original formulation of the amyloid hypothesis was based in part on the discovery that the APP gene is on chromosome 21, implying that individuals with Down's syndrome develop typical Alzheimer neuropathology because they produce too much Aβ lifelong. This supposition has been substantiated by the identification of humans with different segmental microduplications of sub‐regions of chromosome 21. Rare individuals with translocation Down's syndrome involving only the distal part of chromosome 21 telomeric to the APP gene have Down's features but do not get AD (Prasher et al, 1998). Conversely, those rare individuals who have the APP gene micro‐duplicated but not the rest of the chromosome do not have Down's syndrome but get AD, typically in their mid‐50s (Rovelet‐Lecrux et al, 2006). These findings show conclusively that lifelong overexpression of wild‐type APP causes AD. Even more remarkable has been the identification of an APP missense mutation (A673T) at the second amino acid of the Aβ region that results in a lifelong decrease in APP cleavage by β‐secretase (Jonsson et al, 2012). Moreover, this benefit may be compounded, because the mutant Aβ peptide that is generated has altered aggregation properties (Benilova et al, 2014; Maloney et al, 2014; Zheng et al, 2015). A673T carriers have a lower risk of clinical AD and even of age‐related cognitive decline without clinical AD (Jonsson et al, 2012), and they may not show plaque deposition at age 100 (Kero et al, 2013). The reduced amyloid deposition resulting from this AD‐protective mutation strongly supports the amyloid hypothesis.
Improved modeling of the amyloid hypothesis in rodent and cellular systems
Concern has been expressed about the limitations of available rodent and cellular models of β‐amyloid pathogenicity (Table 2). Early APP mouse models (e.g. Games et al, 1995; Hsiao et al, 1996) suffered from reliance on high transgene expression to drive plaque deposition and from a lack of tangle cytopathology and neuronal death. Crossing FAD‐mutant APP mice with mutant MAPT (tau) transgenic (tg) mice succeeded in augmenting tau pathology and suggested that tangle‐like changes occur downstream of Aβ accumulation, but this involved transgene overexpression and multiple AD mutations (Lewis et al, 2001). Recently, mice with gradual Aβ plaque accrual have been developed by the judicious use of selective knockin of human mutations into endogenous mouse APP without overexpression (Saito et al, 2014). Moreover, stem cell‐derived human neurons cultured from skin biopsies of FAD subjects have been used to show first Aβ accumulation and then tau alteration in the absence of overexpression (Shi et al, 2012; Choi et al, 2014; Muratore et al, 2014; Moore et al, 2015) suggesting that the lack of tangle formation in early mouse models was related to the absence of human tau. This progress means we are now able to model a substantial part of the amyloid cascade in culture. In both cellular and mouse models, extensive data now suggest that the neurotoxicity of Aβ is in considerable part dependent on expression of human tau (Rapoport et al, 2002; Jin et al, 2011; Roberson et al, 2011).
Table 2.
Toward a more complete modeling of the pathogenesis of AD amyloid
| Year | System | Achievement | Critique | References |
|---|---|---|---|---|
| 1995 | APP transgenic mouse | Plaque Pathology | Overexpression, no downstream pathology | Games et al (1995) |
| 2000 | MAPT mutant transgenic mouse | Tangle Pathology | Overexpression: no plaque pathology | Lewis et al (2000) |
| 2001 | APP X MAPT transgenic mice | Plaque and tangle pathology | Overexpression of both transgenes: artificiality of two mutations | Lewis et al (2001) |
| 2012 | Down's syndrome derived stem cell neurons | Diffuse plaque pathology: evidence for pre‐tangles | Not full pathology | Shi et al (2012) |
| 2014 | Complex APP mutation knockin into mouse genome | Plaque pathology without overexpression | Artificiality of multiple mutations: no downstream pathology | Saito et al (2014) |
| 2014 | Overexpression of APP mutations in human neuronal lines in gel system | Convincing plaque pathology and also tangle pathology | Overexpression | Choi et al (2014) |
| 2015 | APP and PSEN mutant stem cell lines | Diffuse plaque pathology and tau pathology | Moore et al (2015) |
Cell biology of new AD risk genes
Although the importance of ApoE4 as the major risk factor for AD was discovered in 1993 (Corder et al, 1993), it is only since the advent of genomewide association studies and, more recently, exome and genome sequencing that other risk loci for late‐onset disease have been discovered. Whereas the recently described loci are usually much weaker in effect (Lambert et al, 2013) or much rarer (Guerreiro et al, 2013; Jonsson et al, 2013) than ApoE4, they have helped delineate additional biological processes in AD pathogenesis. Three types of processes have emerged as especially important: cholesterol/sterol metabolism; inflammation and the brain's innate immune system; and endosomal vesicle recycling (Jones et al, 2010).
Apolipoprotein E and other components of cholesterol/sterol metabolism
A role for cholesterol in AD has long been suspected, based on the genetic implication of ApoE in the disease as well as the contrasting effects of cholesterol loading or depletion on amyloid pathology in APP tg mice (Refolo et al, 2000, 2001). Work in APP mice expressing different human ApoE alleles has shown that a major pathogenic influence of ApoE involves differential isoform effects on the clearance of Aβ (Castellano et al 2011: discussed below). The ABCA7 lipid transporter has also been identified as a genetic locus for the disease (Hollingworth et al, 2011), and loss‐of‐function mutations increase AD risk about threefold (Steinberg et al, 2015). ABCA7 is expressed in neurons, microglia, and peripheral macrophages, and it normally promotes the efflux of lipids from cells to apolipoproteins and also regulates phagocytosis. Crossing ABCA7 knockout mice to mutant hAPP mice caused a doubling of insoluble Aβ levels and amyloid plaques without changing APP processing, suggesting that like ApoE, ABCA7 is involved in Aβ clearance (Kim et al, 2013). However, the biochemical details through which both ApoE and ABCA7 influence the development of Aβ pathology need to be pinpointed.
The innate immune system in Alzheimer's disease
Neuropathologists have long suggested that the brain's innate immune system, including the microglial response to plaque formation, was an important factor in AD pathogenesis. For example, the early observation of multiple elements of the classical complement cascade in and around neuritic plaques (McGeer et al, 1989) was prescient. In the last few years, genetic variability in that system has emerged as a compelling determinant of AD risk, implicating many components of innate immunity and the complement cascade as risk factors in the disease (Jones et al, 2010). Three such risk genes have been investigated in some detail: Complement Receptor 1 (CR1; Lambert et al, 2009), CD33 (Bertram et al, 2008), and TREM2, and all three appear to be involved either directly or indirectly in the response of microglia to Aβ deposition. Blockade of CR1 inhibits microglial activation and potentiates microglial phagocytosis (Crehan et al, 2013). Inactivation of CD33 in primary microglia also potentiates microglial uptake of Aβ (Griciuc et al, 2013), and TREM2 is responsible for sustaining microglial phagocytosis of Aβ (Wang et al, 2015). Thus, all three genetically implicated microglial proteins may be involved in helping to maintain the AD microglial phenotype of phagocytosing Aβ deposits. Accordingly, these 3 genes undergo increased expression during plaque development (Griciuc et al 2013, Wang et al, 2015; Matarin et al, 2015) and CSF TREM2 levels go up as plaque load increases, suggesting it may be a useful biomarker (Suárez‐Calvet et al, 2016).
TREM2 is emerging as a key molecular determinant of the CNS response to Aβ accumulation (Forabosco et al, 2013; Zhang et al, 2013; Matarin et al, 2015). However, the biology of TREM2, a Type 1 single‐transmembrane receptor which is principally but not exclusively expressed in microglia and undergoes ADAM/γ‐secretase processing (Wunderlich et al, 2013; Kleinberger et al, 2014), is incompletely understood [reviewed in (Lue et al, 2015)]. The most studied mutation, R47H, may increase the risk of AD to the same extent that ApoE4 does although it is much rarer (Guerreiro et al, 2013; Jonsson et al, 2013). The upregulation of TREM2 in a subset of microglia in amyloid plaques of hAPP tg mice (e.g., Guerreiro et al, 2013) suggests that the known function of TREM2 in phagocytosis is compromised during plaque development. A current hypothesis is that R47H and other AD‐associated TREM2 mutations confer loss of function in microglia. Deleting one TREM2 allele in hAPP tg mice significantly decreased the number of microglia associated with Aβ deposits (Ulrich et al, 2014). Conversely, TREM2 overexpression in hAPP tg mice decreased amyloid plaque burden, neuroinflammation, synapse loss, and spatial memory deficits (Jiang et al, 2014). And TREM2 mutations can alter its transport to the cell surface and shedding, associated with impaired phagocytic function (Kleinberger et al, 2014). The latter work has led to evidence that levels of the shed ectodomain in extracellular fluid and CSF are lower in AD cases associated with TREM2 mutations.
Endosomal vesicle recycling in Alzheimer's disease
The final set of recently identified loci for late‐onset AD map to processes regulating endosomal vesicle recycling (Jones et al, 2010). This category includes SORL1, BIN1, and PICALM (Rogaeva et al, 2007; Lambert et al, 2013; Zhao et al, 2015). SORL1 had previously been shown to be directly involved in the processing of APP (Andersen et al, 2005), and work in human stem cell‐derived neurons carrying the SORL1 risk haplotype confirmed this association (Young et al, 2015). Likewise, PICALM appears to be involved directly in endosomal APP processing (Kanatsu et al, 2014). In addition, PICALM has been implicated in the transport of brain Aβ across the blood–brain barrier: induced pluripotent stem cell (iPSC)‐derived human endothelial cells carrying an AD‐protective allele exhibited higher PICALM levels and enhanced Aβ clearance (Zhao et al, 2015).
In summary, mechanistic studies linking several of the recently identified risk genes for late‐onset (previously “sporadic”) AD to aspects of Aβ homeostasis provide new support for the amyloid hypothesis as a driving factor in AD pathogenesis. They also suggest new avenues for therapeutic intervention, such as intervening in brain cholesterol metabolism and modulating the response of the innate immune system to amyloid deposition.
Recent findings help resolve controversies about the role of Aβ
Connecting plaques and tangles: Aβ can drive tau alteration
The temporal sequence of the two canonical lesions Alois Alzheimer noted in his 1906 index case has been debated ever since. An elegant histopathological staging system created by Braak and Braak (1991) is now widely used to establish the severity of AD neuropathology. This scale principally described the progression of AD‐type cytoskeletal changes, that is, neurofibrillary tangles and dystrophic neurites, in unrelated humans of increasing age (it could not yet include assays for accrual of oligomeric forms of Aβ). The detection of modest amounts of neurofibrillary change in limbic regions of young or middle‐aged individuals dying of other causes does not imply that such individuals would necessarily have developed AD had they lived longer. Instead, human genetic and biomarker studies have provided the answer to the sequence of Aβ and tau accumulation in AD. Inherited mutations in APP and presenilin (i.e., in the substrate and the protease for Aβ generation) cause early‐onset Aβ deposition (Lemere et al, 1996a,b; Bateman et al, 2012) followed by accumulation of tangles/neurites containing filaments of wild‐type tau, so amyloid can clearly precede tangles in humans. In contrast, mutations in the tau gene lead to a form of frontotemporal dementia without subsequent accrual of Aβ. Thus, Aβ accumulation can lead to progressive tau deposition, but the converse has not been clearly demonstrated in humans.
Laboratory studies support this sequence. Crossing hAPP tg mice with hTau tg mice significantly enhances tau deposition without changing Aβ deposition (Lewis et al, 2001). Crossing an APP tg mouse to a tau knockout mouse leads to substantially less behavioral deficits in the offspring than when tau is expressed (Roberson et al, 2011). Treating normal rat neurons in culture with soluble Aβ oligomers isolated from AD cortex causes neuritic dystrophy and AD‐type tau hyperphosphorylation, but no dystrophy ensues if tau is first knocked down (Jin et al, 2011). Several similar studies suggest that Aβ—particularly soluble oligomers of Aβ42 (Shankar et al, 2008)—can trigger AD‐type tau alterations, supporting the sequence that human genetics has indicated. The expression of human tau seems to be “permissive”, enabling certain downstream neuronal consequences of progressive Aβ accrual to occur (Maruyama et al, 2013).
How ApoE4 promotes AD: chronically decreased Aß clearance
Humans expressing the ApoE4 protein develop more plaque and vascular β‐amyloid deposits than those expressing only ApoE3 (Rebeck et al, 1993), and this has been confirmed in genetically engineered mice (Holtzman et al, 2000). A detailed quantitative study of Aβ homeostasis using in vivo microdialysis in hAPP × hApoE crossed mice has shown that Aβ clearance (but not Aβ production) is decreased by ApoE4 > E3 > E2, closely paralleling the degree of Aβ deposition in such mice (Castellano et al, 2011). The decrease in clearance of soluble Aβ was observed in young mice well before any amyloid deposition. The results strongly suggest that ApoE contributes to AD risk at least in part by differentially regulating soluble Aβ clearance, emphasizing Aβ clearance pathways as a major therapeutic target. In accord, one of the numerous risk genes for late‐onset AD is PICALM, and AD‐promoting alleles or knockdown of this gene has been shown to decrease Aβ clearance across the brain endothelium (Zhao et al 2015). As a potentially related effect on Aβ homeostasis, the three ApoE isoforms have also been shown to bind Aβ differentially and modulate its fibrillogenesis (Ma et al, 1994; Wisniewski et al, 1994; Evans et al, 1995). Other potential mechanisms have been suggested, including an adverse effect of ApoE4 on the processing of tau in neurons (Andrews‐Zwilling et al, 2010; Huang & Mahley, 2014).
Synaptic loss, Aβ, and amyloid plaques
Decreased synapse number has long been recognized as perhaps the strongest quantitative neuropathological correlate of dementia in AD. Numerous laboratory studies in the past decade have shown that Aβ oligomers impair both synaptic function (e.g., long‐term potentiation) and synaptic structure (e.g., dendritic spines). Of particular, disease relevance is evidence that soluble oligomers (but not monomers) of Aβ42 isolated directly from AD cortex can dose‐dependently decrease synaptic function and number and can impair the memory of a learned behavior in healthy adult rats (Shankar et al, 2008). Amyloid plaque cores isolated from the same AD brains and washed extensively in vitro do not impair LTP, but the diffusible Aβ42 oligomers that can subsequently be released from them with harsh denaturants do so (Shankar et al, 2008). The latter findings fit nicely with evidence in hAPP tg mice that plaques in situ have a penumbra of soluble Aβ oligomers in which synaptic density is low; synapse number rises toward normal the farther one measures from the edge of the plaque core (Koffie et al, 2009).
The intimate association of diffusible oligomers with fibrillar plaques that such studies imply has been elegantly supported by quantifying Aβ oligomers with a selective ELISA in postmortem brain tissue of subjects who were either clinically normal (Clinical Dementia Rating of 0) or mildly demented (CDR of 1) shortly before death. These brains were selected to have similar plaque densities, but the oligomer‐specific ELISA revealed that the non‐demented plaque‐rich subjects had much lower oligomer‐to‐plaque ratios than the mildly demented plaque‐rich patients (Esparza et al, 2013). Indeed, this ratio completely distinguished (without overlap) the “high‐pathology control” brains from the AD brains. This striking result addresses an oft‐cited “Achilles heel” of the amyloid hypothesis: apparently normal people who have abundant plaques may actually have low plaque‐associated oligomer levels (Table 1). We have hypothesized that plaques can sequester soluble oligomers until they slowly reach a physical limit, after which excess oligomers can diffuse onto surrounding synaptic membranes and other hydrophobic cell surfaces (Hong et al, 2014).
A heterogeneity of Aβ species in AD brain
While Aβ1–42 peptides appear to be the earliest form to accumulate in the brain and their free levels in CSF drop long before clinical symptoms [e.g., Bateman et al (2012)], this initial species can be modified over time into a complex array of truncated, isomerized, and/or phosphorylated peptides. One well‐studied variant that is highly amyloidogenic (Nussbaum et al, 2012) is the “p3E” Aβ peptide that is truncated over time of Asp1 and Ala2 and then cyclized at Glu3 (Mori et al, 1992). DeMattos et al (2012) have shown in hAPP transgenic mice that this variant accumulates rather late and in small amounts, but targeting it with a specific antibody promotes a kind of “bystander clearance” by microglia of also earlier deposited Aβ species, making p3E an attractive target for Aβ immunotherapy despite its low abundance. At the opposite end of Aβ, the variant Aβ43 is highly prone to aggregation (Saito et al, 2011), and it is unclear how long soluble oligomers of this peptide can be present as such in the brain before they deposit as insoluble amyloid plaques. Given the complexity of Aβ species in humans, a worthy goal for future clinical research is to routinely quantify all Aβ peptides in plasma or CSF of pre‐symptomatic and symptomatic AD subjects.
Recent studies have uncovered further heterogeneity of both Aβ and other proteolytic products of APP processing. One example is the detection in certain cell lines expressing pathogenic mutant APP, of Aβ monomers and dimers as well as N‐terminally extended Aβ monomers that begin some ~35–40 resides before the Asp1 of Aβ (Welzel et al, 2014). Levels of these extended monomers rise dramatically when β‐secretase inhibitors are applied to the cells, indicating that they arise from an alternative protease(s) which cleaves APP upstream of the Met‐Asp bond. The N‐terminally extended monomers inhibit LTP, presumably because they contain the Aβ region but in a misfolded form that can bind to synaptic membranes (Welzel et al, 2014). A second and distinct set of APP fragments arises from a novel η (“eta”)‐secretase processing pathway, involving cleavage by a matrix metalloproteinase 92 amino acids upstream of the Met‐Asp bond (Willem et al, 2015). The resultant η‐CTFs can then be processed by α‐ or β‐secretase to generate Aη‐α and Aη‐β peptides. Again, β‐secretase inhibitors elevate these alternative APP products. Single‐cell calcium imaging shows that neuronal activity is attenuated by Aη‐α, suggesting a physiological function of this new pathway but not yet implicating it in AD (no η‐derived fragments containing the intact Aβ region were described; Willem et al, 2015).
Is AD in humans a prion‐like disease of “pathogenic spread”?
In the last decade, the neurodegenerative field has become increasingly intrigued by the hypothesis that the progression of the cytopathological lesions in AD, Parkinson's disease, Frontotemporal dementia, and other age‐related diseases involves a physical spread of the specific offending proteins from neuron to neuron. Initially, this hypothesis was based in considerable part on three observations. First, the Braak staging of tangles (in AD) and Lewy lesions (in Parkinson's disease) in postmortem brains from humans of increasing age was interpreted as a physical spread of the responsible protein aggregates (tau and α‐synuclein, respectively) from one brain region to the next. Second, the development over many years of some Lewy bodies in fetal neurons implanted into the striata of a few advanced Parkinson patients was interpreted as a physical spread of a pathogenic form of α‐synuclein from the diseased neurons of the host into the healthy neurons of the implant. Third, experimental studies in rodents suggested that the extracellular injection of fibrils of tau or α‐synuclein could induce still‐healthy neurons to form the respective intracellular lesions, suggesting a physical spread as well as a “prion‐like” templating of the normal protein by the abnormal (misfolded) protein.
These findings provide circumstantial evidence for the spread of misfolded proteins from neuron to neuron, although the details of the cell biological mechanisms remain unclear. The extent to which the Braak staging in AD (as well as in Parkinson's disease and other neurodegenerative diseases) represents a selective temporal vulnerability of neurons in different brain regions rather than an actual physical spread has not been elucidated. It remains a challenge to distinguish cell autonomous from non‐cell autonomous mechanisms of protein aggregation in vivo, particularly in humans with pathogenic mutations in tau or α‐synuclein, where 50% of the protein in all neurons is mutant and could aggregate without the need for inter‐neuronal spread. Clearly, these are important unresolved issues: understanding the biological mechanisms for the spread of cytopathology could offer new therapeutic targets and may underlie the observed beneficial effects of administered antibodies on lesion clearance.
A separate question from that of neuron‐to‐neuron spread is whether the misfolded Aβ and tau aggregates in AD brain could be true “proteinaceous infectious particles” and thus transmissible between humans. Attempts by Gadjusek and colleagues more than 30 years ago to transmit AD to lower primates by inoculation of AD brain extracts were deemed unsuccessful (Brown et al, 1994). However, Ridley et al (2006) reported that inoculation of marmosets with AD brain extracts induced modest cerebral β‐amyloidosis: Aβ‐immunoreactive deposits were detected in 16 of 18 animals aged < 10 years and 8 of 9 aged > 10 years, whereas spontaneous cerebral amyloid deposition was found in 0 of 11 non‐injected marmosets < 10 years and 5 of 29 > 10 years (Ridley et al, 2006). Neurofibrillary tangles were not detected in any animals, so it seems that inoculation with AD brain extracts accelerated Aβ (but not tau) deposition in these primates. This acceleration is entirely consistent with the long‐standing concept of the seeded polymerization of Aβ in kinetic models of β‐amyloid formation (Jarrett & Lansbury, 1993).
Recently, four patients dying between ages 36 and 51 of iatrogenic Creutzfeld–Jakob disease after childhood treatment for short stature with prion‐contaminated cadaveric pituitary extracts were reported to also have substantial Aβ‐immunoreactive plaques and microvessels in their brains (Jaunmuktane et al, 2015). Since pituitary glands in AD subjects can have Aβ deposits, these cases were interpreted as providing evidence of human‐to‐human transmission of Aβ seeds. No tangles or AD‐type neuritic/microglia‐rich plaques were described, so this was not transmission of AD per se, consistent with an earlier negative study (Irwin et al, 2013). Locating some of the original pituitary extracts used for inoculation and showing that they had abnormal Aβ forms will be necessary to prove that these cases represent definite human transmission of Aβ seeds. At present, there are no clear clinical concerns arising from this unusual iatrogenic event as regards AD risk in the general population.
Biomarkers: approaching the natural history of AD in human subjects
For many years, reaching a correct understanding of the pathogenic sequence in AD patients was hampered by the difficulty of detecting this sequence directly in living humans. The problem has been substantially lessened by three developments: (i) robust assays to quantify soluble Aβ monomers and tau in CSF (Vigo‐Pelfrey et al, 1995); (ii) imaging fibrillar amyloid burden (but not yet soluble Aβ oligomers) by PET scanning, initially with the thioflavin T derivative, Pittsburgh compound B (Klunk et al, 2004); and (iii) the ability to analyze APP metabolism in healthy and diseased humans by quantifying heavy isotope‐labeled Aβ peptides by mass spectrometry in fresh CSF collected continuously through a lumbar intrathecal catheter (Bateman et al, 2006; Mawuenyega et al, 2010).
In vivo APP labeling
The use of 13C (heavy) leucine infusions to label all newly synthesized proteins, including APP, in pre‐symptomatic subjects with presenilin mutations and their non‐carrier siblings confirmed the extensive data in cultures and mouse models that these AD‐causing mutations increase relative Aβ42 production (Potter et al, 2013). Further, during the period of amyloid deposition, the Aβ42 monomer declines in CSF in a manner that suggests it becomes bound to developing plaques (Blennow et al, 2015). And the use of the in vivo labeling approach in ApoE3 vs. E4 carriers showed that E4 subjects had lower rates of Aβ monomer clearance [see (Castellano et al, 2011)]. Together, these human data support the conclusions that presenilin mutation carriers produce relatively more Aβ42 and that E4 carriers clear it less efficiently.
Amyloid imaging and CSF biomarkers
Numerous families carrying APP, PSEN1, or PSEN2 mutations have been studied collectively to determine the time course of fluid biomarker, neuroimaging, and clinical changes prior to the expected onset of AD symptoms, which is based on the age of symptom onset in a parent with the same mutation. Initial analyses of a familial AD cohort [the Dominantly Inherited Alzheimer Network (DIAN)] suggest that Aß42 levels in CSF may first be somewhat elevated (vs. normal) and then begin to decline as early as 25 years before expected symptom onset (Bateman et al, 2012). This is followed by the appearance of fibrillar amyloid deposits in the brain (as detected by PiB‐PET), increased levels of tau in CSF, and progressive brain atrophy roughly 15 years before expected symptom onset (Bateman et al, 2012). Neuronal hypometabolism and subtly impaired episodic memory seem to begin some 10 years or so before expected symptoms (Bateman et al, 2012). If this time course is generally similar to that of “sporadic” AD, and the AIBL study (Villemagne et al, 2013) suggests that it is, then Aβ deposition may begin up to two decades or more before clinically noticeable cognitive decline. A key lesson which emerges from such dynamic analyses of pre‐symptomatic AD is that therapeutic interventions directed only at the mild‐to‐moderate clinical stage may be too late to ameliorate progression.
Overall, brain imaging and CSF biomarker studies in humans suggest that the sequence of AD pathogenic steps currently measurable in vivo broadly follows the schema proposed by Jack and colleagues (Jack & Holtzman, 2013; Jack et al, 2013; Fig 3). These data are consistent with early studies of AD neuropathology in Down's syndrome, which documented an initial accumulation of diffuse Aβ deposits that precedes microglial and astrocytic activation, tangle formation, and neurodegeneration (e.g., Lemere et al, 1996a,b; Mann et al, 1992). The recent development of imaging agents for tangles (Chien et al, 2013; Liang et al, 2014) will help define the time course of accrual of the two major lesions, although tangles are somewhat non‐specific in that they occur increasingly with “normal” aging and in several neurodegenerative processes besides AD.
Figure 3. A hypothetical temporal model integrating Alzheimer's disease biomarkers.
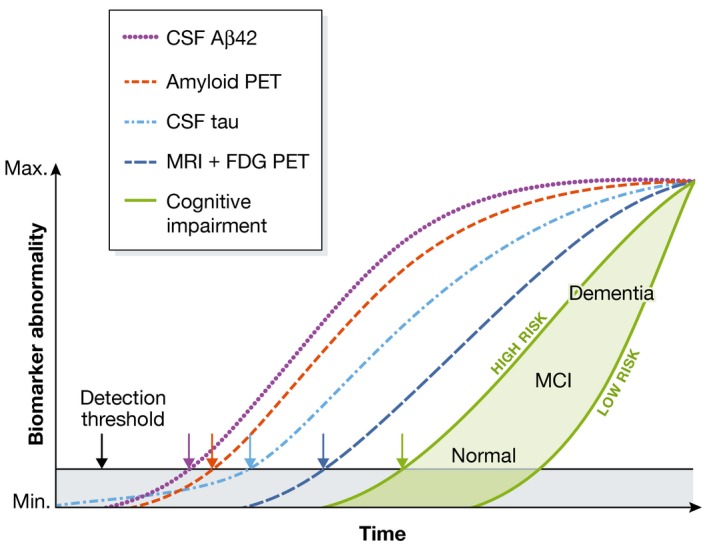
The threshold for the first detection of biomarkers associated with pathophysiological changes is denoted by the black horizontal line. The gray area denotes the zone in which abnormal pathophysiological changes lie below this biomarker detection threshold. In this model, the occurrence of tau pathology can precede Aβ deposition in time, but only early on at a sub‐threshold biomarker detection level. Aβ deposition occurs independently and rises above the biomarker detection threshold (purple and red arrows). This induces acceleration of tauopathy, and CSF tau then rises above the detection threshold (light blue arrow). Later still, changes in FDG PET and MRI (dark blue arrow) rise above the detection threshold. Finally, cognitive impairment becomes evident (green arrow), with a wide range of cognitive responses that depend on the individual's risk profile (light green‐filled area). Note that while CSF Aβ42 alteration is plotted as a biomarker (purple), this represents a decrease in CSF Aβ42 levels and is a surrogate for an increase in parenchymal Aβ42 and changes in other Aβ peptides in the brain tissue. Aβ, amyloid β‐protein; FDG, fluorodeoxyglucose; MCI, mild cognitive impairment. (Adapted from Fig 6 of Jack et al, 2013.)
Recent progress in AD clinical trials
None of the Aβ‐targeted phase 3 clinical trials in Alzheimer's disease has shown statistically significant benefit on its pre‐specified clinical endpoints. Several of these trials, however, were misdesigned in terms of patient selection, choice of agent, target engagement, and/or dose, or they had to be halted because of side effects that may not have been target‐related (De Strooper, 2014; Karran & Hardy, 2014).
Semagacestat was neither an effective nor safe γ‐secretase inhibitor
Inhibiting the β‐ or γ‐secretases is an attractive goal, but the recognition that they have many substrates besides APP makes selectivity an enormous challenge. The discovery that Aβ is normally secreted by cells throughout life (Haass et al, 1992) led to widespread compound screening on cultured cells, and most Aβ‐lowering “hits” that emerged inhibited γ‐secretase. The only such compound to reach Phase 3 testing was semagacestat, but the trial was terminated after ~12 months of dosing due to adverse events (Doody et al, 2013). This may be explained by its low therapeutic index: the IC50 for Notch cleavage was only twofold to threefold higher (or less) than that for APP cleavage. Direct proof that semagacestat was an effective Aβ‐lowering agent in humans was not obtained, and this trial should not have led to the curtailment of research to develop safer inhibitors of γ‐secretase with better substrate selectivity (De Strooper, 2014). Another agent, avagacestat, had a better therapeutic index but still not good enough to avoid certain side effects and be advanced to Phase 3 trials (Coric et al, 2012).
The fundamental catalytic mechanism of this first‐in‐class intramembrane aspartyl protease is incompletely understood, although substantial progress should ensue from biochemical studies that take advantage of the atomic structure of the whole γ‐secretase complex that has recently been solved (Bai et al, 2015). While further research on selective inhibition of the protease is needed, the field now favors modulators of γ‐secretase that shift the peptide bond cleavage 3–4 residues N‐terminal to the Aβ42 site without blocking proteolysis. Different chemical classes of such γ‐modulators are being developed; whether they can achieve brain penetration and potency to levels needed to lower Aβ chronically remains untested.
Solanezumab: a probable signal in mild AD
Since active and passive immunotherapy to lower amyloid was first conceptualized (Schenk et al, 1999; Bard et al, 2000), antibody trials have taken the lead among putative disease‐modifying therapeutics for AD. The antibody most advanced time‐wise in current human testing is solanezumab, which targets the mid‐region of Aβ and binds principally to soluble monomers and perhaps low‐n oligomers but not to plaques. Two large Phase 3 trials in mild and moderate AD patients failed to achieve their clinical endpoints. Pre‐specified, post hoc analyses of the combined mild subjects of the trials showed a statistically significant ~34% slowing of cognitive decline vs. placebo over 18 months [see tables 3 and 4 in Doody et al, 2014]. The moderate AD patients in the same trials showed no benefit, proving the widely held assumption that anti‐Aβ agents should be started in mild AD or even earlier. The results in the mild subjects suggested a small but statistically significant cognitive benefit of this agent, leading to a third Phase 3 study in only mild subjects that is underway. It is of interest that another antibody, crenezumab, produced similar signs of modest slowing of cognitive decline in mild AD patients in a Phase 2 trial (Cummings et al, 2014).
Aducanumab: a big signal in a small proof‐of‐concept trial
The strongest hint to date of the potential clinical and biomarker benefits of an amyloid‐targeting agent came recently in a Phase 1b trial of a human monoclonal antibody (BIIB‐037 or aducanumab) that emerged from a large screen of B‐cell clones obtained from healthy aged people. All 165 trial subjects underwent PET amyloid imaging at entry to confirm the clinical diagnosis; this had not been done in the completed trials reviewed above, where up to 30% of subjects were later found to lack amyloid. Another difference of the small aducanumab trail is that it was conducted in one country (United States), so all cognitive evaluations were performed in one common language, probably reducing inter‐subject variability in scoring. Three IV doses (1, 3 or 10 mg/kg/mo) were initially compared to placebo after 6 and 12 months. The 3 and 10 mg dose reduced PET amyloid levels at 6 months and more so at 12 months, with the 10 mg dose causing a decline to near the level required for trial entry (Sevigny et al, 2015). This dose‐dependent evidence of target engagement and biomarker movement was accompanied by significantly less decline (vs. placebo) in two tests, the Mini‐Mental State Exam and the Clinical Dementia Rating—Sum of Boxes. In the 10 mg dose group, these scores were almost stable from 6 to 12 months. The only meaningful adverse event was transient ARIA‐E (amyloid‐related imaging abnormality—edema) in ~20% of the subjects receiving aducanumab. As in some prior Aβ antibody trials, ARIA‐E occurred mostly in ApoE4+ patients, was dose‐dependent, and produced no symptoms in 65% of these cases. Both the careful design of this small study and the nature of the human antibody, which apparently binds plaques and oligomers but not monomers, may have contributed to the positive clinical and biomarker outcomes. Aducanumab entered the necessary Phase 3 studies in 2015.
The advent of “secondary prevention” trials
The failures of some anti‐amyloid agents that appeared to engage their targets but did not achieve clinical endpoints in mild‐to‐moderate AD patients have moved the field to attempt pre‐symptomatic or “secondary prevention” trials in subjects shown by PET amyloid imaging and/or CSF Aβ42/tau assays to be at high risk for developing AD dementia. Such prevention trials are now being conducted, respectively, in the world's largest kindred carrying a presenilin‐1 mutation (the API study in rural Colombia (Ayutyanont et al, 2014)) or in many smaller kindreds carrying presenilin‐1 or presenilin‐2 or APP mutations (the DIAN trials). Importantly, the first prevention trial in largely pre‐symptomatic humans at risk of late‐onset AD (ages 65–85) based on abnormal PET amyloid scans is now underway in > 60 centers in the United States, Canada and Australia [A4study.org] (Sperling et al, 2014). All three of these trials are initially administering Aβ antibodies, but other agents targeting Aβ or other factors (tau; neuroinflammation) are planned or underway.
Active vaccines: no longer at the forefront but not forgotten
AD immunotherapy in man began with an active vaccine trial (using AN‐1792, a synthetic Aβ1–42 peptide) that was terminated after 2–3 doses due to the occurrence of a T‐cell mediated meningeal inflammation in 6% of the Phase 2 patients (Gilman et al, 2005). But quantitative neuropathological analyses of a few brains from subjects who had died years after a Phase 1 trial of AN1792 revealed evidence of amyloid clearing and apparent lessening of neuritic dystrophy and synaptic deficits, compared to what would be expected in such advanced AD patients (Serrano‐Pozo et al, 2010). Only 1–2 trials of active Aβ vaccines are underway at this writing, but this approach clearly deserves more study, as the polyclonal antibody response may prove beneficial, and the cost and logistics of distributing passively administered monoclonal antibodies several times per year to the world's AD population are daunting.
β–secretase 1 inhibition in Phase 3: much anticipated
Inhibitors of β‐secretase arrived later than those for γ‐secretase, in part because of the pharmacological challenges of targeting the large active site of this aspartyl protease in intact neurons. But now, several companies have Phase 2 or 3 trials of chemically distinct inhibitors underway. No published data of efficacy and side effects in man are yet available. Nevertheless, the discovery of many new substrates of β‐secretase, including some that are critical for signaling events in both the immature and mature nervous system (Willem et al, 2006; Hemming et al, 2009; Kuhn et al, 2012), raises the possibility of significant adverse events appearing over time in such trials. Speculation abounds about whether lowering Aβ42 monomers with β‐ or γ‐secretase inhibitors/modulators or binding and clearing plaques and diffusible Aβ with antibodies will turn out to be more efficacious. The combined testing of two anti‐Aβ agents is desirable and may not lie too far ahead.
The amyloid hypothesis at 25 years
This review perforce mentions only a fraction of the many studies on the relationship of Aβ accumulation to the other features of the AD syndrome. But the examples we highlight underscore the compelling nature of the extensive preclinical and emerging clinical evidence that Aβ dyshomeostasis is upstream of alterations in other proteins and diverse cell types that contribute to the AD cognitive phenotype (Box 1). We emphasized more than a dozen years ago (Hardy & Selkoe, 2002) that definitive proof of this once controversial concept could only come from clinical trials that selectively target Aβ and produce slowing and ultimately arrest of cognitive decline in typical AD patients. The recent aducanumab Phase 1b data are consistent with such evidence, although we obviously need large, multi‐national trials that show significant amelioration of AD progression over 18–24 months.
Success breeds success, and it appears increasingly likely that exciting progress in the clinic, building upon a 3‐decade record of advances in the laboratory, will provide this proof. The continued push toward a safe and efficacious amyloid therapeutic takes nothing away from the need for alternative agents that target other early features of this complex and devastating syndrome. As others have pointed out (Small & Duff, 2008; De Strooper & Karran, 2016) and we concur, after disease initiation, the complexity of the downstream pathogenic processes increases. Nonetheless, it is likely that therapies aimed at these downstream processes will eventually have a role in the armamentarium against this devastating disease. It is not a question of one hypothesis against another. Rather, we must pursue multiple approaches, leading to a range of therapeutics that may together prevent the looming personal and societal tragedy that Alzheimer's disease has become.
Conflict of interest
DS is a director of Prothena Biosciences and has received consulting fees from Janssen, Roche, and Sanofi and a speaker's fee from Biogen‐Idec. JH consults for Eisai and for Cytox and has received speaking fees from Eli Lilly, Takeda, and Roche.
Pending issues.
What are the toxic species of Aβ and tau?
What is the connection between Aβ and tangle pathology? Is it direct and cell autonomous or does it involve non‐neuronal cells?
What is the mechanism of pathology spread and does understanding this spread provide therapeutic opportunities?
What is the function of APP and does Aβ have a function?
GWA studies have identified cholesterol metabolism, the innate immune system, and endosomal vesicle recycling as important pathogenic processes in AD: how do these relate to each other?
Acknowledgements
DS receives NIH grant support from the National Institute on Aging (AG06173 and AG015379). JH receives grant support for gene expression work from the MRC, for genetic work from ARUK and an Anonymous Foundation, and for work on AD in Down syndrome (LonDowns) from the Wellcome Trust.
EMBO Mol Med (2016) 8: 595–608
See the Glossary for abbreviations used in this article.
References
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X et al (2005) Neuronal sorting protein‐related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 102: 13461–13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews‐Zwilling Y, Bien‐Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, Huang Y (2010) Apolipoprotein E4 causes age‐ and Tau‐dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci 30: 13707–13717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayutyanont N, Langbaum JB, Hendrix SB, Chen K, Fleisher AS, Friesenhahn M, Ward M, Aguirre C, Acosta‐Baena N, Madrigal L et al (2014) The Alzheimer's prevention initiative composite cognitive test score: sample size estimates for the evaluation of preclinical Alzheimer's disease treatments in presenilin 1 E280A mutation carriers. J Clin Psychiatry 75: 652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Yan C, Yang G, Lu P, Ma D, Sun LD, Zhou R, Scheres SH, Shi Y (2015) An atomic structure of human gamma‐secretase. Nature 535: 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson‐Wood K et al (2000) Peripherally administered antibodies against amyloid beta‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919 [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM (2006) Human amyloid‐beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med 12: 856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM et al (2012) Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 367: 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, Gallardo R, Ungureanu AA, Castillo Cano V, Snellinx A, Ramakers M, Bartic C, Rousseau F, Schymkowitz J, De Strooper B (2014) The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid‐beta (Abeta) aggregation. J Biol Chem 289: 30977–30989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I et al (2008) Genome‐wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet 83: 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyreuther K, Masters CL (1991) Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer's disease: precursor‐product relationships in the derangement of neuronal function. Brain Pathol 1: 241–251 [DOI] [PubMed] [Google Scholar]
- Blennow K, Mattsson N, Scholl M, Hansson O, Zetterberg H (2015) Amyloid biomarkers in Alzheimer's disease. Trends Pharmacol Sci 36: 297–309 [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82: 239–259 [DOI] [PubMed] [Google Scholar]
- Brown P, Gibbs CJ, Rodgers‐Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC (1994) Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35: 513–529 [DOI] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, Demattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C et al (2011) Human apoE isoforms differentially regulate brain amyloid‐β peptide clearance. Sci Transl Med 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez‐Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H et al (2012) The mechanism of gamma‐Secretase dysfunction in familial Alzheimer disease. EMBO J 31: 2261–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A, Kolb HC (2013) Early clinical PET imaging results with the novel PHF‐tau radioligand [F‐18]‐T807. J Alzheimers Dis 34: 457–468 [DOI] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J et al (2014) A three‐dimensional human neural cell culture model of Alzheimer's disease. Nature 515: 274–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak‐Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921–923 [DOI] [PubMed] [Google Scholar]
- Coric V, van Dyck CH, Salloway S, Andreasen N, Brody M, Richter RW, Soininen H, Thein S, Shiovitz T, Pilcher G et al (2012) Safety and tolerability of the gamma‐secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol 69: 1430–1440 [DOI] [PubMed] [Google Scholar]
- Crehan J, Hardy J, Pocock J (2013) Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis 54: 139–149 [DOI] [PubMed] [Google Scholar]
- Cummings J, Cho W, Ward M, Friesenhahn M, Brunstein F, Honigberg L, Clayton D, Mortensen D, Ho C, Paul R (2014) paper presented at the Alzheimer's Association International Conference (AAIC), Copenhagen, Denmark, 16 July 2014
- De Strooper B (2014) Lessons from a failed gamma‐secretase Alzheimer trial. Cell 159: 721–726 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164: 603–615 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Gundula G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390 [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F et al (2012) A plaque‐specific antibody clears existing beta‐amyloid plaques in Alzheimer's disease mice. Neuron 76: 908–920 [DOI] [PubMed] [Google Scholar]
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG et al, Siemers E, Sethuraman G, Mohs R (2013) Semagacestat Study, a phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med 369, 341–350 [DOI] [PubMed] [Google Scholar]
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun Z, Aisen PS et al, (2014). Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N Engl J Med 370, 311–321 [DOI] [PubMed] [Google Scholar]
- Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, Brody DL (2013) Amyloid‐beta oligomerization in Alzheimer dementia versus high‐pathology controls. Ann Neurol 73: 104–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans KC, Berger EP, Cho CG, Weisgraber KH, Lansbury PT (1995) Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer disease. Proc Natl Acad Sci USA 92: 763–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MA, Klutkowski JA, Freret T, Wolfe MS (2014) Alzheimer presenilin‐1 mutations dramatically reduce trimming of long amyloid beta‐peptides (Abeta) by gamma‐secretase to increase 42‐to‐40‐residue Abeta. J Biol Chem 289: 31043–31052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, Levine AP, Hardy J, Pocock JM, Guerreiro R et al (2013) Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging 34: 2699–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F et al (1995) Alzheimer‐type neuropathology in transgenic mice overexpressing V717F b‐amyloid precursor protein. Nature 373: 523–527 [DOI] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F et al (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64: 1553–1562 [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW (1984) Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: 885–890 [DOI] [PubMed] [Google Scholar]
- Griciuc A, Serrano‐Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE (2013) Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78: 631–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S et al (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Schlossmacher M, Hung AY, Vigo‐Pelfrey C, Mellon A, Ostaszewski B, Lieberburg I, Koo EH, Schenk D, Teplow D et al (1992) Amyloid beta‐peptide is produced by cultured cells during normal metabolism. Nature 359: 322–325 [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends in Pharmac 12: 383–388 [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356 [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins G (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256: 184–185 [DOI] [PubMed] [Google Scholar]
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ (2009) Identification of beta‐secretase (BACE1) substrates using quantitative proteomics. PLoS ONE 4: e8477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo M, Abraham R, Hamshere ML, Pahwa JS, Moskvina V et al (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 43: 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius L, Mackey B, Olney J, McKeel D, Wozniak D et al (2000) Apolipoprotein E isoform‐dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 97: 2892–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Ostaszewski BL, Yang T, O'Malley TT, Jin M, Yanagisawa K, Li S, Bartels T, Selkoe DJ (2014) Soluble Abeta oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 82: 308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Ekman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aß elevation, and amyloid plaques in transgenic mice. Science 274: 99–102 [DOI] [PubMed] [Google Scholar]
- Huang Y, Mahley RW (2014) Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis 72 (Pt A), 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, Trojanowski JQ (2013) Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver‐derived human growth hormone. JAMA Neurol 70: 462–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Holtzman DM (2013) Biomarker modeling of Alzheimer's disease. Neuron 80: 1347–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD et al (2013) Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12: 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT (1993) Seeding “one‐dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73: 1055–1058 [DOI] [PubMed] [Google Scholar]
- Jaunmuktane Z, Mead S, Ellis M, Wadsworth JDF, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard‐Loendt A, Walker AS et al (2015) Evidence for human transmission of amyloid beta‐pathology and cerebral amyloid angiopathy. Nature 525: 247–250 [DOI] [PubMed] [Google Scholar]
- Jiang T, Tan L, Zhu XC, Zhang QQ, Cao L, Tan MS, Gu LZ, Wang HF, Ding ZZ, Zhang YD et al (2014) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer's disease. Neuropsychopharmacology 39: 2949–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ (2011) Soluble amyloid beta‐protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA 108: 5819–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, Pocklington A, Abraham R, Hollingworth P, Sims R et al (2010) Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS ONE 5: e13950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J et al (2012) A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature 488: 96–99 [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ et al (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsu K, Morohashi M, Suzuki Y, Kuroda H, Watanabe T, Tomita T, Iwatsubo T (2014) Decreased CALM expression reduces Abeta42 to total Abeta ratio through clathrin‐mediated endocytosis of gamma‐secretase. Nat Commun 5: 3386 [DOI] [PubMed] [Google Scholar]
- Karran E, Hardy J (2014) A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol 76: 185–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kero M, Paetau A, Polvikoski T, Tanskanen M, Sulkava R, Jansson L, Myllykangas L, Tienari PJ (2013) Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiol Aging 34(1518): e1511–e1513 [DOI] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E (2007) Abeta40 inhibits amyloid deposition in vivo. J Neurosci 27: 627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WS, Li H, Ruberu Chan KS, Elliott DA, Low JK, Cheng D, Karl T, Garner B (2013) Deletion of Abca7 increases cerebral amyloid‐beta accumulation in the J20 mouse model of Alzheimer's disease. J Neurosci 33: 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suarez‐Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger‐Weinzier A, Mazaheri F et al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6, 243ra286 [DOI] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S et al (2004) Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol 55: 306–319 [DOI] [PubMed] [Google Scholar]
- Koffie RM, Meyer‐Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia‐Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM et al (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA 106: 4012–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretner B, Trambauer J, Fukumori A, Mielke J, Kuhn PH, Kremmer E, Giese A, Lichtenthaler SF, Haass C, Arzberger T et al (2016). Generation and deposition of Aß43 by the virtually inactive presenilin‐1 L435F mutant contradicts the presenilin loss of function hypothesis of Alzheimer's disease. EMBO Mol Med doi:10.15252/emmm.201505952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, Volbracht C, Schepers U, Imhof A, Hoffmeister A et al (2012) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J 31: 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B et al (2009) Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41: 1094–1099 [DOI] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim‐Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStefano AL, Bis JC, Beecham GW, Grenier‐Boley B et al (2013) Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45: 1452–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Blustzjan JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ (1996a) Sequence of deposition of heterogeneous amyloid b‐peptides and Apo E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis 3: 16–32 [DOI] [PubMed] [Google Scholar]
- Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Martinez A, Madrigal L et al (1996b) The E280A presenilin 1 Alzheimer mutation produces increased Ab42 deposition and severe cerebellar pathology. Nature Med 2: 1146–1150 [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn‐Hardy K, Paul Murphy M, Baker M, Yu X et al (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: 402–405 [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D et al (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293: 1487–1491 [DOI] [PubMed] [Google Scholar]
- Liang SH, Yokell DL, Normandin MD, Rice PA, Jackson RN, Shoup TM, Brady TJ, El Fakhri G, Collier TL, Vasdev N (2014) First human use of a radiopharmaceutical prepared by continuous‐flow microfluidic radiofluorination: proof of concept with the tau imaging agent [18F]T807. Mol Imaging 13: doi:10.2310/7290.2014.00025 [DOI] [PubMed] [Google Scholar]
- Lue LF, Schmitz C, Walker DG (2015) What happens to microglial TREM2 in Alzheimer's disease: immunoregulatory turned into immunopathogenic? Neuroscience 302: 138–150 [DOI] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer HB, Das S, Potter H (1994) The amyloid‐associated proteins a1‐antichymotrypsin and apolipoprotein E promote the assembly of the Alzheimer b‐protein into filaments. Nature 372: 92–94 [DOI] [PubMed] [Google Scholar]
- Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, van der Brug M, Liu Y, Ernst JA, Watts RJ et al (2014) Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem 289: 30990–31000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DMA, Yuonis N, Jones D, Stoddart DW (1992) The time course of pathological events in Down's Syndrome with particular reference to the involvement of microglial cells and deposits of b/A4. Neurodegeneration 1: 201–215 [Google Scholar]
- Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang MR, Trojanowski JQ, Lee VM, Ono M et al (2013) Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 79: 1094–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, Nahaboo Solim MA, Moens TG, Paublete RM, Ali SS et al (2015) A genome‐wide gene‐expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep 10: 633–644 [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta‐amyloid in Alzheimer's disease. Science 330: 1774–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Akiyama H, Itagaki S, McGreer EG (1989) Immune system response in Alzheimer's disease. Can J Neurol Sci 16: 516–627 [DOI] [PubMed] [Google Scholar]
- Moore S, Evans LD, Andersson T, Portelius E, Smith J, Dias TB, Saurat N, McGlade A, Kirwan P, Blennow K et al (2015) APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep 11: 689–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Takio K, Ogawara M, Selkoe DJ (1992) Mass spectrometry of purified amyloid b protein in Alzheimer's disease. J Biol Chem 267: 17082–17086 [PubMed] [Google Scholar]
- Muratore MR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LN, Walsh DM, Selkoe DJ, Young‐Pearse TL (2014) The familial Alzheimer's disease APPV717I mutation alters APP processing and tau expression in iPSC‐derived neurons. Hum Mol Genet 23: 3523–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Ronicke R et al (2012) Prion‐like behaviour and tau‐dependent cytotoxicity of pyroglutamylated amyloid‐beta. Nature 485: 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, Takeda M (2013) Gamma‐secretase modulators and presenilin 1 mutants act differently on presenilin/gamma‐secretase function to cleave Abeta42 and Abeta43. Cell Rep 3: 42–51 [DOI] [PubMed] [Google Scholar]
- Potter R, Patterson BW, Elbert DL, Ovod V, Kasten T, Sigurdson W, Mawuenyega K, Blazey T, Goate A, Chott R et al (2013). Increased in vivo amyloid‐beta42 production, exchange, and loss in presenilin mutation carriers. Sci Transl Med 5, 189ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC, Butler AC (1998) Molecular mapping of Alzheimer‐type dementia in Down's syndrome. Ann Neurol 43: 380–383 [DOI] [PubMed] [Google Scholar]
- Qi‐Takahara Y, Morishima‐Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R et al (2005) Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma‐secretase. J Neurosci 25: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta ‐amyloid‐induced neurotoxicity. Proc Natl Acad Sci USA 99: 6364–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck GW, Reiter JS, Strickland DK, Hyman BT (1993) Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron 11: 575–580 [DOI] [PubMed] [Google Scholar]
- Refolo LM, Malester B, LaFrancois J, Bryant‐Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA (2000) Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis 7: 321–331 [DOI] [PubMed] [Google Scholar]
- Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas‐Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS et al (2001) A cholesterol‐lowering drug reduces beta‐amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis 8: 890–899 [DOI] [PubMed] [Google Scholar]
- Ridley RM, Baker HF, Windle CP, Cummings RM (2006) Very long term studies of the seeding of beta‐amyloidosis in primates. J Neural Transm 113: 1243–1251 [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce‐Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2011) Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science 316: 750–754 [DOI] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H et al (2007) The neuronal sortilin‐related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 39: 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovelet‐Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M et al (2006) APP locus duplication causes autosomal dominant early‐onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38: 24–26 [DOI] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC (2014) Single App knock‐in mouse models of Alzheimer's disease. Nat Neurosci 17: 661–663 [DOI] [PubMed] [Google Scholar]
- Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, Matsuba Y, Yamada K, Nilsson P, Takano J et al (2011) Potent amyloidogenicity and pathogenicity of Abeta43. Nat Neurosci 14: 1023–1032 [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson‐Wood K, Khan K et al (1999) Immunization with amyloid‐ß attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature 400: 173–177 [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W et al (1996) Secreted amyloid b‐protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nature Med 2: 864–870 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (1991) The molecular pathology of Alzheimer's disease. Neuron 6: 487–498 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2011) Resolving controversies on the path to Alzheimer's therapeutics. Nat Med 17: 1060–1065 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Mandelkow E, Holtzman DM (2012) The Biology of Alzheimer Disease. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano‐Pozo A, William CM, Ferrer I, Uro‐Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH et al (2010) Beneficial effect of human anti‐amyloid‐beta active immunization on neurite morphology and tau pathology. Brain 133, 1312–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Williams L, Miao X, O'Gorman J (2015) Randomized, double‐blind Phase 1B study of BIIB037, an anti‐amyloid beta monoclonal antibody, in patients with prodromal or mild Alzheimer's disease. Neurogener Dis 15(Suppl 1): 311 [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia‐Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA et al (2008) Amyloid‐beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14: 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ (2007) The presenilin hypothesis of Alzheimer's disease: evidence for a loss‐of‐function pathogenic mechanism. Proc Natl Acad Sci USA 104: 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ (2012) A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci Transl Med 4: 124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Duff K (2008) Linking Abeta and tau in late‐onset Alzheimer's disease: a dual pathway hypothesis. Neuron 60: 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, Aisen P (2014) The A4 study: stopping AD before symptoms begin? Sci Transl Med 6, 228fs213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U et al (2015) Loss‐of‐function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet 47: 445–447 [DOI] [PubMed] [Google Scholar]
- Suárez‐Calvet M, Kleinberger G, Araque Caballero MÁ, Brendel M, Rominger A, Alcolea D, Fortea J, Lleó A, Blesa R, Gispert JD et al (2016) sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early‐stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med 2016 doi: 10.15252/emmm.201506123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda‐Falla D, Lleo A, Ryan NS, Lashley T, Fox NC, Murayama S et al (2015) Qualitative changes in human gamma‐secretase underlie familial Alzheimer's disease. J Exp Med 212: 2003–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima‐Kawashima M, Funamoto S, Ihara Y (2009) gamma‐Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta‐carboxyl terminal fragment. J Neurosci 29: 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM (2014) Altered microglial response to Abeta plaques in APPPS1‐21 mice heterozygous for TREM2. Mol Neurodegener 9: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigo‐Pelfrey C, Seubert P, Barbour R, Blomquist C, Lee M, Lee D, Coria F, Chang L, Miller B, Lieberburg I et al (1995) Elevation of microtubule‐associated protein tau in the cerebrospinal fluid of patients with Alzheimer's disease. Neurology 45: 788–793 [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Szoeke C, Macaulay SL, Martins R, Maruff P et al (2013) Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol 12: 357–367 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson KJ, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH et al (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160: 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, Klyubin I, Rowan MJ, Seubert P, Walsh DM et al (2014) Secreted amyloid beta‐proteins in a cell culture model include N‐terminally extended peptides that impair synaptic plasticity. Biochemistry 53: 3908–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, De Strooper B, Saftig P, Birchmeier C, Haass C (2006) Control of peripheral nerve myelination by the beta‐secretase BACE1. Science 314: 664–666 [DOI] [PubMed] [Google Scholar]
- Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S, Hornburg D, Evans LD, Moore S, Daria A et al (2015) η‐Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526: 433–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Castano AM, Golabek A, Vogel T, Frangione B (1994) Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol 145: 1030–1035 [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999) Two transmembrane aspartates in presenilin‐1 required for presenilin endoproteolysis and g‐secretase activity. Nature 398: 513–517 [DOI] [PubMed] [Google Scholar]
- Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J (2013) Sequential proteolytic processing of the triggering receptor expressed on myeloid cells‐2 (TREM2) protein by ectodomain shedding and gamma‐secretase‐dependent intramembranous cleavage. J Biol Chem 288: 33027–33036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ (2015) Presenilin‐1 knockin mice reveal loss‐of‐function mechanism for familial Alzheimer's disease. Neuron 85: 967–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JE, Boulanger‐Weill J, Williams DA, Woodruff G, Buen F, Revilla AC, Herrera C, Israel MA, Yuan SH, Edland SD et al (2015) Elucidating molecular phenotypes caused by the SORL1 Alzheimer's disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 16: 373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R et al (2013) Integrated systems approach identifies genetic nodes and networks in late‐onset Alzheimer's disease. Cell 153: 707–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G et al (2015) Central role for PICALM in amyloid‐beta blood–brain barrier transcytosis and clearance. Nat Neurosci 18: 978–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Liu D, Roychaudhuri R, Teplow DB, Bowers MT (2015) Amyloid beta‐protein assembly: differential effects of the protective A2T mutation and recessive A2V familial Alzheimer's disease mutation. ACS Chem Neurosci 6: 1732–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]