

Hepatic inflammation is a pathogenic process associated with various types of acute and chronic liver disorders, and it contributes to progressive liver injury and fibrosis. Among the various innate immune cells, hepatic macrophages are the major (80-90%) resident macrophages of the body and are central in regulating the pathogenesis of acute and chronic liver injury. While macrophage-mediated inflammatory responses can be hepatoprotective by promoting resolution and repair of tissue injury, excessive macrophage activation is detrimental by exacerbating liver injury of various conditions including alcoholic and non-alcoholic fatty liver disease, ischemia-reperfusion injury as well as insults of xenobiotics and infection [1].
Autophagy is a catabolic lysosomal degradation process that degrades cytoplasm materials including misfolded proteins and dysfunctional organelles as a cellular adaptation and survival mechanism in response to a variety of stress conditions. In the liver, hepatocyte basal autophagy is critical to maintain liver homeostasis for proteins, lipids and organelles and loss of basal hepatocyte autophagy leads to liver injury, inflammation, fibrosis and tumorigenesis [2,3]. In addition, hepatocyte autophagy has also been shown to protect against alcohol, ischemia-reperfusion, drug- and endotoxin-induced liver injury [3-6]. Compared to hepatocyte autophagy, the role of macrophage autophagy in the liver pathogenesis is less known but emerging evidence suggests that macrophage autophagy attenuates endotoxin-induced acute liver injury, steatohepatitis and fibrosis [7,8].
In this issue of the Journal of Hepatology, Ilyas et al [9] demonstrated that macrophage autophagy inhibited inflammasome-mediated IL-1β generation and secretion to limit acute toxin-induced liver injury. To study the specific role of autophagy in macrophages, Ilyas et al generated a macrophage-specific knockout of the autophagy gene Atg5 by crossing the Atg5 floxed mice with LysM-Cre mice expressing a myeloid cell-specific Cre [9]. To trigger acute liver injury in mice, Ilyas et al [9] injected mice with lipopolysaccharide (LPS) and D-galactosamine (GalN), which is a well-established animal model to induce acute liver injury mediated by macrophage-derived tumor necrosis factor-α (TNF-α). TNF-α induces hepatocyte apoptosis and liver injury through both the extrinsic and intrinsic apoptotic pathways [10] and promotes hepatic neutrophil recruitment and activation, which amplifies the apoptotic injury [11]. The authors found that macrophage-specific Atg5 knockout mice had increased liver injury following administration of LPS/GalN as demonstrated by increased serum alanine aminotransferase activities, hepatic caspase activation, TUNEL positive apoptotic liver cells as well as mortality. Intriguingly, they observed that both wild type and macrophage-specific Atg5 knockout mice had a similarly increased number of hepatic macrophages and elevated serum levels of TNF-α after LPS/GalN treatment. These results suggest that autophagy in macrophage is not essential for regulating TNF-α generation. Since TNF-α is the major player in LPS/GalN-induced hepatocyte death, it suggests that other factors could exacerbate TNF-α-mediated hepatocyte apoptosis and liver injury. Indeed, they found that macrophage-specific Atg5 knockout mice had elevated serum IL-1β, which was due to activation of the Nalp3 inflammasome and caspase-1 resulting in enhanced cleavage of pro-IL-1β in Atg5-deficient macrophages. Elevated serum IL-1β serves as a key amplification factor to exacerbate LPS/GalN-induced liver injury since the elevation of serum IL-1β occurred as early as 2 hours prior to the onset of liver injury, and blocking IL-1β signaling by IL-1 receptor antagonist (IL-1Ra) reduced LPS/GalN-induced liver injury. Increased IL-1β generation seems to be a general event in autophagy-deficient macrophages since elevated serum IL-1β levels have also been observed in CCl4-induced fibrosis in macrophage-specific Atg5 knockout mice [8], and in dextran sulphate sodium-induced colitis in Atg16L-deficient mice [12]. Similar to the LPS/GalN model, an enhanced IL-1-driven inflammatory response aggravated CCl4 –induced liver injury in macrophage-specific Atg5 knockout mice [8]. While Ilyas et al [9] did not determine how loss of autophagy activates the Nalp3 inflammasome and caspase 1 in macrophages in response to LPS/GalN, it has been shown that toll-like receptor adaptor protein TRIF, macrophage K+ efflux and reactive oxygen species (ROS) production but not NF-kB and p38 are required for caspase 1 activation and IL-1β generation in Atg16L-deficient macrophages [12]. It remains to be determined whether TRIF, K+ efflux and ROS would also contribute to caspase-1 activation in LPS/GalN-treated macrophage-specific Atg5 knockout mice. It is well known that one of the major sources for intracellular ROS production is mitochondria. Damaged mitochondria can be selectively removed via mitophagy to reduce ROS production [13]. It will be interesting to determine whether there is any defective mitophagy and increased ROS generation in Atg5-deficient macrophages after LPS treatment in the future.
Although the authors have provided convincing evidence for the role of IL-1β in the enhancement of liver injury in the macrophage-specific Atg5 knockout mice, the mechanism of this aggravated cell injury is less clear. Using IL-1Ra, the authors showed that blocking IL-1 activity reduced IL-1 target gene expression (NOS2, COX2), but not TNF-α, and did not affect hepatic neutrophil recruitment. Although both TNF-α and IL-1 are capable of activating and recruiting neutrophils into the liver [14], in the LPS/GalN model there is substantially more TNF-α produced, which make this cytokine the dominant mediator for this effect. However, using the conditional medium from cultured macrophages isolated from macrophage-specific Atg5 knockout mice and IL-1Ra-treated animals, Ilyas et al further demonstrated that the activation status of neutrophils (CXC chemokine formation) from gene knockout mice appears to be higher, an effect that is dependent on IL-1β [9]. These observations are consistent with previous findings that neutrophil cytotoxicity in this model is dependent on neutrophil extravasation [11,15] and their cytotoxic potential [16]. The main chemotactic event for neutrophil extravasation is apoptotic cell death of hepatocytes [11] not CXC chemokine formation [17]. This raises the question whether IL-1β also enhanced the priming for ROS formation, a critical mechanism for neutrophil cytotoxicity [16]. Since macrophage-specific Atg5 knockout mice also show enhanced apoptotic cell death, part of the aggravated inflammatory injury could also come from more neutrophil extravasation. Thus, the effect of IL-1β could be caused by a dual impact on neutrophils including the direct enhancement of the cytotoxic capacity of individual neutrophils and by facilitating neutrophil transmigration into the parenchyma by promoting apoptotic cell death. However, the mechanism by which IL-1β modulates the signaling pathway of TNF-mediated hepatocyte apoptosis remains to be investigated.
Macrophages are notable for their plasticity and polarization. Depending on the activating stimuli, macrophages can be polarized into pro-inflammatory M1 and anti-inflammatory M2 macrophages. M1 macrophages release pro-inflammatory cytokines such as TNFα, IL-1β and IL-2, and ROS whereas M2 macrophages secrete IL-10, TGF-β, IL-14 and IL-13 to mediate resolution of inflammation. While Ilyas et al did not find significant changes of macrophage polarization in the macrophage-specific Atg5 knockout mice after LPS/GalN, the same group previously reported that macrophages polarized into a more pro-inflammatory M1 phenotype when macrophage-specific Atg5 knockout mice were challenged with a high fat diet (HFD) together with a low dose of LPS [7]. In steatotic livers, HFD and LPS did not activate the Nalp3 inflammasome and caspase-1, and IL-1β levels were not different in wild type and macrophage-specific Atg5 knockout mice [7]. These observations are distinctly different from LPS/GalN despite that LPS is the major driving force for the inflammatory response in both models (Figure 1). While the mechanisms behind these differences are yet to be determined, it is likely that increased free fatty acids and neutral lipids such as triglyceride in steatotic livers may alter the response of autophagy-deficient macrophages to LPS. Despite these differences in response to HFD/LPS and LPS/GalN in macrophages, loss of autophagy in macrophages exacerbated liver injury in both models. These findings may have significant implications for considering macrophage autophagy as a potential novel therapeutic target for treating liver diseases. Pharmacological activation of autophagy in hepatocytes has already been shown to be beneficial for a number of liver diseases including alcohol and non-alcoholic liver disease as well as drug-induced liver injury [4,6,18]. However, it should be noted that activation of autophagy in hepatic stellate cells promotes liver fibrosis by increasing lipid droplet degradation via lipophagy [19]. Therefore, pharmacological systemic activation of autophagy may be a promising approach in treating acute liver injury by not only increasing hepatocellular defense mechanisms against cell death but also limiting inflammation via activating autophagy in macrophages. However, the efficacy of this therapeutic approach may be limited when treating chronic liver diseases with ongoing fibrosis [20].
Figure 1. Different inflammatory responses to LPS/GalN-, HFD/LPS- and CCl4-induced liver injury in mice with impaired macrophage autophagy.
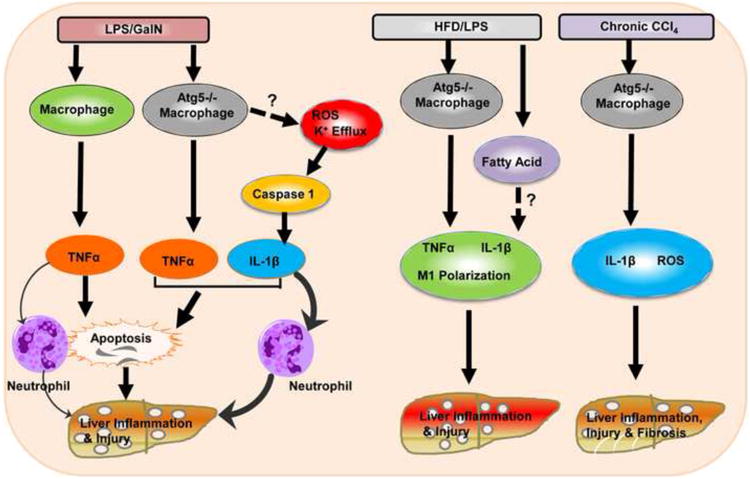
In wild type mice, LPS/GalN activates macrophages to generate TNF-α which induces hepatocyte apoptosis and neutrophil infiltration to the liver resulting in liver injury. In macrophage-specific Atg5 knockout mice, in addition to TNF-α production, LPS/GalN also increased caspase-1-mediated IL-1β production likely via increased ROS and K+ efflux due to the lack of autophagy in macrophages. IL-1β amplifies TNF-α-induced death signals, enhances LPS/GalN-induced apoptosis and induces more neutrophil activation resulting in exacerbated liver injury. In HFD/LPS-challenged macrophage-specific Atg5 knockout mice, in addition to the increased production of TNF-α and IL-1β, autophagy-deficient macrophages also undergo polarization to become more M1 macrophages likely due to the accumulated fatty acids and neutral lipids in the steatotic environment. Macrophage-specific Atg5 knockout mice also have increased IL-1β and ROS production and liver fibrosis after CCl4 administration “?” and dotted-line arrows indicate molecular events that were not investigated in this study.
Acknowledgments
The authors acknowledge funding support from the National Institutes of Health grants R01 AA020518 (W.X.D.), R01 DK102142 (W.X.D. and H.J.) and P20 GM103549-07 (H.J.).
Abbreviations
- GalN
D-galactosamine
- IL-1β
interleukin-1β
- LPS
lipopolysaccharide
- Nalp3
NACHT, LRR and PYD domains-containing protein 3
- ROS
reactive oxygen species
- TNF-α
tumor necrosis factor-α
Footnotes
Conflict of Interest: The authors report no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 2.Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP, et al. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61:617–625. doi: 10.1016/j.jhep.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Czaja MJ, Ding WX, Donohue TM, Jr, Friedman SL, Kim JS, Komatsu M, et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amir M, Zhao E, Fontana L, Rosenberg H, Tanaka K, Gao G, et al. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 2013;20:878–887. doi: 10.1038/cdd.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lodder J, Denaes T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11:1280–1292. doi: 10.1080/15548627.2015.1058473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ilyas G, Zhao E, Liu K, Lin Y, Tesfa L, Tanaka KE, et al. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1 beta. J Hepatol. 2015 doi: 10.1016/j.jhep.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin XM, Ding WX. Death receptor activation-induced hepatocyte apoptosis and liver injury. Curr Mol Med. 2003;3:491–508. doi: 10.2174/1566524033479555. [DOI] [PubMed] [Google Scholar]
- 11.Jaeschke H, Fisher MA, Lawson JA, Simmons CA, Farhood A, Jones DA. Activation of caspase 3 (CPP32)-like proteases is essential for TNF-alpha-induced hepatic parenchymal cell apoptosis and neutrophil-mediated necrosis in a murine endotoxin shock model. J Immunol. 1998;160:3480–3486. [PubMed] [Google Scholar]
- 12.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 13.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajt ML, Farhood A, Jaeschke H. Effects of CXC chemokines on neutrophil activation and sequestration in hepatic vasculature. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1188–1195. doi: 10.1152/ajpgi.2001.281.5.G1188. [DOI] [PubMed] [Google Scholar]
- 15.Essani NA, Fisher MA, Farhood A, Manning AM, Smith CW, Jaeschke H. Cytokine-induced upregulation of hepatic intercellular adhesion molecule-1 messenger RNA expression and its role in the pathophysiology of murine endotoxin shock and acute liver failure. Hepatology. 1995;21:1632–1639. [PubMed] [Google Scholar]
- 16.Gujral JS, Hinson JA, Farhood A, Jaeschke H. NADPH oxidase-derived oxidant stress is critical for neutrophil cytotoxicity during endotoxemia. Am J Physiol Gastrointest Liver Physiol. 2004;287:G243–252. doi: 10.1152/ajpgi.00287.2003. [DOI] [PubMed] [Google Scholar]
- 17.Dorman RB, Gujral JS, Bajt ML, Farhood A, Jaeschke H. Generation and functional significance of CXC chemokines for neutrophil-induced liver injury during endotoxemia. Am J Physiol Gastrointest Liver Physiol. 2005;288:G880–886. doi: 10.1152/ajpgi.00317.2004. [DOI] [PubMed] [Google Scholar]
- 18.Lin CW, Zhang H, Li M, Xiong X, Chen X, Dong XC, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–999. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mallat A, Lodder J, Teixeira-Clerc F, Moreau R, Codogno P, Lotersztajn S. Autophagy: a multifaceted partner in liver fibrosis. Biomed Res Int. 2014;2014:869390. doi: 10.1155/2014/869390. [DOI] [PMC free article] [PubMed] [Google Scholar]