

Increasing antibiotic resistance in bacteria, particularly Gram-negative bacilli, has significantly affected the ability of physicians to treat infections, with resultant increased morbidity, mortality, and health care costs. In fact, some strains of bacteria are resistant to all available antibiotics, such as Acinetobacter baumannii, which is the focus of this report. Therefore, the development of new antibiotics active against these resistant strains is urgently needed. In this study, BfmR is further validated as an intriguing target for a novel class of antibiotics. Successful inactivation of BfmR would confer the multiple benefits of a decreased ability of A. baumannii to survive in human body fluids, increased sensitivity to complement-mediated bactericidal activity and, importantly, increased sensitivity to other antibiotics. Structural studies support the potential for this “druggable” target, as they identify the potential for small-molecule binding at functionally relevant sites. Next-phase high-throughput screening studies utilizing BfmR are warranted.
KEYWORDS: Acinetobacter baumannii, Gram-negative bacilli, antibiotic target, bacterial drug target, drug discovery, essential genes, multidrug resistant
ABSTRACT
Identification and validation is the first phase of target-based antimicrobial development. BfmR (RstA), a response regulator in a two-component signal transduction system (TCS) in Acinetobacter baumannii, is an intriguing potential antimicrobial target. A unique characteristic of BfmR is that its inhibition would have the dual benefit of significantly decreasing in vivo survival and increasing sensitivity to selected antimicrobials. Studies on the clinically relevant strain AB307-0294 have shown BfmR to be essential in vivo. Here, we demonstrate that this phenotype in strains AB307-0294 and AB908 is mediated, in part, by enabling growth in human ascites fluid and serum. Further, BfmR conferred resistance to complement-mediated bactericidal activity that was independent of capsular polysaccharide. Importantly, BfmR also increased resistance to the clinically important antimicrobials meropenem and colistin. BfmR was highly conserved among A. baumannii strains. The crystal structure of the receiver domain of BfmR was determined, lending insight into putative ligand binding sites. This enabled an in silico ligand binding analysis and a blind docking strategy to assess use as a potential druggable target. Predicted binding hot spots exist at the homodimer interface and the phosphorylation site. These data support pursuing the next step in the development process, which includes determining the degree of inhibition needed to impact growth/survival and the development a BfmR activity assay amenable to high-throughput screening for the identification of inhibitors. Such agents would represent a new class of antimicrobials active against A. baumannii which could be active against other Gram-negative bacilli that possess a TCS with shared homology.
IMPORTANCE Increasing antibiotic resistance in bacteria, particularly Gram-negative bacilli, has significantly affected the ability of physicians to treat infections, with resultant increased morbidity, mortality, and health care costs. In fact, some strains of bacteria are resistant to all available antibiotics, such as Acinetobacter baumannii, which is the focus of this report. Therefore, the development of new antibiotics active against these resistant strains is urgently needed. In this study, BfmR is further validated as an intriguing target for a novel class of antibiotics. Successful inactivation of BfmR would confer the multiple benefits of a decreased ability of A. baumannii to survive in human body fluids, increased sensitivity to complement-mediated bactericidal activity and, importantly, increased sensitivity to other antibiotics. Structural studies support the potential for this “druggable” target, as they identify the potential for small-molecule binding at functionally relevant sites. Next-phase high-throughput screening studies utilizing BfmR are warranted.
INTRODUCTION
The incidence of infections due to multidrug-resistant, extensively resistant, and pan-drug-resistant (MDR, XDR, and PDR) Gram-negative bacilli (GNB) is increasing, with Acinetobacter baumannii one of the most problematic species (1–8). Treatment of infections due to these strains has become challenging, with resultant increased morbidity, mortality, and health care costs (9–11). The promise of a postantibiotic era is on the cusp of being fulfilled for A. baumannii (1–8, 12, 13), and a true PDR strain has been reported (14). This reality has been appreciated for a number of years, with the 2004 report by the Infectious Diseases Society of America, Bad Bugs, No Drugs: As Antibiotic R&D Stagnates, a Public Health Crisis Brews, being one of several attempts to increase exposure and enable solutions (15–20). However, development of new antimicrobials is a long, arduous, and expensive process. As a result, many major pharmaceutical companies left the antimicrobial research and development (R&D) business (19, 21–23). The burden of early-stage development has fallen in part to academia and to “start-up” companies. The term ESKAPE pathogens was coined to advertise the major culprits, with Klebsiella pneumoniae, A. baumannii, Pseudomonas aeruginosa, and Enterobacter spp. being the initial GNB implicated. A. baumannii, with its propensity of being XDR and PDR, has been termed the “poster child” for this ever-increasing threat to health care. The need to identify new antimicrobials active against A. baumannii is more pressing than ever. Therefore, as a first step, our group identified A. baumannii essential genes in vivo (i.e., those required for bacterial growth and survival in a host) which represent potential novel drug targets (24), with the long-term goal to develop a new class of antimicrobials. In support of such efforts, new regulatory paths may make the development of focused treatments for XDR pathogens, such as A. baumannii, commercially viable (25).
One of the potential targets identified as essential in vivo was BfmR (24). BfmR is the response regulator of a two-component signal transduction system (TCS) which propagates the signal from its corresponding sensor histidine kinase BfmS and acts as a transcriptional regulator (26). The environmental stimuli that the BfmR/BfmS (also referred to RstAB) system responds to are unknown, other than a direct or indirect effect of sub-MICs of chloramphenicol in the strain ATCC 17978 (27). Such systems have been considered potential drug targets (28), and the histidine kinase QseC has been successfully targeted by using the small organic molecule LED209 (29). Although LED209 inhibited virulence in vivo, it did not inhibit pathogen growth. Since inactivation of BfmR resulted in loss of viability and was essential in vivo (24) and important for in vivo pathogenicity in a murine pneumonia model (30), its successful targeting may result in even greater efficacy than that observed for antimicrobials targeting QseC. Phenotypes conferred by BfmR include increased resistance to complement-mediated bactericidal activity, biofilm formation, and scaffolding of the outer membrane protein OmpA, as well as increased resistance to selected antimicrobials (e.g., carbapenems) (27, 31, 32). In this report, we extend our initial biological observations on BfmR and add structural insights focused on its receiver domain (residues 1 to 130; BfmR1‑130) that further support this protein as a potential drug target in A. baumannii.
RESULTS
Disruption of BfmR synthesis affects growth/survival in human ascites fluid and serum ex vivo, but not in rich laboratory medium.
A critical criterion for a potential drug target is essentiality. We have previously published data that demonstrated BfmR as being essential in vivo in a rat infection model (24). To extend this observation, we performed quantitative growth/survival studies in human serum and ascites fluid samples. AB307.70 (ΔbfmR) underwent a 2-log complement-mediated decrease in growth/survival over 24 h in serum (Fig. 1A). This loss in growth/survival was restored to wild-type (wt) levels when AB307.70 was complemented in trans (AB307.70/pNLAC1[bfmR]). Surprisingly, when AB307.70 was grown in serum in which complement activity had been inactivated (Δ56°C), growth was not restored to wild-type levels, but instead an approximate 1-log loss in growth/survival was observed (Fig. 1A). Importantly, the decrease in growth/survival of AB307.70 was significantly greater in untreated serum than in serum in which complement was inactivated. Likewise, AB307.70 underwent a greater-than-3-log decrease in growth/survival when grown in human ascites fluid, and this loss in growth/survival was again complemented in trans (AB307.70/pNLAC1[bfmR]) (Fig. 1B). These data further support an essential role for BfmR for optimal growth/survival of AB307-0294 in these clinically relevant environments. In contrast and, importantly, AB307.70 is capable of growth in rich laboratory medium, with a slight decrease in log-phase growth but a similar plateau for cell density as with AB307-0294, thereby excluding a generalized growth defect (Fig. 1C). These data demonstrate that BfmR mediates (directly or indirectly) resistance to complement-mediated bactericidal activity. These data also demonstrate that BrmR regulates bacterial factors needed for optimal growth, presumably via nutrient acquisition, since decreased growth of AB307.70 was observed in serum compared to growth of its wild-type parent when complement was inactivated.
FIG 1 .
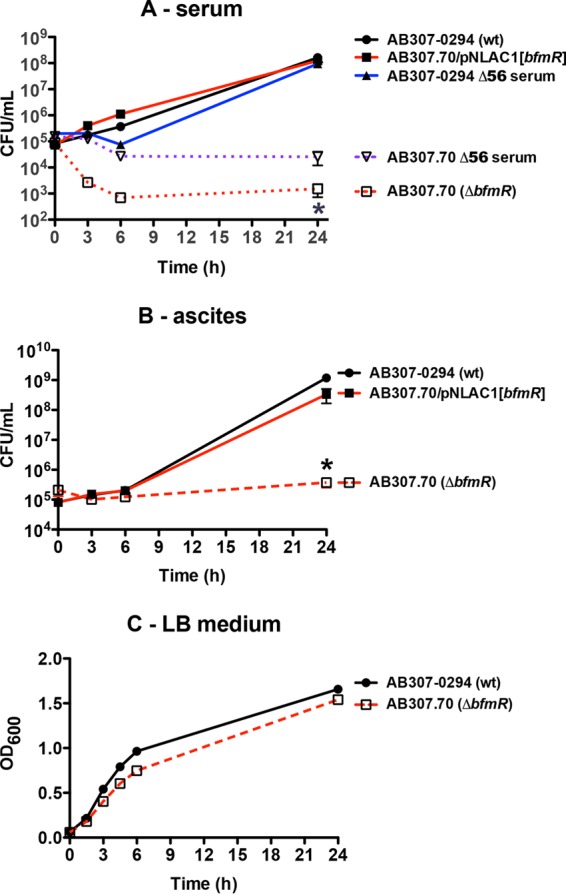
Comparison of the growth/survival of AB307-0294 (wt) and AB307.70 (ΔbfmR) cells in 90% human serum, 100% human ascites fluid, and rich laboratory medium. (A) Growth/survival in serum was assessed by measurement of CFU at 0, 3, 6, and 24 h. AB307-0294 and AB307.70 were also grown in serum that was heated to 56°C for 30 min, which inactivated complement-mediated bactericidal activity (Δ56 serum). AB307.70/pNLAC1[bfmR], a BfmR-complemented derivative of AB307.70, was also tested to confirm that the observed phenotype was BfmR mediated. Data are means ± SEM (n = 6). (B) Growth/survival in ascites fluid was assessed by measurement of CFU at 0, 3, 6, and 24 h. Data are means ± SEM (n = 6 to 8). (C) Growth in LB medium, as measured by the OD600 of four replicate cultures per strain. Data are means ± SEM (n = 4). *, P < 0.05/2 (two-tailed unpaired t test for AB307-0294 compared to AB307.70 in untreated serum and ascites fluid).
Disruption of BfmR synthesis has no effect on capsule production.
Previously published work by our group demonstrated that in A. baumannii the capsular polysaccharide was a major factor contributing to resistance of complement-mediated bactericidal activity (33). Therefore, as a first step in examining the mechanism by which BfmR confers serum resistance, the production of the K1 capsular polysaccharide in AB307-0294 (wt) and AB307.70 (ΔbfmR) was assessed. Both strains were grown in LB medium at either 21°C (room temperature) or 37°C, and cell lysates or supernatant fractions underwent Western blot analysis using the monoclonal antibody (MAb) 13D6, which has been shown to recognize the K1 capsule present on AB307-0294 (33). Surprisingly, capsule was produced in roughly equal amounts by both AB307-0294 and AB307.70 (Fig. 2A). Further, the majority of capsular polysaccharide was cell associated. Next, Western blot analysis was performed to determine if growth at 37°C in minimal medium and human ascites fluid (in which complement was inactivated to enable the growth of AB307.70) affected the production of the K1 capsular polysaccharide in AB307-0294 and AB307.70 (Fig. 2B). These data clearly demonstrated that the loss of BfmR does not decrease capsule production. Taken together, these data do not support BfmR as a positive regulator of capsule production. Further, the mechanism by which BfmR confers resistance to complement-mediated bactericidal activity appears to be independent of capsule production.
FIG 2 .
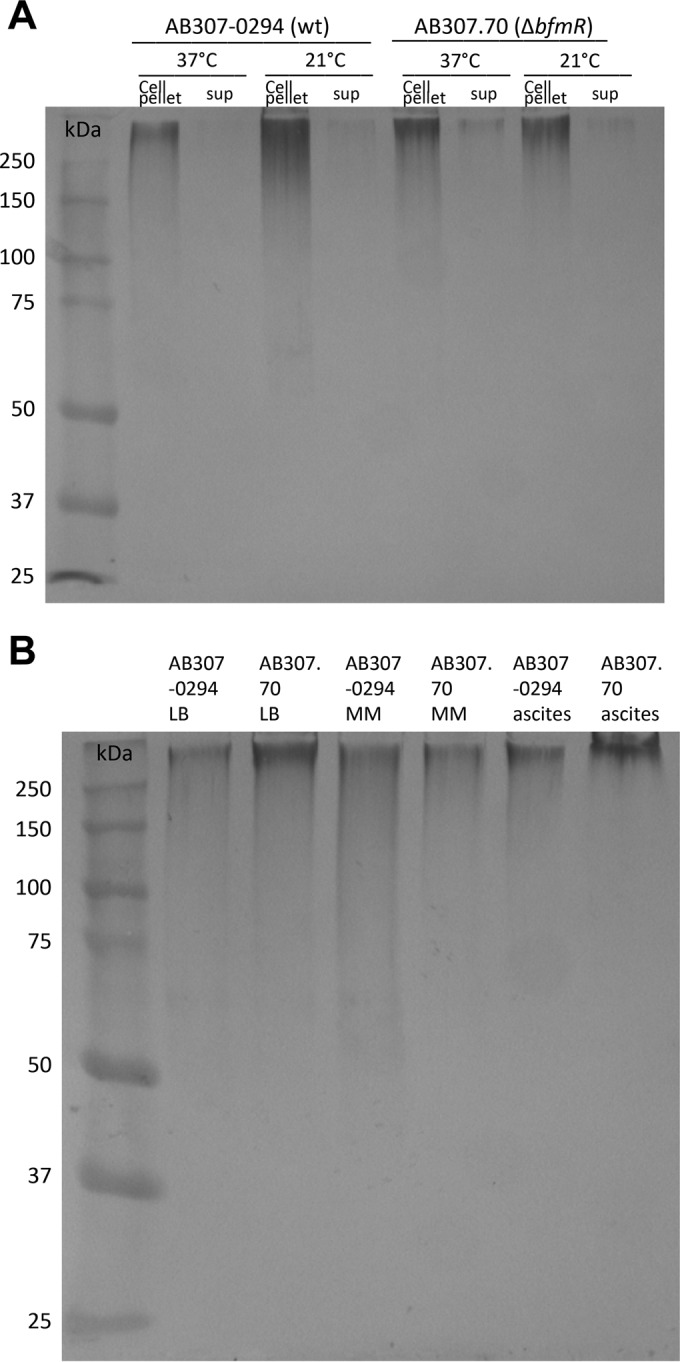
Comparison of capsular polysaccharide production by AB307-0294 (wt) and AB307.70 (ΔbfmR). Capsule production was assessed by Western blotting. The monoclonal antibody 13D6, which has been shown to recognize the K1 capsule produced by AB307-0294, was used to detect capsule. (A) Cell-associated capsule (cell pellet) and capsule in culture supernatant (sup) was assessed after AB307-0294 and AB307.70 were grown in rich Luria-Bertani laboratory medium at 37°C and 21°C. (B) Cell-associated capsule was assessed after AB307-0294 and AB307.70 were grown in rich LB laboratory medium, minimal medium (MM), or human ascites fluid at 37°C.
BfmR increases resistance to the antimicrobials meropenem and colistin.
Previous studies had suggested that BfmR caused increased resistance to colistin (polymyxin E), rifampin, erythromycin, and imipenem (27, 31). However, these studies used the A. baumannii type strain ATCC 17978, which is of unclear clinical significance (34). Therefore, we assessed the sensitivities of AB307-0294 (wt), AB307.70 (ΔbfmR), and AB307.70/pNLAC1[bfmR] to the carbapenem antimicrobials meropenem, colistin, and tigecycline. BfmR did not affect the activity of tigecycline (Fig. 3C). In contrast and consistent with prior studies (27, 31), BfmR significantly increased resistance to meropenem and colistin (Fig. 3A and B). The MIC for meropenem decreased from 1.0 µg/ml to 0.125 µg/ml, and for colistin it decreased from 2.0 µg/ml to 1.0 µg/ml with the loss of BfmR. These data demonstrate that BfmR mediates an increased level of resistance to two important classes of antimicrobials.
FIG 3 .
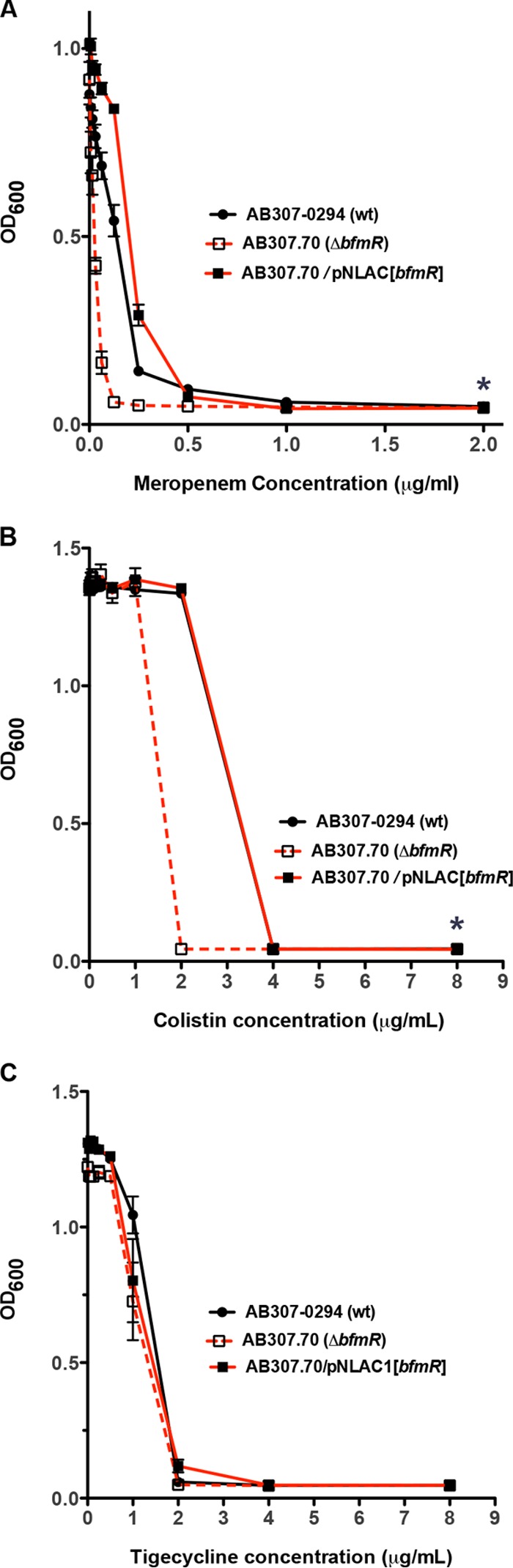
Comparison of the growth/survival of AB307-0294 (wt), AB307.70 (ΔbfmR), and AB307.70/pNLAC[bfmR] after exposure to various concentrations of meropenem (A), colistin (B), or tigecycline (C). Bacterial growth/survival was assessed by measuring the OD600 after overnight growth in Müeller-Hinton broth in the presence or absence of various concentrations of meropenem (0 µg/ml, 0.0078 µg/ml, 0.0156 µg/ml, 0.0312 µg/ml, 0.0625 µg/ml, 0.125 µg/ml, 0.25 µg/ml, 0.5 µg/ml, 1.0 µg/ml, and 2.0 µg/ml) or colistin/tigecycline (0 µg/ml, 0.0312 µg/ml, 0.0625 µg/ml, 0.125 µg/ml, 0.25 µg/ml, 0.5 µg/ml, 1.0 µg/ml, 2.0 µg/ml, 4.0 µg/ml, and 8.0 µg/ml). Data are means ± standard errors of the means (n = 9 to 15 for each strain at each concentration of antimicrobial). *, P < 0.05/2 (two-tailed unpaired t test for AB307-0294 compared to AB307.70).
Disruption of BfmR synthesis in a second A. baumannii clinical isolate (AB908ΔbfmR) does not affect capsule production and decreases growth/survival in human ascites fluid and serum ex vivo.
To confirm that the phenotypic effect of BfmR was not unique to AB307-0294, a site-directed disruption of bfmR was created in the contemporary clinical isolate AB908, resulting in the construct AB908ΔbfmR. As was observed for AB307-0294, disruption of bfmR did not affect capsule production (Fig. 4A), resulted in decreased growth/survival in human ascites fluid and serum (Fig. 4B and C), and demonstrated similar growth in rich laboratory medium compared to its wild-type parent (Fig. 4D). Likewise, as observed for AB307.70, when AB908ΔbfmR was grown in serum in which complement activity was inactivated (Δ56°C), growth was not restored to wild-type levels (Fig. 4B). These data support that the phenotypic effects of the disruption of BfmR synthesis were similar in 2 clinically relevant strains of A. baumannii.
FIG 4 .
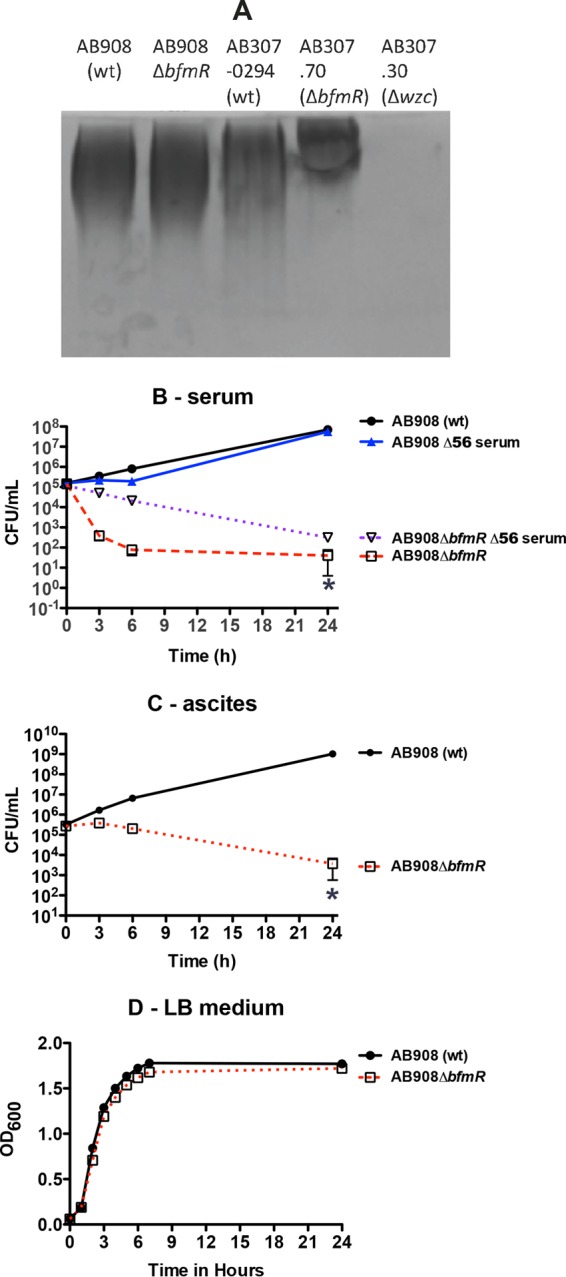
Comparison of capsular polysaccharide production and growth/survival of AB908 (wt) and AB908ΔbfmR in 90% human serum, 100% human ascites fluid, or rich laboratory medium. (A) Capsule production was assessed by alcian blue staining. Cell-associated capsule (cell pellet) was assessed after AB908 (wt) and AB908ΔbfmR were grown in rich Luria-Bertani laboratory medium at 37°C. AB307-0294 (wt, capsule positive), AB307.70 (ΔbfmR, capsule positive), and AB307.30 (Δwzc, capsule negative) served as positive and negative controls. (B) Growth/survival in serum was assessed by measurement of CFU at 0, 3, 6, and 24 h. AB908 (wt) and AB908ΔbfmR were also grown in serum that was heated to 56°C for 30 min, which inactivated complement-mediated bactericidal activity (Δ56 serum). Data are means ± SEM (n = 5 to 7). (C) Growth/survival in ascites fluid was assessed by measurement of CFU at 0, 3, 6, and 24 h. Data are means ± SEM (n = 4). (D) Growth in LB medium, as measured by the OD600. Data are means ± SEM (n = 4). *, P < 0.05/2 (two-tailed unpaired t test for AB908 [wt] compared to AB908ΔbfmR in untreated serum and ascites fluid).
BfmR is not homologous to human proteins and is conserved among isolates of A. baumannii.
A desirable trait of a potential antimicrobial drug target is a lack of homology to human proteins in order to enable target specificity. An amino acid sequence similarity search against the human proteome revealed a lack of any human proteins homologous to BfmR. This was an expected result, as TCSs are not known to be present in humans and other mammals (35, 36).
A second desirable trait of an antimicrobial target is overall protein conservation, and perhaps more importantly, conservation of the targeted drug binding site across bacterial species and strains of interest. BfmR consists of 238 amino acids, has a predicted molecular mass of 27.1 kDa, and has a predicted pI of 4.98. A BLASTp search of all nonredundant A. baumannii protein sequences (14 July 2015 data set) identified 908 proteins that were ≥99% identical (majority, 100%; over ≥99% coverage) to AB307-0294 BfmR. This analysis was complemented using the OMA orthologue database (September 2014 release) (37), which employs only high-quality complete genomic sequences. BfmR was 100% prevalent with fully identical sequences in the eight A. baumannii strains annotated in the OMA (Orthologous MAtrix) database (AB307-0294, ATCC 17978, 0057, AYE, ACICU, SDF, 1656-2, and TCDC-AB0715) plus the two A. baumannii strains sequenced by our group in addition to AB307-0294: AB853 (blood isolate from Iraq) and AB979 (environmental isolate from Iraq) (unpublished data). Additionally, two other Acinetobacter species present within OMA, A. baylyi ADP1 (a nonpathogenic soil bacterium) and A. calcoaceticus PHEA-2 (an industrial wastewater isolate) both contained a BfmR orthologue with fully identical amino acid sequences. These data support that BfmR is conserved across A. baumannii strains.
BfmR1-130 crystal structure.
The ability to determine a crystal structure is an important tool for characterizing ligand-protein interactions during downstream drug development. A. baumannii BfmR is predicted to belong to the OmpR/PhoB response regulator family based on sequence analysis, and it contains a receiver domain (BfmR1-130) and a DNA binding effector domain (BfmR131-238). BfmR1-130 was selected for structural studies, as conformational changes within the receiver domain dictate DNA binding activity of the effector domain. BfmR1-130 was a promiscuous crystallizer, with multiple hits observed in the automated 1536 condition crystallization screen. A selection of chemically diverse crystallization conditions were evaluated for optimization, but in each case the resulting BfmR1-130 crystals were relatively small. Evaluation of X-ray diffraction by crystals grown under a condition that was inherently cryoprotective demonstrated that the rod-like crystals of 0.15 to 0.20 mm length but only ~0.03 mm in diameter, with a hexagonal cross-section, produced data usable for the structure determination (Table 1).
TABLE 1 .
X-ray data collection and refinement statistics
| Characteristic | Valuea |
|---|---|
| PDB code | 5E3J |
| Data collection | |
| Space group | P 65 |
| Unit cell parameters (Å) | a = b = 52.0, c = 197.9 |
| Resolution range (Å) | 50.00–2.10 (2.18–2.10) |
| Completeness (%) | 99.3 (94.3) |
| Total no. of reflections | 189,638 |
| No. of unique reflections | 17,580 (1,668) |
| Multiplicity | 10.8 (6.5) |
| Rmerge | 0.086 (0.401) |
| 〈 I/σ(I)〉 | 20.5 (4.4) |
| Wilson B factor (Å2) | 24.1 |
| Refinement | |
| Resolution range (Å) | 29.73–2.10 (2.18–2.10) |
| No. of reflections, working set | 16,282 (1,737) |
| No. of reflections, test set | 1,229 (132) |
| Rcryst/Rfree | 0.1651 (0.2200)/0.1938 (0.2420) |
| No. of non-H atoms | |
| Protein/water | 1,929/181 |
| Model geometry (root-mean-square deviations from ideal) | |
| Bonds (Å)/angles (°) | 0.004/0.83 |
| Average B factors (Å2) | |
| Protein/water | 31.3/40.5 |
| Ramachandran plot (%)b | |
| Favored/allowed/outlier | 100/0/0 |
| MolProbity clashscoreb | 1.26 |
| Rotamer outliers (%)b | 0.94 |
Values in parentheses are for the highest resolution shell.
The BfmR1-130 crystal structure was readily determined by molecular replacement, using the PhoP receiver domain from Bacillus subtilis (PDB 1MVO; protein sequence 50% identical and 73% similar over 119 residues) as the search model. The BfmR1-130 crystal structure asymmetric unit contained two protomers, associated as a dimer by a noncrystallographic 2-fold rotation (root mean square [RMS] deviation for the C-α atom superposition of the two protomers is 0.18 Å). The space group was determined to be P65, with symmetry-related reflections scaling much poorer in the higher-symmetry P6522 space group (Rmerge, 8.6% versus 35.2%, respectively, to 2.10-Å resolution). The final refined model possessed a defining electron density for Lys6-Thr125 for both protomers, plus 181 modeled solvent molecules. The fold of an individual BfmR1-130 protomer is highly similar to the α5-β5 doubly wound motif observed for other OmpR/PhoB family receiver domains, as expected (38).
BfmR1-130 homodimerization.
The OmpR/PhoB family of response regulators is activated by autophosphorylation of a conserved aspartate residue in the receiver domain (Asp58 in BfmR). Phosphorylation induces an equilibrium shift favoring the active conformation, which includes formation of a 2-fold symmetrical homodimer involving the α4-β5-α5 face of the receiver domain. This homodimeric conformation facilitates the effector domain to bind DNA and modulate transcription (39, 40). A small molecule that interrupts this activation in BfmR would be a potential drug-lead candidate. In the BfmR1-130 crystal structure described here, Asp58 was not subjected to phosphorylation or the presence of a phosphorylation mimic (e.g., BeF3−). Moreover, the divalent cation (Mg2+ or Mn2+) required for autophosphorylation was not observed in the electron density in the cleft neighboring Asp58. Thus, BfmR1-130 is expected to be in an inactive state (i.e., a monomer or a nonproductive homodimer complex) within this crystal structure.
The BfmR1-130 crystal structure exhibited a noncrystallographic 2-fold symmetric homodimer in the asymmetric unit, formed by the α1-β2-α2 face of each protomer (Fig. 5). This α1-β2-α2 interface buried a total of 906 Å2 (6.7% of the total surface area). A search of the PDB revealed that this specific homodimer interface has not been previously observed for response regulator receiver domains. Moreover, the BfmR residues that mediate this interface are poorly to modestly conserved in other OmpR/PhoB family receiver domains (Fig. 6). A second BfmR1-130 noncrystallographic 2-fold symmetric homodimer was also present, unexpectedly exhibiting the α4-β5-α5 interface typical of OmpR/PhoB family receiver domains in the active phosphorylated conformation. This homodimer was comprised of a protomer A and a protomer B from two neighboring asymmetric units. This interface buried a total of 2,051 Å2 (or 15.3% of the total surface area), similar to that observed in crystal structures of receiver domains activated using BeF3− (41, 42). Furthermore, the amino acid composition of the BfmR1-130 α4-β5-α5 interface was highly similar to that present in other OmpR/PhoB family receiver domains (Fig. 6). For example, comparison of the BfmR1-130 structure with the analogous interface in BeF3−-activated Escherichia coli PhoB (PDB 1ZES) revealed that of the 21 interface residues per protomer, 12 (57%) are identical and 5 (24%) are similar in PhoB. By comparison, the overall residue identity between these two receiver domains is 39%. The four residue positions that are significantly different at this interface are BfmR His78, Gln79, Gln93, and Val113 versus PhoB Asp76, Ile77, Arg91, and Glu111, respectively. The BfmR His78-Gln79/PhoB Asp76-Ile77 residues participate at their respective dimer interfaces through formation of hydrogen bonds between their main chain carbonyl oxygens to the guanidinium group of BfmR Arg124′/PhoB Arg122′, respectively, where the prime symbol indicates a residue belongs to the partner protomer within the dimer. In PhoB, a salt bridge is formed across the dimer interface by Arg91 and Glu111′. The positionally equivalent residues in BfmR, Gln93 and Val113′, are incapable of participating in an analogous interfacial salt bridge. However, to partially compensate for the loss of this stabilizing interaction in BfmR, Gln93 participates in a hydrogen bond with the guanidinium group of Arg117′ across the interface.
FIG 5 .
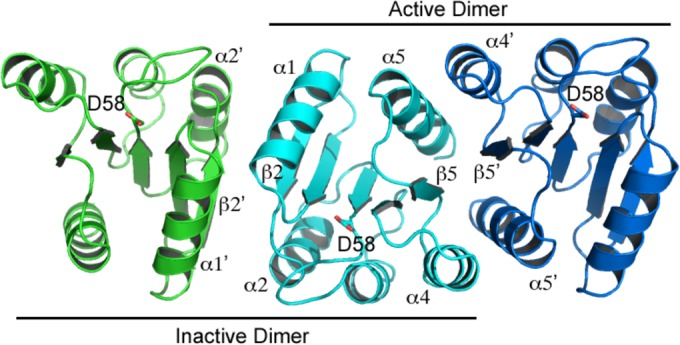
The BfmR1-130 crystal structure exhibits two distinct homodimer interfaces. The OmpR/PhoP family canonical active homodimer (α4-β5-α5 interface; cyan and blue chains) was observed despite the unphosphorylated state of Asp58. A unique and presumably inactive homodimer (α1-β2-α2 interface; cyan and green chains) was also observed.
FIG 6 .

Alignment of the BfmR1-130 sequence against representative OmpR/PhoP family receiver domains. Fully conserved residues are contained in red filled boxes, and highly conserved residues are in blue outlined boxes. Secondary structure elements are based on the A. baumannii BfmR1-130 crystal structure. Black and cyan boxes denote residues participating in the α1-β2-α2 and α4-β5-α5 homodimer interfaces, respectively; blue triangles denote residues with interface interactions limited to main chain atoms, and green circles denote residues participating in interprotomer salt bridges in A. baumannii BfmR1-130. Sequence abbreviations: abBfmR, A. baumannii BrmR; kpRstA, K. pneumoniae RstA; ecPhoB, E. coli PhoB; ecPhoP, E. coli PhoP; bsPhoP, B. subtilis PhoP.
The BfmR1-130 α4-β5-α5 dimer interface is also three-dimensionally similar to the typical OmpR/PhoB family receiver domain active homodimer structure. The RMS deviations for superimposed C-α atoms from BfmR1-130 and E. coli PhoB (PDB 1ZES) individual protomers range between 0.96 and 1.08 Å, versus 1.12 Å for the superimposed α4-β5-α5 homodimers. In addition to homodimerization, the canonical activation mechanism involves a characteristic shift of a serine or threonine switch residue toward the phosphorylated aspartate, such that the side chain hydroxyl group forms a hydrogen bond to the phosphate (42). An aromatic residue (tyrosine or phenylalanine) on the β5 strand undergoes a rotamer change to shift its bulky side chain away from the α4-β5-α5 dimerization interface and into the volume previously occupied by the Ser/Thr side chain. The BfmR1-130 crystal structure exhibits the active conformation of these two residues (Thr85 and Tyr104), further suggesting that this structure represents the active form of the BfmR receiver domain, despite the absence of phosphorylation or a phosphorylation mimic (Fig. 7).
FIG 7 .

Stereoview of cartoon representations of A. baumannii BfmR1-130 (cyan) superimposed upon the activated E. coli PhoB receiver domain (PDB 1ZES; green). The conserved active site and conformational switch residues are displayed as sticks. Labels are in the following format: BfmR residue/PhoB residue. Residues present only in the E. coli PhoB structure are BeF3−, which mimics phosphorylation of D53, and the active site Mg2+ ion. The BfmR1-130 conformational switch residues T85 and Y104 are in the canonical active conformation, similar to those (T83 and Y102) in activated PhoB. The inactive side chain conformations of these two PhoB residues are displayed in gray.
Prediction of small-molecule binding hot spots.
Computational predictions (via the FTMap server) of small-molecule binding hot spots (i.e., surface regions with major contributions to ligand binding free energy) on BfmR1-130 were performed to estimate the likelihood that small-molecule inhibitors acting specifically through the BfmR receiver domain can be identified (43). This analysis predicted 11 binding hot spots on the BfmR1-130 protomer surface, which could be classified as very strong (1 predicted hot spot), strong (3 predicted hot spots), medium (3 predicted hot spots), or weak (4 predicted hot spots). It was hypothesized that a BfmR inhibitor would likely bind at the α4-β5-α5 face, thereby preventing productive formation of the active BfmR homodimer, or it would bind near Asp58 to inhibit phosphorylation. Six binding hot spots were identified at these locations (Fig. 8A). A medium binding hot spot was present in the cleft near Asp58, occupying the volume where phosphorylation would occur. A single weak hot spot was predicted on the middle of the α4-β5-α5 face, in a shallow cleft between strand β5 and helix α5, and one medium and two strong hot spots were predicted on the periphery of this dimerization face. A sixth predicted binding hot spot was located neighboring the phosphorylation site, between helix α1 and the N terminus of helix α5.
FIG 8 .
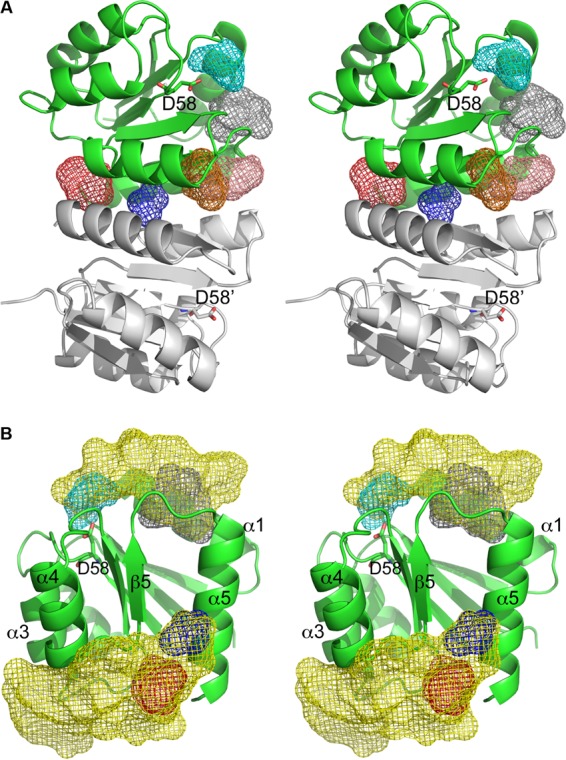
Stereoviews of BfmR1-130 structural features as potential antimicrobial targets. (A) Results of the FTMAP prediction of ligand-binding hot spots at functionally important locations, displayed as mesh-enclosed volumes, on the surface of a BfmR1-130 protomer (represented in green). The binding hot spots are color-coded according to the predicted importance (red, strong; salmon, strong; orange, medium; cyan, medium; blue, weak), which correlated to the number of bound small molecules predicted to cluster at that location. A sixth predicted binding hot spot neighboring the phosphorylation site is represented by gray mesh. The second protomer (gray ribbon cartoon) illustrates the homodimer organization, but it was excluded from the binding hot spot calculations. For clarity, only predicted hot spots near the functionally important α4-β5-α5 face and Asp58 are displayed. (B) Stereoview of preferential docking locations of drug-like molecules at functionally important regions of the BrmR1-130 protomer. The yellow mesh encompasses volumes where drug-like molecule preferentially docked on the BfmR1-130 protomer surface. These preferential docking locations encompass several of the binding hot spots identified by FTMAP (red and blue mesh on the α4-β5-α5 face and cyan and gray mesh at and near the phosphorylation site).
Screening of drug and drug-like virtual libraries.
The binding hot spot predictions were based upon docking of chemically simple probes composed of 2 to 8 nonhydrogen atoms. The potential for BfmR to bind more elaborate drug-like small molecules was further evaluated through a blind docking strategy. This in silico analysis was performed by docking virtual libraries representative of FDA-approved, NCI diversity 3 and LOPAC1280 compounds over the entire BfmR receptor domain protomer surface to predict preferential binding sites. Two clusters of high-scoring docked molecules were present about a subset of the FTMap-predicted binding hot spots at functionally important locations, indicative of locations likely to be suitable for binding drug-like molecules (Fig. 8B). Specifically, one cluster was at the dimer interface occupying both the binding hot spot at the cleft formed by the N terminus of strand β5 and the C terminus of α5 (red mesh in Fig. 8B) and the hot spot at the center of the α4-β5-α5 face (blue mesh in Fig. 8B). Binding interactions for this cluster also extended under the C-terminal ends of helices α3 and α4. A second cluster occupied a binding hot spot in the cleft between helix α1 and the N terminus of helix α5 (gray mesh in Fig. 8B). Several molecules in this docking cluster extended to also occupy the binding hot spot located in the phosphorylation site (cyan mesh in Fig. 8B).
DISCUSSION
Findings from this report further support BfmR as a potential drug target of A. baumannii. An important and highly appealing characteristic of BfmR is that its inhibition would have the dual benefit of significantly decreasing in vivo survival as well as increasing the sensitivity of A. baumannii to other antimicrobials.
A biological property that supports this concept is the previous demonstration that BfmR is essential for survival in vivo (24). Here, we have demonstrated a mechanism responsible for this phenotype. BfmR enhances growth/survival in human ascites fluid and serum, which was mediated, at least in part, by conferring resistance to complement-mediated bactericidal activity (Fig. 1A and B and 4B and C). A recent report that studied A. baumannii strain ATCC 17978 also demonstrated that BfmR increased resistance to complement-mediated bactericidal activity and that this phenotype was mediated by decreased capsule production (27). However, the mechanism by which BfmR mediates resistance to complement-mediated bactericidal activity in AB307-0294 and AB908, the strains assessed in this study, appears to be different. Although our group previously demonstrated that capsular polysaccharide confers resistance to complement-mediated bactericidal activity (33), in AB307-0294 BfmR does not affect K1 capsule production (Fig. 2). An alternate explanation would be the production of a second, non-K1 capsular polysaccharide. However, surface polysaccharide was purified by extraction with pyridine acetate (44) from AB307-0294 (wt) and AB307::Tn17 (both capsule positive) and mutant constructs with disruptions in either a K1 nucleotide-sugar biosynthetic gene (gna; AB307::Tn69) or a K1 capsule export gene (wzc; AB307.30) (both K1 capsule negative) (33, 45), resolved by SDS–8% polyacrylamide gel electrophoresis and subjected to alcian blue staining. A capsule was observed for AB307-0294 and AB307::Tn17, but not AB307::Tn69 or AB307.30, thereby excluding a cryptic second capsule (data not shown). Therefore, the mechanism by which BfmR confers resistance to complement-mediated bactericidal activity is independent of capsule production. This difference is likely strain dependent. AB908 is a contemporary clinical isolate, and the model pathogen AB307-0294 is a blood isolate that was frozen from the primary clinical microbiology laboratory culture, thus maintaining its genetic integrity. It possesses a complete capsule, is resistant to 90% human serum, and is virulent in rat pneumonia and soft tissue infection models and a mouse systemic infection model (46). Disruption of the capsule transport gene wzc results in the loss of capsule but is not lethal (33), and capsule is primarily cell associated (this study). In contrast, the ATCC 17978 strain produces a thin capsule (27), is relatively sensitive to complement-mediated bactericidal activity (34), disruption of wzc appears to be a lethal mutation (27), significant amounts of capsule are detected in culture supernatant (27), and it is less virulent in both mouse and Galleria mellonella infection models (34, 47). Therefore, ATCC 17978 does not appear to be an optimal clinical representative of A. baumannii, making the translational implications of studies using this strain unclear.
Although the effect of BfmR on capsule production was discordant between AB307-0294 and ATCC 17978, its effect on conferring increased resistance to selected antimicrobials was similar (27, 31). Previous published studies demonstrated that BfmR increased resistance to colistin, imipenem, rifampin, and erythromycin, but not chloramphenicol, ceftazidime, amikacin, or ciprofloxacin (27, 31). These data are consistent with the concept that BfmR confers increased resistance to certain antimicrobials. Limitations of those studies were the use of ATCC 17978, which is not an optimal representative of A. baumannii, and the fact that erythromycin and chloramphenicol are not antimicrobials usually considered for the treatment of infections due to A. baumannii. In this report, we used a clinically relevant strain and antimicrobials. The mechanism that mediates this effect is unclear; however, a previous report demonstrated that BfmR affected the scaffolding of OmpA (31). It is tempting to speculate that alterations in outer membrane proteins could affect the sensitivity to certain antimicrobials. Regardless of the mechanism, these data add further support to BfmR as an antimicrobial target, since its inactivation would also have the additional beneficial effect of increasing its sensitivity to other antimicrobials.
Lastly, although we did not assess the effect of BfmR on biofilm formation, other investigators have demonstrated that it mediates increased biofilm production (32). Biofilms can directly contribute to infection (e.g., intravascular access or implanted devices). Therefore, targeting BfmR may have further benefit for these difficult-to-treat infections.
Understanding the structure of a potential drug target may provide valuable information for guiding drug development. For TCS response regulators, structure-function insights may identify possible ligand binding sights that will modulate formation of the active homodimer. BfmR exhibited several interesting structural features. The active homodimeric conformation of BfmR1-130 was observed despite not undergoing phosphorylation or phosphorylation mimicked by BeF3− or other means. This unexpected active form has been observed previously for other OmpR/PhoB family receiver domains. It is thought that the high protein concentration present during crystallization may shift the equilibrium from monomer (or a nonproductive dimer) to the active homodimeric form (41, 42, 48, 49). Conversely, the crystallization conditions and resulting crystal lattice packing may favor the active BfmR homodimer.
The computational prediction of small-molecule binding hot spots on BfmR1-130 supports the possibility of identifying small-molecule inhibitors of BfmR activity that act by specific binding to the receiver domain. Binding hot spots on the BfmR1-130 protomer were predicted on the protomer face that participates in the active homodimer interface and at the phosphorylation site. It is possible that ligand binding at other hot spots outside these two regions may inhibit BfmR activity through an allosteric mechanism, but that is beyond the scope of this study, as is consideration of the possibility of inhibitor binding to the DNA-binding domain.
A predicted strong binding hot spot (red mesh in Fig. 8A) lies between the N terminus of strand β5 and the C terminus of α5, and the Arg124 side chain fits into this niche on the partner protomer of the homodimer. A small molecule bound in this niche would likely interrupt the Arg124 hydrogen bond to the His78′ carbonyl oxygen and the salt bridge to the Asp102′ carboxyl group. A second predicted strong hot spot (orange mesh in Fig. 8A) and the predicted weak hot spot (blue mesh in Fig. 8A) neighbor the Arg117 side chain, and a small molecule bound in either location would likely interfere with formation of the Arg117-Asp103′ salt bridge across the interface. Given the conserved nature of the OmpR/PhoB family receiver domain α4-β5-α5 face, it may be possible to develop a small molecule that inhibits activation of a broad range of response regulators. Interestingly, an in silico ligand-screening study for the E. coli PhoP receiver domain focused upon binding sites near PhoP residues Arg111 (analogous to BfmR Arg117) and Arg118 (analogous to BfmR Arg124), as these residues are key determinants of PhoP homodimerization and activation (50). Importantly, ligand binding hot spot prediction has been demonstrated to be less sensitive to protein conformational changes than the in silico prediction of a specific binding site for a specific ligand (43).
Blind docking results supported that several but not all predicted binding hot spots may be suitable for participating in binding interactions with drug-like molecules that will interfere with BfmR activation. However, this analysis also suggested that successful binding of drug-like molecules likely requires interactions with two binding hot spots or neighboring regions. For example, the phosphorylation site by itself was not a promising drug-like molecule binding site. Rather, docked drug-like molecules bound at the phosphorylation site also required binding interactions at the neighboring binding hot spot between helix α1 and the N terminus of helix α5. The phosphorylation site pocket is comprised primarily of charged and polar residues. A general feature of drug-protein complexes is the presence of hydrophobic in addition to charged or polar amino acids at the binding site (51), and so the phosphorylation site is not optimal for interacting with drug-like molecules but may participate in a larger binding site.
Computational methods predicted that BfmR is likely a druggable target. However, development of robust BfmR activity assays are required prior to further experimental exploration to identify and characterize hit or lead molecules for therapeutic development. Cell permeability and efflux pumps are major barriers in the identification of tractable Gram-negative antimicrobial lead molecules (52). Thus, an assay to evaluate efficacy of a compound with a BfmR-specific mechanism of action in the context of live A. baumannii is required. Furthermore, an assay to determine the threshold of BfmR inhibition necessary to result in therapeutically relevant antimicrobial activity is also needed, as a small-molecule drug may not completely inhibit the BfmRS signaling pathway in vivo. Previous efforts to identify small-molecule inhibitors of response regulator activation lacked assays to evaluate if targeted TCSs were effectively and specifically downregulated and thereby conferred significant biological effects (50, 53).
A. baumannii BfmR is an appealing antimicrobial target, because its inhibition would significantly decrease in vivo survival and increase sensitivity to antimicrobials. In addition, it has a lack of significant sequence similarity to any human protein, and BfmR belongs to a protein family absent from humans and other mammals. Lastly, the BfmR sequence is highly conserved across A. baumannii strains. However, several important questions remain to be addressed before BfmR can be classified as an established target. Thus far, no receiver domain from the OmpR/PhoB family has been demonstrated as a druggable target (i.e., activity can be modulated by a drug-like small molecule). Likewise, determining the degree of BfmR inhibition needed to impact growth/survival, an efficient BfmR activity assay for high-throughput screening BfmR activity against a small-molecule library, and an assay for determining specificity of the mechanism of action are needed for progress of BfmR as a potential new antimicrobial target. Additionally, major impediments for development of an antimicrobial effective against GNB are permeability and efflux, and a small-molecule inhibiting BfmR activity must overcome these barriers. Despite these challenges, an intriguing aspect of TCS response regulators as antimicrobial targets is the possibility of developing drugs effective against either a narrow or broad spectrum of bacteria. Additionally, the development of small-molecule tool compounds that modulate activities of response regulators will allow chemical biology dissection of bacterial signaling pathways responsive to environmental stimuli. The OmpR/PhoB family receiver domains in response regulators are widely present in bacterial pathogens and possess a degree of shared homology at the dimer interface and phosphorylation site. Therefore, it is possible that a drug inhibiting BfmR activity through its receiver domain will also inhibit the activity of additional TCS response regulators. This multitarget approach may increase durability and overcome a major concern in drug development, the development of resistance. Taken together, data presented in this report support further studies designed to develop BfmR as a drug target, including development of assays required for further target validation.
MATERIALS AND METHODS
Bacterial strains.
A. baumannii strain AB307-0294 (blood isolate; sequence type 15, clonal group 1, based on the methods of Ecker et al. [54]) has a K1 capsular serotype (55) and was isolated from a patient hospitalized at Erie County Medical Center, Buffalo, NY, in 1994 (56). The genome of AB307-0294 has been fully sequenced, and it contains 3.76 Mbp with 3,531 predicted open reading frames (57). Previous comparisons with A. baumannii strains in the public domain support AB307-0294 as being clinically appropriate, representative of contemporary A. baumannii strains, and an ideal background to identify new or unrecognized antimicrobial targets of interest (24).
AB307.70 is an isogenic BfmR-minus derivative of AB307-0294 generated by transposon mutagenesis, and it possesses a disruption in bfmR (nucleotides 1 to 717) due to the transposon insertion after nucleotide 592. bfmR putatively encodes a response regulator protein, BfmR (24). Polar effects were excluded, as described previously (24), and the complemented derivative, AB307.70/pNLAC[bfmR], was generated for this study to confirm that the observed phenotypic differences between AB307-0294 and AB307.70 were due to BfmR. A second BfmR-minus derivative was created by site-directed mutagenesis of the clinically relevant isolate AB908 (obtained from Walter Reed Medical Hospital) by using a one-step chromosomal gene inactivation method described elsewhere (58). In brief, a linear PCR-generated amplicon (forward primer, 5′-ATGAGCCAAGAAGAAAAGTTACCAAAGATTCTGATCGTTGAAGACGACGAGCGTTTAGCGCGATTAACTCAAGAATATTTAATCCGTAATGGTTTGGAAGTTGGTGTAGAAACCGATGGTAACCGTAAAGCCACGTTGTGTCTCAAAATC-3′; reverse primer, 5′-TTACAATCCATTGGTTTCTTTAACAAACAAGTAACCTTTACTACGTACAGTTTTAATACGTTTTGGATTTTCAGGATCATCGCCAATTTTTGGACGAATACGTGAAATACGTACGTCAATTGAACGCATTATTCCCTCCAGGTATTAGAA-3′; bold sequence portions are homologous to the kanamycin resistance gene sequence) that contained a kanamycin resistance cassette flanked by the first and last 125 bp of bfmR was electroporated into AB908/pAT02 (58), and recombinants were selected on Luria-Bertani (LB) plates containing kanamycin. The successful disruption of bfmR in AB908ΔbfmR was confirmed by sequence analysis, and a derivative cured of pAT02 was used for subsequent studies. AB307.30 and AB307::Tn69 are isogenic derivatives generated by transposon mutagenesis that possess disruptions in wzc (a capsule transport gene) and gna (a capsule biosynthetic gene), respectively (33, 45). AB307.30 and AB307::Tn69 were used in studies to exclude the presence of a second, non-K1 capsule. AB307-0294 (wt) and AB307::Tn17 (ΔlpsB) (59), which possesses a disruption in a glycosyltransferase involved in lipopolysaccharide core biosynthesis, are both capsule positive and served as positive controls. Strains were maintained at −80°C in 50% LB medium (5 g of yeast extract, 10 g of tryptone, and 10 g of NaCl per liter) and 50% glycerol.
Media.
The procedures for obtaining human ascites fluid and human serum were reviewed and approved by the Western New York Veterans Administration Institutional Review Board. Informed consent was obtained from healthy volunteers for blood collection. Blood underwent coagulation at room temperature for 15 min, followed by refrigeration at 4°C for 60 min to enable clot retraction. Serum was obtained from the blood after subsequent centrifugation at 4°C for 15 min at 3,000 × g. For the collection of ascites fluid, the Western New York Veterans Administration Institutional Review Board waived informed consent because the ascites fluid was collected from deidentified patients who were undergoing therapeutic paracentesis for symptoms due to abdominal distension. These individuals were not being treated with antimicrobials and were not infected with human immunodeficiency virus, hepatitis B virus, or hepatitis C virus. The ascites fluid was cultured to confirm sterility, divided into aliquots, and stored at −80°C. Each batch was obtained from a different patient and was designated by the date of removal. A single ascites fluid batch (obtained on 18 October 2012) was used for growth studies. For some experiments, human serum and ascites fluid were incubated at 56°C for 30 min to inactivate complement-mediated bactericidal activity (Δ56°C). BBL Müeller-Hinton II cation-adjusted (MH) broth consisted of 22 g of powder per liter (Becton, Dickinson). Minimal medium consisted of 200 ml of solution A [2.0 g (NH4)2SO4, 6.0 g Na2HPO4, 3.0 g KH2PO4, 3.0 g NaCl, 0.011 g Na2SO4], 800 ml of solution B (0.2 g MgCl2, 0.0132 g CaCl2 ⋅ 2H2O, 0.0005 g FeCl3 ⋅ 7H2O, 2.9241 g citrate [trisodium salt dehydrate]), and 3 g Casamino Acids. AB307.70 was grown in the presence of 40 µg/ml of kanamycin, and AB307.70/pNLAC[bfmR] was grown in the presence of 40 µg/ml of kanamycin and 200 µg/ml of carbenicillin.
In vitro and ex vivo growth in LB medium, human ascites fluid, and human serum.
Growth experiments in LB medium, human ascites fluid, and human serum were performed as described elsewhere (56), with aliquots removed for bacterial enumeration at various times. For growth studies, 100% LB medium, 90% ascites fluid–10% 1× phosphate-buffered saline (PBS; pH 7.4), and 90% serum–10% 1× PBS were used.
Analysis of capsule production.
AB307-0294 (wt) and AB307.70 (ΔbfmR) cells were grown overnight in LB medium, minimal medium, or human ascites fluid. The next day, an aliquot was removed for enumeration and bacterial cells were concentrated by centrifugation and resuspended in 50 µl/1 ml of culture lysis buffer (2% SDS, 10% glycerol in 1 M Tris [pH 6.8], to which fresh 4% β-mercaptoethanol was added). Cells in lysis buffer were boiled for 10 min. Next, the lysate was treated with 20 µl/50 µl of lysate with proteinase K (1.25 mg/ml) for 120 min at 60°C. Supernatant was generated by precipitation with 5 volumes of ice-cold 75% ethanol, which was placed at 4°C for 18 h. The precipitant was recovered by centrifugation at 8,000 × g for 30 min at 4°C. The supernatant was discarded and the tubes were air dried. The dried precipitates were resuspended in lysis buffer at the corresponding volume of the concordant pellet, boiled for 10 min, and treated with proteinase K in the same manner as the pellets. The cell pellet and supernatant volumes used for gel loading were normalized to a bacterial titer of 3.9 × 106 CFU by dilution in lysis buffer. Capsular polysaccharide was resolved by SDS–8% polyacrylamide gel electrophoresis and subjected to Western blot analysis with MAb 13D6, which is directed against the AB307-0294 capsule (33), or were stained with alcian blue.
Antimicrobial susceptibility testing.
A. baumannii strains were grown for 18 h in MH broth at 37°C. Bacteria were diluted 1:100 in the same medium, and titers were determined by serial 10-fold dilutions and enumerated on LB plates with or without appropriate antibiotics. Assays were performed in 96-well microtiter plates. Wells contained either 0 µg/ml, 0.0078 µg/ml, 0.0156 µg/ml, 0.0312 µg/ml, 0.0625 µg/ml, 0.125 µg/ml, 0.25 µg/ml, 0.5 µg/ml, 1.0 µg/ml, or 2.0 µg/ml of meropenem or 0 µg/ml, 0.0312 µg/ml, 0.0625 µg/ml, 0.125 µg/ml, 0.25 µg/ml, 0.5 µg/ml, 1.0 µg/ml, 2.0 µg/ml, 4.0 µg/ml, or 8.0 µg/ml of colistin or tigecycline in 100 µl of MH broth to which 100 µl of the same medium containing bacteria was added. Final concentrations of bacteria were 2.4 × 105 CFU/ml for AB307-0294, 5.0 × 105 CFU/ml for AB307.70, and 1.8 × 105 CFU/ml for AB307.70/pNLAC[bfmR] for meropenem studies; 6.2 × 105 CFU/ml for AB307-0294, 8.9 × 105 CFU/ml for AB307.70, and 6.8 × 105 CFU/ml for AB307.70/pNLAC[bfmR] for colistin studies; and 7.1 × 105 CFU/ml for AB307-0294, 7.9 × 105 CFU/ml for AB307.70, and 1.3 × 106 CFU/ml for AB307.70/pNLAC[bfmR] for tigecycline studies. Control wells contained MH broth only. The plates were incubated for 18 h at 37°C. The optical density at 600 nm (OD600) of each well was measured in a microplate spectrophotometer (SpectraMax 190; Molecular Devices) at 26°C. Two independent experiments, each with an n of 6, were performed for each strain and antimicrobial concentration.
Bioinformatic analysis of BfmR sequence and structure.
The protein sequences for BfmR orthologues were generated from respective bfmR gene sequences. The OMA orthologue database (September 2014 release) was searched to evaluate BfmR conservation across A. baumannii strains and additional Acinetobacter species (37). BLASTp (60), as implemented on the NCBI server (http://blast.ncbi.nlm.nih.gov), and Clustal Omega (61) were used to align sequences, and ESPript 3.0 (62) was used to display protein sequences. FTMap was used to predict locations of small-molecule binding hot spots on the exposed surface of the BfmR receiver domain, using the default set of small organic molecular probes (16 chemicals; mass range, 30.1 to 106.1 Da) (43). AutoDock Vina v. 1.1.2 and PyRx v. 0.8 were used for in silico screening of the BfmR receiver domain against a virtual library of drug and drug-like molecules (63, 64). The virtual molecular libraries represented small-molecule chemicals approved for clinical trials or clinical use (3,180 chemicals; mass range, 60.1 to 829.0 Da), the NCI Diversity 3 collection (1,880 chemicals; mass range, 114.1 to 697.1 Da), and the LOPAC1280 collection (1,280 chemicals; mass range, 59.1 to 836.4 Da), with molecular descriptor files obtained from the ZINC database (65).
Recombinant protein expression and purification.
E. coli strain DH5α (Invitrogen) was used during construction of the bfmR expression vector, and strain BL21(DE3) (Novagen) was used for recombinant protein expression. PCR amplification of the portion of the bfmR open reading frame (NCBI accession number ACJ59134), coding for the N-terminal receiver domain (BfmR1-130), was acccomplished using purified AB307-0294 chromosomal DNA (RefSeq NC_011595.1) as the template and the primers forward, 5′-GCGCGACATATGATGAGCCAAGAAGAAAAGTTA-3′; and reverse, 5′-ATATATCTCGAGTTATTCAACAGTTTTATCCGTACG-3′, which included an NdeI and an XhoI cleavage site, respectively. Gel-purified PCR products were restriction enzyme digested and ligated using T4 DNA ligase (Promga) into a linearized customized pET15b-TEV (tobacco etch virus) vector containing an N-terminal His5 affinity tag plus a TEV protease cleavage site replacing the usual thrombin site (pET15b-TEV-bfmR_rec). The integrity of the expression cassette was verified by DNA sequencing (Roswell Park Cancer Institute Sequencing Facility).
E. coli BL21(DE3) cells transformed with pET15b-TEV-bfmR_rec were grown to an OD600 of 0.8 at 30°C in LB medium supplemented with 100 µg/ml ampicillin. Protein expression was induced with 1 mM β-d-1-thiogalactopyranoside (IPTG). The culture was incubated for an additional 5 h prior to harvesting by centrifugation. Frozen cell pellets were thawed and resuspended in lysis buffer (20 mM Tris-HCl [pH 8.0], 200 mM NaCl, 10 mM imidazole, and 1 mM β-mercaptoethanol). Cells were lysed using a Microfluidizer (Microfluidics). Filtered lysate supernatant was loaded onto a HisTrap immobilized metal ion affinity chromatography (IMAC) column (GE) and eluted with a 10 mM-to-300 mM imidazole gradient plus lysis buffer. Elution fractions containing His5-BfmR1-130 were dialyzed into lysis buffer plus 60 mM imidazole. TEV protease was added to the fusion protein at a 1:100 mass ratio, and the mixture was incubated overnight at 4°C (66). The sample was reapplied to a HisTrap IMAC column to separate the cleaved His5 affinity tag from BfmR1-130. Eluted BfmR1-130 was dialyzed against 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, and 1 mM dithiothreitol and then was concentrated (final concentration, 11.25 mg/ml) by using Centricon-3 centrifugal concentrators (Amicon). Small aliquots were flash-frozen in liquid nitrogen and stored at −80°C. Protein purity was verified via SDS-PAGE.
Crystallization.
Crystallization screening via microbatch under oil in 1,536-well microtiter plates was conducted using the high-throughput robotics at the Hauptman-Woodward Institute (67). Selected hits were optimized via manual sitting-drop vapor diffusion. Crystals used for diffraction data collected were obtained by equilibration at 20°C of a crystallization drop containing 2 µl of purified BfmR1-130 at 11.25 mg/ml plus 2 µl of reservoir solution against 1 ml of reservoir (20% [vol/vol] polyethylene glycol 400, 20% [vol/vol/] glycerol, 100 mM Tris-HCl [pH 8.5], and 150 mM NaCl).
X-ray diffraction data collection and processing.
Crystals were harvested using a nylon loop and then subsequently flash-cooled in liquid nitrogen. No additional cryoprotectant was required due to the composition of the reservoir solution. Crystals were shipped to the Stanford Synchrotron Radiation Lightsource (SSRL), where diffraction data were collected remotely at −173°C by using a MARmosaic 325 charge-coupled-device detector on beamline 9-2. Data were collected using an X-ray wavelength of 0.97946 Å, a crystal-to-detector distance of 330 mm, a φ range of 180.4° by 0.55° increments, with 5-s exposures per frame. Data were indexed, integrated, and scaled using HKL2000 (68).
Statistical analyses.
Continuous data were assessed for normality and are presented as means ± standard errors of the means (SEM.) P values of 0.05/n (n = the number of comparisons) were considered statistically significant based on the Bonferroni correction for multiple comparisons. To normalize ex vivo growth/survival data (Fig. 1), log10-transformed values were utilized, the area under each curve was calculated, and these areas were compared using two-tailed unpaired t tests (Prism 4 for MacIntosh; GraphPad Software, Inc.).
Structure solution and refinement.
The receiver domain from the response regulator PhoP from Bacillus subtilis (PDB 1MVO) was used as the search model in molecular replacement, following removal of associated solvent and ions. The Auto-Rickshaw server (69) was used to determine the molecular replacement solution, which was automatically fed to Arp/wARP (70) to produce an initial BfmR model. Refinement was conducted using Phenix, including TLS refinement to model anisotropic displacements and noncrystallographic symmetry (NCS) restraints (71). Manual model building and analysis were performed using Coot (72). Structure analysis and validation made use of PyMol (Schrödinger, LLC), Phenix (71), PISA (73), and the validation tools present in the wwPDB deposition tool (http://deposit.wwpdb.org/deposition).
Protein structure accession number.
The refined coordinates and scaled diffraction data have been deposited in the PDB (PDB code 5E3J).
ACKNOWLEDGMENTS
We thank Leah Herzog and Wendy Franke for assistance in expression vector and protein production and L. Wayne Schultz (HWI; presently with QuaDPharma) for helpful discussions. The TEV protease expression vector was a kind gift of David Waugh (NCI).
This research is based in part upon work supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (IBX000984A, T.A.R.). This work was also supported in part by Telemedicine and Advance Technical Research Center (TATRC) cooperative agreement W23RYX1055N607 and an interdisciplinary grant from the University at Buffalo (T.A.R. and T.C.U.). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including grant P41GM103393).
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Funding Statement
The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the U.S. Department of Energy Office of Biological and Environmental Research and by the National Institute of General Medical Sciences, National Institutes of Health (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
REFERENCES
- 1.Garnacho-Montero J, Amaya-Villar R. 2010. Multiresistant Acinetobacter baumannii infections: epidemiology and management. Curr Opin Infect Dis 23:332–339. doi: 10.1097/QCO.0b013e32833ae38b. [DOI] [PubMed] [Google Scholar]
- 2.Manchanda V, Sanchaita S, Singh N. 2010. Multidrug resistant Acinetobacter. J Glob Infect Dis 2:291–304. doi: 10.4103/0974-777X.68538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim BN, Peleg AY, Lodise TP, Lipman J, Li J, Nation R, Paterson DL. 2009. Management of meningitis due to antibiotic-resistant Acinetobacter species. Lancet Infect Dis 9:245–255. doi: 10.1016/S1473-3099(09)70055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snitkin ES, Zelazny AM, Montero CI, Stock F, Mijares L, NISC Comparative Sequence Program, Murray PR, Segre JA. 2011. Genome-wide recombination drives diversification of epidemic strains of Acinetobacter baumannii. Proc Natl Acad Sci U S A 108:13758–13763. doi: 10.1073/pnas.1104404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Towner KJ. 2009. Acinetobacter: an old friend, but a new enemy. J Hosp Infect 73:355–363. doi: 10.1016/j.jhin.2009.03.032. [DOI] [PubMed] [Google Scholar]
- 6.Sengstock DM, Thyagarajan R, Apalara J, Mira A, Chopra T, Kaye KS. 2010. Multidrug-resistant Acinetobacter baumannii: an emerging pathogen among older adults in community hospitals and nursing homes. Clin Infect Dis 50:1611–1616. doi: 10.1086/652759. [DOI] [PubMed] [Google Scholar]
- 7.Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 51:3471–3484. doi: 10.1128/AAC.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fishbain J, Peleg AY. 2010. Treatment of Acinetobacter infections. Clin Infect Dis 51:79–84. doi: 10.1086/653120. [DOI] [PubMed] [Google Scholar]
- 9.Spellberg B, Bonomo RA. 2014. The deadly impact of extreme drug resistance in Acinetobacter baumannii. Crit Care Med 42:1289–1291. doi: 10.1097/CCM.0000000000000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villar M, Cano ME, Gato E, Garnacho-Montero J, Miguel Cisneros J, Ruíz de Alegría C, Fernández-Cuenca F, Martínez-Martínez L, Vila J, Pascual A, Tomás M, Bou G, Rodríguez-Baño J. 2014. Epidemiologic and clinical impact of Acinetobacter baumannii colonization and infection: a reappraisal. Medicine (Baltimore) 93:202–210. doi: 10.1097/MD.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. CDC, Atlanta, GA: http://www.cdc.gov/drugresistance/threat-report-2013. [Google Scholar]
- 12.Sirijatuphat R, Thamlikitkul V. 2014. Preliminary study of colistin versus colistin plus fosfomycin for treatment of carbapenem-resistant Acinetobacter baumannii infections. Antimicrob Agents Chemother 58:5598–5601. doi: 10.1128/AAC.02435-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Napier BA, Burd EM, Satola SW, Cagle SM, Ray SM, McGann P, Pohl J, Lesho EP, Weiss DS. 2013. Clinical use of colistin induces cross-resistance to host antimicrobials in Acinetobacter baumannii. mBio 4:e00021-13. doi: 10.1128/mBio.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottig S, Gruber TM, Higgins PG, Wachsmuth M, Seifert H, Kempf VAJ. 2014. Detection of pan drug-resistant Acinetobacter baumannii in Germany. J Antimicrob Chemother 69:2578–2579. doi: 10.1093/jac/dku170. [DOI] [PubMed] [Google Scholar]
- 15.Boucher HW, Talbot GH, Benjamin DK, Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D, Infectious Diseases Society of America . 2010. 10 x '20 progress: development of new drugs active against resistant Gram-negative bacilli. Infectious Disease Society of America 49th Annual Meeting 2011. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 17.Talbot GH, Bradley J, Edwards JE Jr, Gilbert D, Scheld M, Bartlett JG, Antimicrobial Availability Task Force of the Infectious Diseases Society of America . 2006. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis 42:657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 18.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J Jr, Infectious Diseases Society of America . 2008. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 19.Spellberg B, Powers JH, Brass EP, Miller LG, Edwards JE Jr.. 2004. Trends in antimicrobial drug development: implications for the future. Clin Infect Dis 38:1279–1286. doi: 10.1086/420937. [DOI] [PubMed] [Google Scholar]
- 20.Boucher HW, Talbot GH, Benjamin DK Jr., Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D, Infectious Diseases Society of America . 2013. 10 x '20 progress—development of new drugs active against Gram-negative bacilli. An update from the Infectious Diseases Society of America. Clin Infect Dis 56:1685–1694. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wenzel RP. 2004. The antibiotic pipeline—challenges, costs, and values. N Engl J Med 351:523–526. doi: 10.1056/NEJMp048093. [DOI] [PubMed] [Google Scholar]
- 22.Butler MS, Blaskovich MA, Cooper MA. 2013. Antibiotics in the clinical pipeline in 2013. J Antibiot (Tokyo) 66:571–591. doi: 10.1038/ja.2013.86. [DOI] [PubMed] [Google Scholar]
- 23.Brown ED. 2013. Is the GAIN act a turning point in new antibiotic discovery? Can J Microbiol 59:153–156. doi: 10.1139/cjm-2013-0089. [DOI] [PubMed] [Google Scholar]
- 24.Umland TC, Schultz LW, Macdonald U, Beanan JM, Olson R, Russo TA. 2012. In vivo-validated essential genes identified in Acinetobacter baumannii by using human ascites fluid overlap poorly with essential genes detected on laboratory media. mBio 3:e00113-12. doi: 10.1128/mBio.00113-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rex JH, Goldberger M, Eisenstein BI, Harney C. 2014. The evolution of the regulatory framework for antibacterial agents. Ann N Y Acad Sci 1323:11–21. doi: 10.1111/nyas.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laub MT, Goulian M. 2007. Specificity in two-component signal transduction pathways. Annu Rev Genet 41:121–145. doi: 10.1146/annurev.genet.41.042007.170548. [DOI] [PubMed] [Google Scholar]
- 27.Geisinger E, Isberg RR. 2015. Antibiotic modulation of capsular exopolysaccharide and virulence in Acinetobacter baumannii. PLoS Pathog 11:e1004691. doi: 10.1371/journal.ppat.1004691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stephenson K, Hoch JA. 2004. Developing inhibitors to selectively target two-component and phosphorelay signal transduction systems of pathogenic microorganisms. Curr Med Chem 11:765–773. doi: 10.2174/0929867043455765. [DOI] [PubMed] [Google Scholar]
- 29.Rasko DA, Moreira CG, Li de R, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei S, Roth M, Hughes DT, Huntley JF, Fina MW, Falck JR, Sperandio V. 2008. Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080. doi: 10.1126/science.1160354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang N, Ozer EA, Mandel MJ, Hauser AR. 2014. Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. mBio 5:e01163-14. doi: 10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liou ML, Soo PC, Ling SR, Kuo HY, Tang CY, Chang KC. 2014. The sensor kinase BfmS mediates virulence in Acinetobacter baumannii. J Microbiol Immunol Infect 47:275–281. doi: 10.1016/j.jmii.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Tomaras AP, Flagler MJ, Dorsey CW, Gaddy JA, Actis LA. 2008. Characterization of a two-component regulatory system from Acinetobacter baumannii that controls biofilm formation and cellular morphology. Microbiology 154:3398–3409. doi: 10.1099/mic.0.2008/019471-0. [DOI] [PubMed] [Google Scholar]
- 33.Russo TA, Luke NR, Beanan JM, Olson R, Sauberan SL, MacDonald U, Schultz LW, Umland TC, Campagnari AA. 2010. The K1 capsular polysaccharide of Acinetobacter baumannii strain 307-0294 is a major virulence factor. Infect Immun 78:3993–4000. doi: 10.1128/IAI.00366-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruhn KW, Pantapalangkoor P, Nielsen T, Tan B, Junus J, Hujer KM, Wright MS, Bonomo RA, Adams MD, Chen W, Spellberg B. 2015. Host fate is rapidly determined by innate effector-microbial interactions during Acinetobacter baumannii bacteremia. J Infect Dis 211:1296–1305. doi: 10.1093/infdis/jiu593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lyon GJ, Muir TW. 2003. Chemical signaling among bacteria and its inhibition. Chem Biol 10:1007–1021. doi: 10.1016/j.chembiol.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Roychoudhury S, Zielinski NA, Ninfa AJ, Allen NE, Jungheim LN, Nicas TI, Chakrabarty AM. 1993. Inhibitors of two-component signal transduction systems: inhibition of alginate gene activation in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 90:965–969. doi: 10.1073/pnas.90.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altenhoff AM, Škunca N, Glover N, Train CM, Sueki A, Piližota I, Gori K, Tomiczek B, Müller S, Redestig H, Gonnet GH, Dessimoz C. 2015. The OMA orthology database in 2015: function predictions, better plant support, synteny view and other improvements. Nucleic Acids Res 43:D240–D249. doi: 10.1093/nar/gku1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stock AM, Mottonen JM, Stock JB, Schutt CE. 1989. Three-dimensional structure of CheY, the response regulator of bacterial chemotaxis. Nature 337:745–749. doi: 10.1038/337745a0. [DOI] [PubMed] [Google Scholar]
- 39.Mack TR, Gao R, Stock AM. 2009. Probing the roles of the two different dimers mediated by the receiver domain of the response regulator PhoB. J Mol Biol 389:349–364. doi: 10.1016/j.jmb.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbieri CM, Wu T, Stock AM. 2013. Comprehensive analysis of OmpR phosphorylation, dimerization, and DNA binding supports a canonical model for activation. J Mol Biol 425:1612–1626. doi: 10.1016/j.jmb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bent CJ, Isaacs NW, Mitchell TJ, Riboldi-Tunnicliffe A. 2004. Crystal structure of the response regulator 02 receiver domain, the essential YycF two-component system of Streptococcus pneumoniae in both complexed and native states. J Bacteriol 186:2872–2879. doi: 10.1128/JB.186.9.2872-2879.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bachhawat P, Stock AM. 2007. Crystal structures of the receiver domain of the response regulator PhoP from Escherichia coli in the absence and presence of the phosphoryl analog beryllofluoride. J Bacteriol 189:5987–5995. doi: 10.1128/JB.00049-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozakov D, Grove LE, Hall DR, Bohnuud T, Mottarella SE, Luo L, Xia B, Beglov D, Vajda S. 2015. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc 10:733–755. doi: 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pelkonen S, Häyrinen J, Finne J. 1988. Polyacrylamide gel electrophoresis of the capsular polysaccharides of Escherichia coli K1 and other bacteria. J Bacteriol 170:2646–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kenyon JJ, Hall RM. 2013. Variation in the complex carbohydrate biosynthesis loci of Acinetobacter baumannii genomes. PLoS One 8:e62160. doi: 10.1371/journal.pone.0062160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russo TA, Beanan JM, Olson R, MacDonald U, Luke NR, Gill SR, Campagnari AA. 2008. Rat pneumonia and soft-tissue infection models for the study of Acinetobacter baumannii biology. Infect Immun 76:3577–3586. doi: 10.1128/IAI.00269-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gebhardt MJ, Gallagher LA, Jacobson RK, Usacheva EA, Peterson LR, Zurawski DV, Shuman HA. 2015. Joint transcriptional control of virulence and resistance to antibiotic and environmental stress in Acinetobacter baumannii. mBio 6:e01660-15. doi: 10.1128/mBio.01660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toro-Roman A, Mack TR, Stock AM. 2005. Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the alpha4-beta5-alpha5 face. J Mol Biol 349:11–26. doi: 10.1016/j.jmb.2005.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toro-Roman A, Wu T, Stock AM. 2005. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci 14:3077–3088. doi: 10.1110/ps.051722805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang YT, Gao R, Havranek JJ, Groisman EA, Stock AM, Marshall GR. 2012. Inhibition of bacterial virulence: drug-like molecules targeting the Salmonella enterica PhoP response regulator. Chem Biol Drug Des 79:1007–1017. doi: 10.1111/j.1747-0285.2012.01362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Copeland RA. 2013. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. John Wiley & Sons, Hoboken, NJ. [PubMed] [Google Scholar]
- 52.Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. 2015. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov 14:529–542. doi: 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- 53.Thompson RJ, Bobay BG, Stowe SD, Olson AL, Peng L, Su Z, Actis LA, Melander C, Cavanagh J. 2012. Identification of BfmR, a response regulator involved in biofilm development, as a target for a 2-aminoimidazole-based antibiofilm agent. Biochemistry 51:9776–9778. doi: 10.1021/bi3015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ecker JA, Massire C, Hall TA, Ranken R, Pennella TT, Agasino Ivy C, Blyn LB, Hofstadler SA, Endy TP, Scott PT, Lindler L, Hamilton T, Gaddy C, Snow K, Pe M, Fishbain J, Craft D, Deye G, Riddell S, Milstrey E, Petruccelli B, Brisse S, Harpin V, Schink A, Ecker DJ, Sampath R, Eshoo MW. 2006. Identification of Acinetobacter species and genotyping of Acinetobacter baumannii by multilocus PCR and mass spectrometry. J Clin Microbiol 44:2921–2932. doi: 10.1128/JCM.00619-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Russo TA, Beanan JM, Olson R, MacDonald U, Cox AD, St Michael F, Vinogradov EV, Spellberg B, Luke-Marshall NR, Campagnari AA. 2013. The K1 capsular polysaccharide from Acinetobacter baumannii is a potential therapeutic target via passive immunization. Infect Immun 81:915–922. doi: 10.1128/IAI.01184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Russo TA, MacDonald U, Beanan JM, Olson R, MacDonald IJ, Sauberan SL, Luke NR, Schultz LW, Umland TC. 2009. Penicillin-binding protein 7/8 contributes to the survival of Acinetobacter baumannii in vitro and in vivo. J Infect Dis 199:513–521. doi: 10.1086/596317. [DOI] [PubMed] [Google Scholar]
- 57.Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H, Jamison JJ, MacDonald IJ, Martin KM, Russo T, Campagnari AA, Hujer AM, Bonomo RA, Gill SR. 2008. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J Bacteriol 190:8053–8064. doi: 10.1128/JB.00834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tucker AT, Nowicki EM, Boll JM, Knauf GA, Burdis NC, Trent MS, Davies BW. 2014. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. mBio 5:e01313-14. doi: 10.1128/mBio.01313-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luke NR, Sauberan SL, Russo TA, Beanan JM, Olson R, Loehfelm TW, Cox AD, St Michael F, Vinogradov EV, Campagnari AA. 2010. Identification and characterization of a glycosyltransferase involved in Acinetobacter baumannii lipopolysaccharide core biosynthesis. Infect Immun 78:2017–2023. doi: 10.1128/IAI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 61.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. 2013. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res 41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dallakyan S, Olson AJ. 2015. Small-molecule library screening by docking with PyRx. Methods Mol Biol 1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- 64.Trott O, Olson AJ. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. 2012. Zinc: a free tool to discover chemistry for biology. J Chem Inf Model 52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tropea JE, Cherry S, Waugh DS. 2009. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol 498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 67.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. 2003. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol 142:170–179. doi: 10.1016/S1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 68.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 69.Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. 2005. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr D Biol Crystallogr 61:449–457. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- 70.Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. Phenix: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krissinel E. 2015. Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res 43:W314–W319. doi: 10.1093/nar/gkv314. [DOI] [PMC free article] [PubMed] [Google Scholar]