

Abstract
The clinical correlations linking diabetes with accelerated atherosclerosis, cardiomyopathy, and increased post-MI fatality rates are increasingly understood in mechanistic terms. The multiple mechanisms discussed in this review seem to share a common element: prolonged increases in ROS production in diabetic cardiovascular cells.
Intracellular hyperglycemia causes excessive ROS production. This activates nuclear poly(ADP ribose) polymerase (PARP), which inhibits GAPDH, shunting early glycolytic intermediates into pathogenic signaling pathways. ROS and PARP also reduce sirtuin, PGC1α, and AMPK activity. These changes cause decreased mitochondrial biogenesis, increased ROS production, and disturbed circadian clock synchronization of glucose and lipid metabolism. Excessive ROS production also facilitates nuclear transport of pro-atherogenic transcription factors, increases transcription of the neutrophil enzyme initiating NETosis, PAD4, and activates the NRLP3 inflammasome.
Insulin resistance causes excessive cardiomyocyte ROS production by increasing fatty acid flux and oxidation. This stimulates overexpression of the nuclear receptor PPARα and nuclear translocation of FOXO1, which cause cardiomyopathy. ROS also shift the balance between mitochondrial fusion and fission in favor of increased fission, reducing the metabolic capacity and efficiency of the mitochondrial electron transport chain and ATP synthesis.
Mitochondrial oxidative stress also plays a central role in angiotensin II-induced gap junction remodeling and arrhythmogenesis. ROS contribute to sudden death in diabetics after MI by increasing post-translational protein modifications which cause increased ryanodine receptor phosphorylation and downregulation of SERCA2a transcription. Increased ROS also depress autonomic ganglion synaptic transmission by oxidizing the nAch receptor α3 subunit, potentially contributing to the increased risk of fatal cardiac arrhythmias associated with diabetic cardiac autonomic neuropathy.
Keywords: Diabetes mellitus, insulin resistance, atherosclerosis, heart failure, reactive oxygen species
Subject Terms: Obesity; Cardiovascular Disease; Diabetes, type 2; Vascular Biology; Myocardial Biology; Pathophysiology; Metabolism; Oxidant Stress
INTRODUCTION
We are in the midst of a global diabetes epidemic. Since 1985, the number of people with diabetes has increased from 30 million to nearly 400 million, and the number of people with diabetes is increasing rapidly in every country. Predictably, as the number of people with diabetes has increased, a world-wide epidemic of diabetic complications has followed. Cardiovascular disorders are by far the leading cause of death in people with diabetes, reducing the median life expectancy for diabetic adults aged 55–64 years by 8 years1. Unlike microvascular complications, which are unique to diabetes, diabetic cardiovascular disorders are clinically similar to cardiovascular diseases in people without diabetes. However, there are important differences with major clinical implications.
First, diabetes causes accelerated atherosclerosis, with greater inflammatory infiltrate (macrophages and T lymphocytes), larger necrotic core size, and more diffuse atherosclerosis in the coronary arteries2. In the general U.S. population, deaths due to coronary artery disease have declined substantially over the past decades in the general population. In contrast, in people with diabetes the reduction in deaths due to CAD has been much less dramatic3. Second, diabetes increases both diastolic heart failure with preserved ejection fraction (HFpEF) and systolic heart failure with reduced ejection fraction (HFrEF). Even after adjustment for standard risk factors, diabetes increases heart failure risk four-fold. Contributing factors include diabetes-induced cardiomyocyte dysfunction (cardiomyopathy), impaired microvascular perfusion due to defective endothelial function, increased collagen deposition with fibrosis, and maladaptive remodeling after myocardial infarction, leading to both diastolic and systolic heart failure4. Third, diabetes increases both early (30 days) and late (1 yr) post-MI fatality rates5. The fatality rate for people with diabetes is nearly twice the rate for people without diabetes at both time points. A major cause of post-MI mortality is ventricular arrhythmia.
Diabetes-induced downregulation of SERCA2a transcription and increased phosphorylation of the ryanodine receptor by activated CaMKII6, 7 increase intracellular Ca++, contributing to potentially fatal arrhythmias such as premature ventricular complexes and delayed afterdepolarizations, and activation of a mitochondrial/oxidized-CaMKII pathway8 contributes to increased sudden death in diabetics after myocardial infarction. Cardiac autonomic neuropathy, present in almost 50% of patients with diabetes and CAD, is associated with a significantly increased risk for fatal cardiac arrhythmias9.
BIOCHEMICAL, MOLECULAR, AND CELLULAR MECHANISMS
Conceptual Overview
Virtually all the data to be discussed in this section comes from studies of murine models and of cultured cells. Murine models are valuable tools for defining the pathogenesis of diabetic cardiovascular disorders. They have significant limitations, however, and it is important to recognize some of their limits. First, no diabetic animal model, regardless of genetic background, recapitulates the structural and functional alterations of human diabetic cardiovascular disease. Rodent models of diabetes do not develop coronary atherosclerosis with complex plaque formation and plaque rupture, nor do they develop the degree of fibrosis seen in human hearts with late stage diabetes-associated heart failure. Second, most of the mechanistic data currently available comes from studies of the earliest stages of each complication. Murine atherosclerosis, for example, is limited to foam cell accumulation and fatty streaks in a very restricted anatomical distribution. Mechanisms dominant in the pathogenesis of fibrous plaques, complicated lesions, and plaque rupture are likely quite different from those dominant in fatty streak formation. Cultured cell experiments also have important limitations. Cells from species that do not develop diabetic cardiovascular disease are unlikely to respond to hormonal and metabolic perturbations in the same way as human cells. Immortalized human cell lines metabolize glucose and other relevant substrates quite differently from primary human cells. Even primary human cell studies are limited by the lack of important interactions with other cell types within and between different tissues that occur in human cardiovascular disease. For example, the development of human atherosclerosis involves cross-talk among endothelial, smooth muscle, inflammatory and phagocytic cells in the arterial wall, as well as interactions with cells whose functions are altered by diabetes in the bone marrow, visceral fat, and liver. Despite these limitations, however, there has been enormous recent progress in understanding the biochemical, molecular, and cellular mechanisms involved in the pathogenesis of diabetic cardiovascular disorders. Many of these are described in several excellent recent reviews10–12.
Hyperglycemia and insulin resistance are the two major consequences of diabetes responsible for cardiovascular disorders in patients with diabetes, and the mechanisms associated with each are presented in the following sections. The multiple mechanisms discussed in this review seem to share a common element: prolonged increases in ROS production in diabetic cardiovascular cells. Physiologic levels of ROS (H2O2) are signaling molecules essential for normal cardiovascular cell homeostasis. However, ROS production at too high a level, for too long, or at an inappropriate subcellular location, leads to impaired cellular function and cardiovascular pathology.
Mechanisms of Hyperglycemia-Induced Cardiovascular Damage
Increased aldose reductase (AKR1B1) substrate conversion
Aldose reductase (AKR1B1), a member of the large aldo-keto reductase superfamily, catalyzes the reduction of a wide variety of hydrophobic and hydrophilic carbonyl-containing compounds– including glucose and several glycolytic intermediates– to their corresponding alcohols. This enzyme is cytosolic, and requires NADPH as a co-factor. In some cell types, where glucose is converted to the sugar alcohol sorbitol, sorbitol is then converted to fructose by another enzyme, sorbitol dehydrogenase, using NAD+ as a cofactor. This series of reactions, termed the polyol pathway, has been implicated in the pathogenesis of several diabetic complications including diabetic cardiovascular disease13. In diabetic apolipoprotein E knockout mice, overexpression of human aldose reductase (hAR) accelerated atherosclerosis and pharmacological inhibition of the enzyme prevented this14. In the same model, diabetes caused sustained activation of the C2H2-type zinc-finger transcription factor Egr-1, with subsequent induction of its downstream targets, the pro-thrombotic protein tissue factor (TF) and the pro-inflammatory vascular cell adhesion molecule-1 (VCAM-1)15. In contrast, in mice expressing physiologic levels of aldose reductase, knockout or pharmacological inhibition of the enzyme unexpectedly caused increased early atherosclerotic lesion size in both diabetic and non-diabetic mice16. Differences in total enzyme activity, cofactor levels, and levels of alternative intracellular substrates, coupled with known differences in enzyme kinetics for different substrates, likely explain these seemingly paradoxical observations.
Increased intracellular formation of the major AGE-precursor methylglyoxal
The post-translational modifications of proteins called Advanced Glycation Endproducts (AGEs) are formed by glucose-derived dicarbonyls reacting with amino groups of unprotonated lysine and arginine residues of proteins. Methylglyoxal, formed by the non-enzymatic fragmentation of the glycolytic intermediate triose-phosphate, accounts for the majority of hyperglycemia-induced increase in AGE adducts in diabetic tissues17. Intracellular methylglyoxal is detoxified by the glyoxalase system18. The enzyme glyoxalase I, together with glyoxalase II and a catalytic amount of glutathione, reduces this highly reactive α-oxoaldehyde to D-lactate. In cells, methylglyoxal reacts with unprotonated arginine residues to form the major methylglyoxal-derived epitope MG-H1. Intracellular production of AGE precursors damages target cells by three general mechanisms. First, AGE modification of intracellular proteins changes their function. Second, AGE-modification of extracellular matrix components alters their interaction with other matrix components and with integrin matrix receptors. Third, intracellular methylglyoxal increases expression of both the pattern recognition receptor RAGE (Receptor for AGEs) and its major endogenous ligands, the proinflammatory S100 calgranulins19. Ligation of these ligands with RAGE causes cooperative interaction with the innate immune system signaling molecule toll-like receptor 4 (TLR4)20. Expressions of RAGE, S100A8, S100A12, and HMGB1 are all increased by high levels of glucose in cell culture and in diabetic animals. This hyperglycemia-induced overexpression is mediated by ROS-induced increases in methylglyoxal, which increase binding of the transcription factors NFκB and activator protein 1 (AP-1) to the promoters of RAGE and of RAGE ligands, respectively19.
Recent work has identified increased methylglyoxal as an important element in the pathogenesis of both diabetic atherosclerosis and diabetic cardiomyopathy. In non-diabetic Apo E null mice, increasing plasma methylglyoxal levels to diabetic levels using a Glo1 inhibitor caused endothelial inflammation and atherogenesis similar to that induced by diabetes21. In human atherosclerotic plaques, MG-H1 levels were associated with rupture-prone plaques having increased levels of the inflammatory mediators IL-8 and MCP-1, and higher MMP-9 activity. MG-H1 was primarily found in lesion macrophages surrounding the necrotic core, and co-localized with cleaved caspase-322. In the diabetic heart, methylglyoxal preferentially reacts with both ryanodine receptor 2, the major myocardial intracellular mediator of calcium-induced calcium release, and with sarco-endoplasmic reticulum Ca++-ATPase (SERCA2a), which is responsible for the synchronized reuptake of released intracellular calcium23. This coordinated process of calcium cycling is critical for efficient cardiac contractions, and diabetes-induced defects caused by increased methylglyoxal adduct formation and increased O-GlcNAcylation likely contribute to impaired systolic function. Increased methylglyoxal production also appears to be responsible for poor cardiac stem cell-mediated repair and angiogenic capacity24.
Cardiac stem cells from biopsies of hearts from human diabetics were less able to repair post-infarction damage in immunodeficient mice than cardiac stem cells from non-diabetic patients, and conditioned medium from these cells had less angiogenic capacity. Culture of non-diabetic murine cardiac stem cells in high glucose induced the same cardiac repair and angiogenesis defects seen in human diabetic cells. In both human and mouse cells, overexpression of glyoxalase 1 restored the angiogenic defects24. In diabetic mice with defective post-ischemia hind limb revascularization, overexpression of the methylglyoxal-metabolizing enzyme glyoxalase-1 (GLO1) exclusively in bone marrow cells (BMCs) was sufficient to restore BMC function and neovascularization of ischemic tissue in diabetes25.
Increased methylglyoxal also activates the unfolded protein response (UPR) in cardiomyocytes26. While transient activation of the UPR relieves ER stress, prolonged activation of the UPR in CVD triggers apoptosis, mediated by the downstream effector CHOP (C/EBP-homologous protein). CHOP plays a critical role in macrophage apoptosis, a process involved in plaque necrosis in advanced atheromata. In Chop−/−Apoe −/− mice, lesion area plaque necrosis was reduced by 50%. In high fat fed Apo E −/− and LDL receptor −/− mice, CHOP promoted plaque growth, apoptosis, and plaque necrosis27. In cardiomyocytes, methylglyoxal also induces apoptosis via CHOP26. Infusion of methylglyoxal in non-diabetic mice induced cardiomyocyte apoptosis, inflammation, and a significant reduction in LV fractional shortening and LV ejection fraction. Each of these adverse effects was prevented in CHOP−/− mice. In the hearts of diabetic mice, overexpression of GLO1 in the vasculature (Vuleseveic, B. et al., Diabetes 2016, in press) prevented diabetes-induced reduction of myocardial capillary density, increased apoptosis, and loss of cardiac function. Neuregulin production, which transduces signals between the heart’s microvasculature and cardiomyocytes28, eNOS dimerization, and Bcl-2 expression were also maintained in the diabetic GLO1 transgenic hearts.
Activation of protein kinase C β, δ, and θ
Protein kinase C is a family of protein kinase enzymes with 15 isoforms that are involved in the regulation of protein function. Nine of these 15 PKC isoforms are activated by a lipid second-messenger, diacylglycerol (DAG). Elevated intracellular glucose levels increase DAG levels in a variety of diabetic target tissues, including arterial smooth muscle cells and cardiomyocytes29–31 by de novo synthesis. Hyperglycemia primarily activates the β and δ isoforms of PKC, but increases in activity of several other isoforms have also been found. These PKC isoforms can also be activated by intracellular ROS in the absence of DAG or Ca++. The regulatory domain of these PKC isoforms contains two pairs of zinc fingers with six cysteine residues and two zinc atoms, which can be oxidized by intracellular ROS. Oxidation alters zinc finger conformation and activates PKC32. Many cellular abnormalities involved in diabetic cardiovascular disease have been linked to PKC activation. These include endothelial dysfunction, increased vascular permeability, impaired angiogenesis and increased apoptosis. Molecular mechanisms affected by diabetes-induced PKC activation include alterations in functionally significant enzymatic activities such as mitogen-activated protein kinase (MAPK), cytosolic phospholipase A2, and Na+–K+–ATPase, and alterations in several transcription factors33.
Hyperglycemia-induced activation of PKCβ promotes vascular inflammation and acceleration of atherosclerosis in diabetic ApoE null mice by augmenting expression of inflammatory mediators. In addition, it increased macrophage expression of CD11c (integrin, alpha X complement component 3 receptor 4 subunit), chemokines (C-C motif) ligand 2), and interleukin-1β via increased extracellular signal-regulated kinase 1 and 2 (ERK1/2) and Jun-N-terminus kinase-mitogen-activated kinase (JNK kinase)34. In this same diabetic model, PKCβ activation increased transcription of the proinflammatory cytokine IL-18 and inhibited transcription of IL-18-binding protein in the aorta. Diabetic mice showed increased plaque formation, cholesteryl ester content and macrophage infiltration. Treatment with a PKCβ inhibitor prevented these35. PKC-β2 in endothelial cells from transgenic ApoE null mice overexpressing PKCβ decreased insulin-stimulated Akt/eNOS activation and increased basal and angiotensin-induced expression of the vasoconstrictor endothelin-1. These dual effects increased endothelial dysfunction and accelerated atherosclerosis in this model compared with apoE−/− mice36.
PKC activity has also been linked to myocardial dysfunction, causing cardiomyopathy and cardiac failure. Ruboxistaurin, a PKC inhibitor, improved the metabolic gene profile and reduced PKC activity in diabetic hearts without altering levels of circulating metabolites. Selective overexpression of PKC-β2 in the myocardium of diabetic mice increased expression of connective tissue growth factor (CTGF) and TGF-β1, cardiomyopathy and cardiac fibrosis37. More recently, activation of PKCα/β in diabetic hearts has been shown to mediate reactivation of fetal splicing programs in diabetic hearts by phosphorylation and upregulation of the RNA binding proteins CELF1 and Rbfox238. Chronic activation of PKC isozymes α, β, and δ promotes diastolic and systolic dysfunction, fibrosis, cardiomyocyte hypertrophy, and apoptosis39. Another PKC isoform, PKC θ, plays crucial roles in the proliferation, differentiation and activation of mature T-cells via activation of several transcription factors in the nuclei of T-cells, including NFAT, c-Jun, c-Fos and AP-1. Diabetes-induced cardiac interstitial fibrosis, reduced contractility, reduced expression of the tight junction maintaining protein ZO-1, and T-cell infiltration were all improved by treatment with an isoform-specific PKC θ inhibitor.
Increased protein modification by O-GlcNAc
The hexosamine pathway causes reversible post-translational modification of intracellular protein serine and threonine residues by N-acetylglucosamine. In cells damaged by hyperglycemia, excess intracellular glucose provides increased fructose-6-phosphate (Fruc-6-P) which is converted to glucosamine 6-phosphate (Glc-6-P) by the rate limiting enzyme glutamine:fructose-6-phosphate amidotransferase (GFAT). Glucosamine-6-phosphate (Glc-6-P) is further converted to N-acetylglucosamine-6-phosphate (GlcNAc-6-P) and finally to UDP-N-acetylglucoseamine (UDP-GlcNAc). The enzyme O-GlcNAc transferase (OGT) uses UDP-GlcNAc to transfer N-acetylglucosamine to a variety of proteins, resulting in increased protein modification by N-acetylglucosamine. Another enzyme, N-acetylglucosaminidase (O-GlcNAcase or OGA), removes this protein modification. Alternative splicing of the genes encoding the O-linked GlcNAc cycling enzymes OGT and OGA yields isoforms targeted to discrete sites in the nucleus, cytoplasm, and mitochondria. O-GlcNAc serves as a nutrient/stress sensor regulating cellular homeostasis by altering signaling, transcription, metabolism, organelle biogenesis, cytoskeletal dynamics and apoptosis40, 41.
The role of the hexosamine pathway in CVD has been reviewed recently42. Studies have linked chronically elevated O-GlcNAc levels to diabetic cardiovascular complications. Adverse cardiac effects of chronically increased O-GlcNAcylation include decreased mitochondrial function, decreased autophagic signaling, and decreased contractile function. In mouse coronary endothelial cells (MCECs) isolated from diabetic mice, O-GlcNAcase protein expression was significantly decreased compared with control MCECs. In contrast, OGT protein expression was markedly increased43. The resultant increased protein modification by O-GlcNAc was responsible for decreased endothelium-dependent relaxation of the coronary arteries and reduced capillary density in the left ventricle. Both of these defects were restored by overexpression of O-GlcNAcase.
Decreased endothelium-dependent relaxation of the coronary arteries and reduced capillary density both reflect inhibition of endothelial nitric oxide synthase (eNOS), which is required for endothelium-dependent arterial relaxation and for mobilization of stem and progenitor cells from the bone marrow compartment44. In human arterial endothelial cells, activation of eNOS by phosphorylation at Serine 1177 is inhibited directly by hyperglycemia-induced O-GlcNAcylation at this site45, and indirectly by reduced insulin-stimulated phosphatidylinositol 3-kinase (PI3-K) and Akt activity by O-GlcNAc modification. eNOS activity is also affected by several other post-translational modifications, but the effect of diabetes on these has not yet been determined46. Carotid plaques from diabetic patients have a marked increase of O-GlcNAcylation in both cytoplasm and nuclear compartments of endothelial cells compared with non-diabetic subjects47. Increased O-GlcNAcylation may also contribute to diabetic accelerated atherosclerosis by increasing ubiquitination and proteasomal degradation of the anti-inflammatory NF-κB inhibitory protein A20 in coronary endothelial and smooth muscle cells48.
Chronically elevated O-GlcNAc levels also adversely affect myocardial function. Ventricular contraction and relaxation are controlled mainly by release and uptake of Ca2+ by the sarcoplasmic reticulum Ca++-ATPase (SERCA2) pump. In hypertrophied and failing myocardium, SERCA2 protein level and its Ca2+ uptake function are depressed. Overexpression of O-GlcNAc-transferase (OGT) significantly reduced transcription of SERCA2, causing decreased calcium re-uptake and impaired diastolic relaxation49. High glucose also increased O-GlcNAc modification of the calcium/calmodulin-dependent protein kinase IIδ (CaMKIIδ), an enzyme critical for Ca2+homeostasis and reuptake in cardiomyocytes. O-GlcNAc-modified CaMKII at Ser 279 is increased in the heart of diabetic humans and rats7, causing autonomous activation of CaMKII. Thus, CaMKII remains activated even after intracellular Ca++ declines. This contributes to decreased cardiac contractility and potentially fatal arrhythmias such as premature ventricular complexes and delayed afterdepolarizations (Figure 1). Delayed afterdepolarizations are associated with the initiation of long QT-interval arrhythmias such as torsade de pointes. Overexpression of GlcNAcase or inhibition of GlcNAc modification increased expression of SERCA2a, ablated sarcoplasmic reticulum Ca++ leak, improved cardiac contractility, and reduced arrhythmic events. Increased levels of ROS also cause autonomous activation of CaMKII by oxidation of adjacent methionine residues in its regulatory domain50. Activation of this mitochondrial ROS-oxidized CaMKII pathway increased mortality after myocardial infarction in diabetic mouse models8.
Figure 1. Hyperglycemia-induced myocardial protein modification by O-GlcNAc causes increased intracellular Ca++ and delayed afterpolarizations.
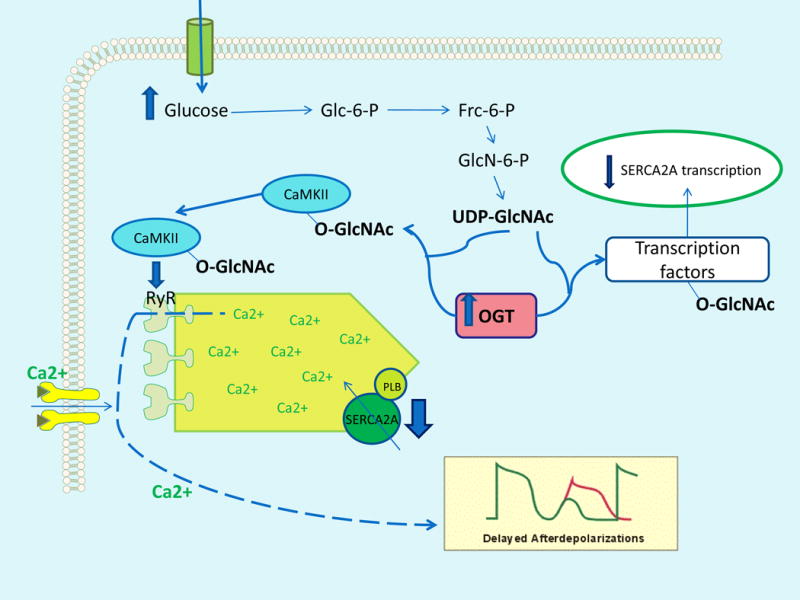
Increased intracellular glucose flux provides more substrate for the enzyme O-GlcNAc-transferase (OGT). This increases O-GlcNAc modification of calcium/calmodulin-dependent protein kinase IIδ (CaMKII), causing autonomous CaMKII activation. CaMKII increases intracellular Ca++ by phosphorylating ryanodine receptor 2 (RyR). OGT also modifies transcription complex factors regulating expression of sarcoplasmic reticulum Ca2+-ATPase (SERCA2), reducing SERCA2A expression and contributing toincreased intracellular Ca++. Increased O-GlcNAc modification of these proteins causes delayed afterdepolarizations in cardiomyocytes. PLB, phospholamban.
Mitochondrial O-GlcNAc transferase (OGT) is increased in diabetic cardiac mitochondria, while O-GlcNAcase (OGA) is reduced, causing increased O-GlcNAcylation of cardiac mitochondrial proteins. Inhibition of OGA and the resulting increased mitochondrial protein modification by O-GlcNAc increases oxygen consumption and reduces reserve capacity6. Reduced bioenergenic reserve capacity makes cells more sensitive to stress and cell death.
Different hyperglycemia-induced pathogenic mechanisms reflect a single upstream process: overproduction of ROS
A single upstream hyperglycemia-induced process — overproduction of superoxide by the mitochondrial electron transport chain—activates all four mechanisms described in the previous sections51, 52 (Figure 2). Enhanced intracellular glucose transport and oxidation leads to mitochondrial overproduction of superoxide52–54. This can, in turn, activate other superoxide production pathways that may amplify the original damaging effect of hyperglycemia55. Examples of amplification mechanisms include ROS-mediated uncoupling of eNOS dimers to eNOS monomers in endothelial cells, activation of various NADPH oxidase isoforms in cardiovascular cells, and increased mitochondrial fission mediated by the rho-associated protein kinase ROCK156, 57. The initiating role of mitochondrial ROS is suggested by the observation that cells lacking mitochondrial electron transport chain function (ρ0 cells)53 fail to increase ROS production in response to high glucose.
Figure 2. Four hyperglycemia-induced pathogenic mechanisms are activated by overproduction of ROS.
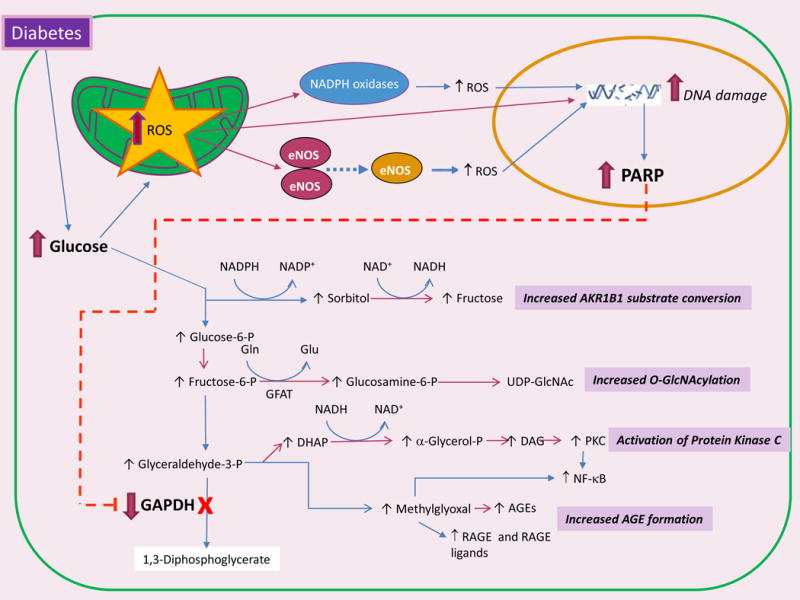
Increased intracellular glucose flux causes mitochondrial overproduction of reactive oxygen species (ROS), which can further amplify ROS production by activating NADPH oxidases and uncoupling eNOS. Stable ROS species diffuse into the nucleus, where they cause DNA damage and activation of poly(ADP ribose) polymerase (PARP). PolyADP-ribosylation of glyceraldehyde-3-dehydrogenase (GAPDH) by PARP reduces GAPDH activity, which causes upstream accumulation of early glycolytic intermediates which are diverted into four pathogenic signaling pathways. AKR1B1, aldose reductase; Gln, glucosamine; GFAT, glutamine fructose-6-phosphate amidotransferase; UDP-GlcNAc, uridine diphosphate N-acetylglucosamine; DHAP, dihydroxyacetone phosphate; DAG, diacylglycerol; PKC, protein kinase C; NF-κB, nuclear factor-κB; AGEs, advanced glycation end-products; RAGE, Receptor for Advanced Glycation Endproducts.
In mitochondria, increased superoxide causes the release of Fe2+ from ferritin and iron sulfur cluster–containing proteins. Interaction of this released free iron with diffused mitochondrial superoxide-derived hydrogen peroxide forms hydroxyl radicals, the only ROS species capable of cleaving bonds in macromolecules58. This results in ROS-mediated DNA double strand breaks in the nucleus which activate DNA repair mechanisms, including the enzyme poly(ADP ribose) polymerase 1 (PARP1). Activation of PARP1 inhibits the key glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by polyADP-ribosylation, and depletes intracellular NAD+ by degrading it to ADP-ribose and nicotinamide. Inhibition of GAPDH activity causes upstream accumulation of early glycolytic intermediates which are diverted into the four pathogenic signaling pathways52, 59. Diversion of glucose increases polyol pathway flux, while diversion of fructose-6-phosphate increases hexosamine pathway activity. Diversion of glyceraldehyde-3-phosphate to α-glycerol phosphate activates PKC, and reduced activity of GAPDH diverts glyceraldehyde-3-phosphate to the highly reactive α-dicarbonyl methylglyoxal, which increases expression of the receptor for advanced glycation end-products (RAGE) and its activating ligand S100A8/9. Together, these diversions and pathway activations lead to cellular dysfunction, inflammation, apoptosis, and fibrosis in cells exposed to excessive glucose flux. The central importance of ROS in initiating each of these processes is illustrated by the fact that each can be prevented when hyperglycemia-mediated ROS generation is curtailed by transgenic expression of the enzyme superoxide dismutase52, 59.
In arteries from patients undergoing coronary artery bypass surgery, the levels and activity of NAD(P)H oxidase protein subunits (p22phox, p67phox, and p47phox) were significantly increased. The endothelium was an additional net source of superoxide production because of dysfunctional endothelial NO synthase60. Overexpression of the mitochondrial isoform of SOD also prevents hyperglycemia-induced inhibition of the antiatherogenic enzyme prostacyclin synthase53. In diabetes, inhibition of prostacyclin synthase causes the common prostacyclin and thromboxane precursor prostaglandin H2 to be shunted towards thromboxane synthesis. Activation of thromboxane receptors triggers vasoconstriction, platelet aggregation, increased expression of leukocyte adhesion molecules, and apoptosis61. In mice overexpressing the mitochondrial isoform of the superoxide scavenging enzyme catalase (mCAT) in macrophages, lesional macrophage accumulation was successfully suppressed, causing a significant reduction in lesional area. The mCAT lesions had fewer monocyte-derived cells, fewer Ly6c(hi) monocyte infiltration into lesions, and lower levels of monocyte chemotactic protein-1(MCP-1). The decrease in lesional MCP-1 was associated with the suppression of other markers of inflammation and with decreased phosphorylation of RelA (NF-κB p65), indicating decreased activation of the proinflammatory NF-κB pathway. Thus, mitochondrial overproduction of ROS in lesional macrophages amplifies atherosclerotic lesion development by promoting NF-κB-mediated entry of monocytes and other inflammatory processes62. Nox4 expression and activity are also increased in cardiomyocytes exposed to high glucose63 and in the heart of diabetic mouse models64. Transgenic overexpression of the antioxidant enzymes Mn-SOD and catalase reduced ROS, and prevented diabetes-induced abnormalities in cardiac contractility in an animal model of diabetic cardiomyopathy65, 66.
Physiologic ROS production is essential for normal intracellular signaling
In normal cardiovascular physiology, ROS production is coupled to circadian clocks and metabolic networks, and ROS species (H2O2) function as signaling molecules essential for normal cellular homeostasis67, 68. Physiologic ROS (H2O2) signaling is essential for normal intracellular communication, cell differentiation, autophagy, response to insulin and growth factor stimulation, and the generation of physiologic inflammatory responses67. Enhanced production of H2O2 from a mitochondrial source of superoxide is observed when flow rate is increased in human coronary resistance vessels. Hydrogen peroxide (H2O2) hyperpolarizes and dilates human coronary arterioles through opening of Ca2+-activated K+ channels. Catalase, a scavenger of H2O2, greatly inhibited this flow-induced dilation69. Similarly, H2O2 exerted a beneficial effect on vasodilator function and reduced blood pressure in transgenic mice with endothelium-targeted Nox4 overexpression70.
In the heart, low levels of hydrogen peroxide induce proliferation of mouse embryonic stem (ES) cells as well as neonatal cardiomyocytes, and ROS induce expression of cardiac-specific genes, transcription factors and growth factors in ES cells. These effects are dampened by free radical scavengers. ROS also act as transducers of mechanical strain-induced cardiovascular differentiation of embryonic stem cells (ES)71. ROS-dependent activation of integrins and subsequent induction of PI3K/Akt signaling are also involved in cyclic strain-mediated cardiomyogenesis72. Localized ROS production appears to play a role in stretch-induced augmentation of cardiac contractile activity as well73. Physiologic excitation-contraction coupling in heart muscle may also involve ROS signaling. Prosser et al. showed that physiologic stretch rapidly activates NOX2 located on sarcolemmal and t-tubule membranes in cardiomyocytes. The local ROS produced sensitizes ryanodine receptors (RyRs) in the sarcoplasmic reticulum (SR). This triggers a burst of Ca2+ sparks, thereby increasing Ca2+ signaling sensitivity in healthy cardiomyocytes.
Thus, ROS production at an inappropriate place or time, for too long, at too high a level or of inappropriate forms lead to impaired cellular function and pathological gain of function, while ROS production at the right time, place, level, and duration plays a crucial role in physiological homeostasis74.
Decreased Nrf2 activity
Mechanisms which can damage the cardiovascular system are normally counterbalanced by protective mechanisms that maintain homeostasis. In diabetes, however, the hyperglycemia, quantitative and qualitative changes in lipids, and insulin resistance which together promote tissue injury often inhibit these protective mechanisms39. The number of recognized ROS-regulating enzymes has increased in recent years74. Examples include superoxide dismutases, catalases, glutathione peroxidases, glutathione reductase, thioredoxins, thioredoxin reductases, methionine sulfoxide reductases, and peroxiredoxins74. The activity of these enzymes is largely determined by ROS-induced changes in their transcription. Increased transcription of many of these antioxidant enzymes is mediated by the transcription factor Nrf2 (nuclear erythroid–related factor 2), a member of the cap ‘n’ collar (CNC) subfamily of basic region leucine zipper (bZip) transcription factors75. By regulating oxidant levels and oxidant signaling, Nrf2 participates in the control of inflammasome signaling, the unfolded protein response, apoptosis, mitochondrial biogenesis and stem cell regulation.
Nrf2 also increases transcription of glyoxalase 1 (Glo1), the rate-limiting enzyme of the glyoxalase system which prevents posttranslational modification of proteins by methylglyoxal, the major AGE precursor76. It also increases transcription of the rate-controlling enzyme in the non-oxidative branch of the pentose phosphate pathway, transketolase. Activation of transketolase by the lipid-soluble thiamine derivative benfotiamine inhibits three of the major hyperglycemia-driven pathways implicated in the pathogenesis of vascular diabetic vascular damage (the DAG-protein kinase C (PKC) pathway, the methylglyoxal-advanced glycation end product (AGE) formation pathway, and the hexosamine pathway) and inhibits hyperglycemia-induced NFκB activation77. Transketolase activation by benfotiamine also prevented high glucose-induced arterial endothelial cell death78, improved diastolic and systolic function, prevented left ventricular end-diastolic pressure increase and chamber dilatation, improved cardiac perfusion, and reduced cardiomyocyte apoptosis and interstitial fibrosis79.
Nrf2 is expressed constitutively, and its intranuclear levels are controlled post-translationally. In the absence of inducers, Nrf2 associates with the redox-sensitive protein Keap1 (Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1), where it is rapidly polyubiquinated by Keap1-associated cullin-3 (Cul3)–RING E2 ubiquitin ligase proteins and degraded by proteasomes. ROS oxidation of critical cysteine thiols of Keap1 or the reaction of these thiols with ROS-generated electrophiles such as methylgloxal from glycolysis-derived triose phosphate and the lipid peroxidation product 4-hydroxynonenal causes the release of bound Nrf2 protein. Phosphorylation of Nrf2 by protein kinases such as CK2 may help target Nrf2 to the nucleus. After forming heterodimers with small Maf proteins, Nrf2 binds to the antioxidant response element (ARE) to induce transcription of its target genes. Export of Nrf2 from the nucleus is controlled by phosphorylation75. Src family members such as Fyn phosphorylate Nrf2 at Tyr568, causing export from the nucleus and degradation80. Reduction of Nrf2 protein in the cytosolic compartment is mediated by β-transducin repeat-containing protein (β-TrCP), a substrate adaptor for the S-phase kinase-associated protein-1 (Skp1)–Cul1–F-box protein (SCF) E3 ubiquitin ligase which targets Nrf2 phosphorylated byGSK3β to the proteosome81 (Figure 3).
Figure 3. Diabetes reduces Nrf2 protein in diabetic heart.
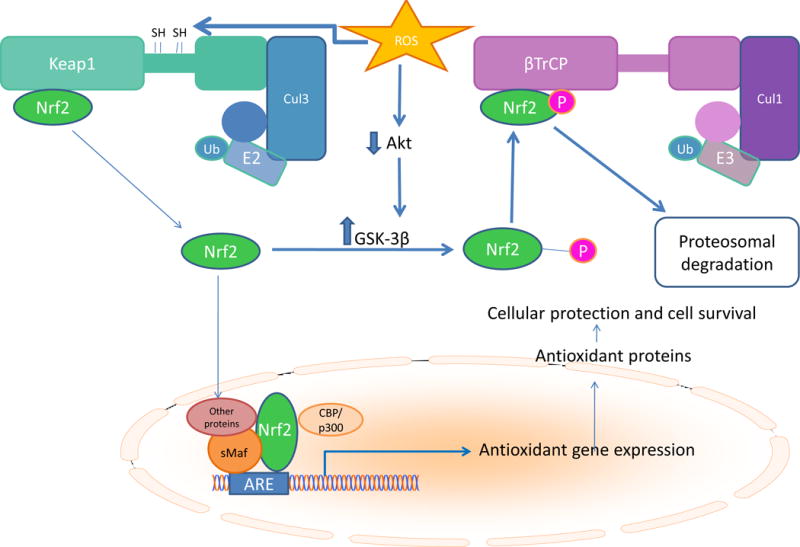
Nuclear erythroid–related factor 2 (Nr2), the master regulator of antioxidant gene expression, associates with the redox-sensitive protein Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1(Keap1), where it is rapidly polyubiquinated by Keap1-associated cullin-3 (Cul3)–RING E2 ubiquitin ligase proteins and degraded by proteasomes. ROS oxidation of critical cysteine thiols of causes release of bound Nrf2 protein. Phosphorylation of Nrf2 by protein kinases such as CK2 help target Nrf2 to the nucleus. Nrf2 forms heterodimers with small Maf proteins which bind to the antioxidant response element (ARE) in its target gene promoters. After export of Nrf2 from the nucleus, cytosolic Nrf2 is phosphorylated by GSK3β. This phosphorylated Nrf2 recognized by β-transducin repeat-containing protein (β-TrCP), a substrate adaptor for the S-phase kinase-associated protein-1 (Skp1)–Cul1–F-box protein (SCF) E3 ubiquitin ligase which targets Nrf2 phosphorylated by GSK3β to the proteosome.
Modification of critical Keap1 cysteine thiols by the dietary isothiocyanate sulforaphane also releases Nrf282. In endothelial cells, sulforaphane prevented hyperglycemia-induced activation of the hexosamine and PKC pathways and prevented increased cellular accumulation and excretion of the major AGE precursor methylglyoxal. In the aortae of diabetic mice, sulforaphane treatment restored aortic levels of Nrf2 and Nrf2-dependent antioxidant gene expression, preventing diabetes-induced increases in wall thickness, fibrosis, inflammation (TNFα and VCAM-1 expression), apoptosis, and increased cell proliferation83. Diabetic cardiomyopathy was also prevented in mouse models by sulforaphane treatment84. Restoration of Nrf2 activity prevented these, as well as preventing diabetes-associated inflammation, fibrosis, and cardiac lipid accumulation. In addition, decreased autophagy was restored.
In hearts from diabetic patients, Nrf2 protein is dramatically reduced85. In mice, cardiac Nrf2 protein was similarly reduced after 5 months of diabetes85. What mechanisms are responsible for this long-term decrease in Nrf2 protein content and function in diabetic heart? Since hyperglycemia-induced mitochondrial overproduction of ROS has been shown to increase GSK3β activity by inhibiting Akt1-dependent phosphorylation of GSK3β at Serine 958, increased proteasomal degradation of cytosolic Nrf2 phosphorylated by GSK3β mediated by the β-transducin repeat-containing protein (β-TrCP)/Skp1−Cul1−F-box protein (SCF) E3 ubiquitin ligase is currently the only mechanism supported by experimental data81.
NFAT activation
The transcription factor Nuclear Factor of Activated T Cells (NFAT) has been implicated in the development of diabetic cardiovascular complications. Many of the cell types involved in diabetic cardiovascular disease express one or more of the four calcium-dependent NFAT isoforms, NFATc1-NFATc4. In resting cells, NFAT proteins are phosphorylated and are located in the cytoplasm. In diabetes, intracellular calcium is increased by increased ROS. Increased intracellular calcium then activates NFAT by increasing NFAT dephosphoryation by the Ca2+/calmodulin-dependent serine/threonine phosphatase calcineurin, which facilitates NFAT translocation into the nucleus. Once in the nucleus, NFAT interacts with coregulators to achieve optimal NFAT activation86. In the nucleus, ADP-ribosylation mediated by PARP-1 acts as a molecular switch to positively regulate NFAT-dependent cytokine gene transcription87. In diabetes, nuclear PARP-1 is activated by intracellular hyperglycemia via ROS-induced DNA strand breaks (Figure 4).
Figure 4. Increased mitochondrial oxidation of glucose or fatty acids activates NFAT-mediated transcription of genes promoting diabetic atherosclerosis and heart failure.
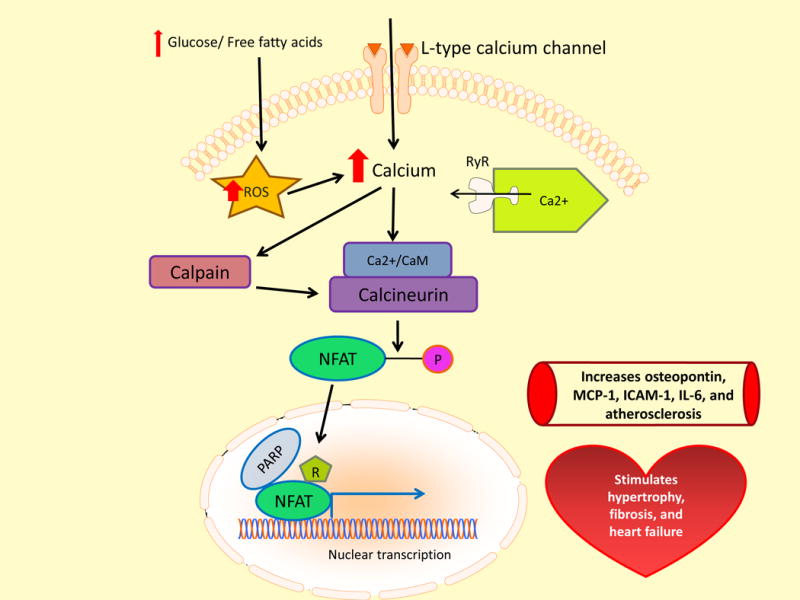
Mitochondrial overproduction of ROS causes increased intracellular Ca++, which activates the calcium-activated neutral cysteine protease calpain. Calpain then activates the Ca2+/calmodulin-dependent /CaM) (Ca2+)serine/threonine phosphatase calcineurin. Dephosphorylation facilitates nuclear translocation of the transcription factor Nuclear Factor of Activated T Cells (NFAT). In the nucleus, NFAT interacts with polyADP-ribose polymerase (PARP), which increases NFAT transcriptional activity via NFAT polyADP ribosylation. MCP-1, monocyte chemoattractant protein-1; ICAM-1, Intercellular Adhesion Molecule 1; IL-6, Interleukin-6.
In diabetic mice, activated NFATc3 plays a role in accelerated atherosclerosis. Activated NFATc3 induces arterial cell expression of the pro-inflammatory matrix protein osteopontin (OPN), a cytokine that promotes atherosclerosis and diabetic vascular disease. NFAT inhibition effectively reduced osteopontin, IL-6, monocyte chemotactic protein 1, intercellular adhesion molecule 1, CD68 and tissue factor expression in the arterial wall, and lowered plasma IL-6 in diabetic mice88. In diabetic ApoE−/− mice, inhibition of NFAT-signaling completely suppressed a 2.2-fold increase in atherosclerotic plaque area. Inhibition of NFAT also reduced lipid content in the plaque of diabetic mice independent of plasma glucose and lipid levels89. NFATc3 activated by increased mitochondrial ROS production also increases arterial vasoconstrictor reactivity to endothelin-190.
NFAT activation also appears to play a role in cardiac hypertrophy, fibrosis, and cardiomyocyte apoptosis91. In the diabetic heart, NFAT is activated by the calcium-activated neutral cysteine protease calpain, which in turn activates calcineurin. In cardiomyocytes, high glucose increases calpain activity, which activates NFAT-dependent cardiac hypertrophy and heart failure92. In two mouse models of diabetes, cardiac-specific deletion of calpain reduced myocardial hypertrophy and fibrosis, leading to the improvement of myocardial function. Calpain activation correlated with increased activity of NFAT and NFκB, consistent with calpain’s role in activation of calcineurin and degradation of the cytosolic NF-κB inhibitor, NF-κB inhibitor alpha (IκBα)93, 94.
Increased PAD4 and NETosis activation
Atherosclerotic lesions begin with the deposition of cholesterol-rich lipoproteins in the artery wall, followed by the entry of inflammatory leukocytes into lesions. Early lesions are characterized by infiltration of neutrophils and lipid-filled monocyte-derived macrophages. In CAD, inflammation becomes chronic, and lesions progress rather than resolve. During lesion progression, neutrophils and macrophages continue to accumulate, and recruit pro-inflammatory IL-17-producing T-cells. Smooth muscle cell proliferation and altered matrix production occur. Advanced lesions containing a variety of inflammatory cell types accumulate a necrotic core, which contains dead macrophage foam cells, prothrombotic molecules, and matrix proteases. A large necrotic core predisposes to release of thrombogenic material due to protease erosion or rupture of the plaque95.
Recently, Warnatsch et al96 showed that neutrophils prime macrophages for pro-inflammatory responses in atherosclerotic plaques. This priming is mediated by neutrophil extracellular traps (NETs), extracellular webs of DNA bound to cytotoxic histones which are released by activated neutrophils97. This process, called NETosis, appears to follow a coordinated multi-step process: histone citrullination, chromatin decondensation, migration of elastase and other granule enzymes into the nucleus, disintegration of the nuclear membrane and release of DNA, histones and granule proteins into the extracellular space98. The release of NETs prime macrophages to produce pro-IL-1β, which is cleaved to mature pro-inflammatory IL-1β by caspase 1. Caspase 1, in turn, is secreted by macrophages in response to activation of the NLRP3 (NOD-like receptor family, pyrin domain-containing 3) inflammasome (discussed in the following section)96 (Figure 5).
Figure 5. Diabetes increases neutrophil extracellular traps (NETs), priming macrophages for inflammation.
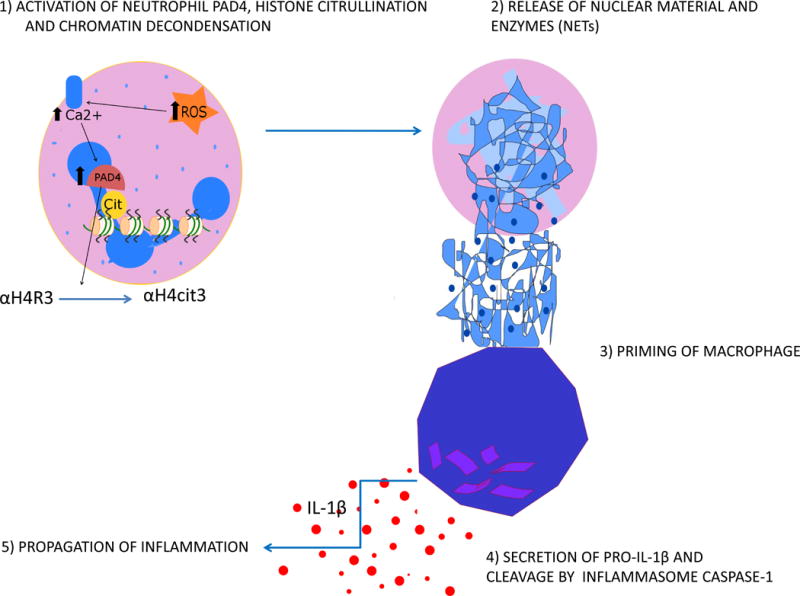
Increased ROS increase transcription and activation of peptidylarginine deiminase 4 (PAD4), the enzyme which initiates formation and release of NETs by citrullination of histones. Released NETs prime macrophages to produce pro-IL-1β, which is cleaved to mature pro-inflammatory IL-1β by caspase 1 secreted by macrophages in response to NLRP3 inflammasome activation. Cit, citrullination; αH4R, αhistone 4 Arginine; αH4cit, αhistone 4 with arginine residues converted to citrulline.
Apoe−/− mice with deletions of two serine proteases that localize to NETs, neutrophil elastase and proteinase-3 (Apoe−/−/Ela2−/−/Prtn3−/−), developed dramatically smaller atherosclerotic lesions compared to Apoe−/− control animals, despite similar lipid concentrations and leukocyte counts in blood. Triple mutant mice had no NETs, lower systemic IL-1β concentration, and fewer lesional IL-17 producing T cells99. NETs are also pro-thrombotic96. Neutrophils from type 1 and type 2 diabetic humans as well as mice are primed to produce NETs, and have increased expression of peptidylarginine deiminase 4 (PAD4), the enzyme critical for histone citrullination-mediated chromatin decondensation and NET formation100, 101. Increased PAD4 transcription is driven by NFκB102, which is chronically activated in diabetes103. Hyperglycemia-induced ROS likely activate PAD4 as well, by increasing intracellular Ca++ concentration. Other consequences of increased intracellular ROS such as PKC activation increase levels of the NETosis-priming cytokine tumor necrosis factor-α 7. Normal resolution of inflammation caused by infiltration of neutrophils and macrophages involves a switch from synthesis of arachidonic acid–derived prostaglandins and leukotrienes to synthesis of lipoxins, which stop neutropil recruitment. At the same time, increased production of resolvins and protectins from omega-3 polyunsaturated fatty acids induce neutrophil apoptosis. Phagocytosis of these apoptotic neutrophils by macrophages causes a switch to an anti-inflammatory macrophage phenotype, which secretes anti-inflammatory and reparative cytokines104. The mechanism responsible for defective inflammatory lesion resolution in atherosclerosis and diabetes is not known.
NLRP3 inflammasome activation
Increased expression of the NRLP3 inflammasome components Nlrp3 and ASC was found in monocytes from new, untreated patients with type 2 diabetes105. Along with increased expression, there was increased inflammasome activation. Consistent with this, the drug-naïve type 2 diabetic patients had significantly higher serum levels of the pro-inflammatory cytokines IL-1β and IL-18 than did healthy subjects105. In a Type 2 diabetic rat model, excessive activation of NLRP3 was associated with cardiac inflammation, cell death, disorganized ultrastructure, and fibrosis. NLRP3 gene silencing ameliorated cardiac inflammation, apoptosis, fibrosis, and left ventricular cardiac dysfunction106. The NLRP3 inflammasome is formed by oligomerization of inactive NLRP3, associated with apoptosis-associated speck-like protein (ASC), and procaspase-1. This complex, in turn, catalyzes the conversion of procaspase-1 to caspase-1, which contributes to the production and secretion of mature pro-inflammatory IL-1β and IL-18107. The transcription factor NF-κB, which is chronically active in mononuclear cells from diabetic patients and in vascular endothelial cells of diabetic rats103 promotes transcription of NLRP3, proIL-1β, and proIL-18108. These proteins remain in the cytoplasm in inactive forms. A second signal activates the NLRP3 inflammasome by facilitating the oligomerization of inactive NLRP3, apoptosis-associated speck-like protein (ASC), and procaspase-1.
Many, but not all reported activators of the NRLP3 inflammasome converge on excessive production of ROS (Figure 6). The ROS-induced reduction of intracellular NAD+ levels, discussed previously, reduces the activity of SIRT2, causing accumulation of acetylated α-tubulin109. Acetylated α-tubulin regulates the transport of mitochondria and helps form an efficient interaction between the adaptor protein ASC and NLRP3. ROS also activate NLRP3 by opening the cell membrane calcium channel TRPM2, increasing Ca2+ influx. The mitochondrial membrane phospholipid cardiolipin, discussed in the following section, also activates the NLRP3 inflammasome after translocation to the outer mitochondrial membrane, where it binds to NLRP3110. It is currently not known what causes cardolipin to move to the outer mitochondrial membrane, but ROS-induced cardiolipin remodeling may be one explanation111, 112. Activation of NLRP3 in diabetic heart and artery may also reflect the increased cell surface CD 36 induced by insulin-resistance (discussed in the following section), which facilitates internalization of oxidized LDL (ox-LDL) and intracellular conversion of ox-LDL to cholesterol crystals.
Figure 6. NLRP3 inflammasome activation in diabetic atherosclerosis.
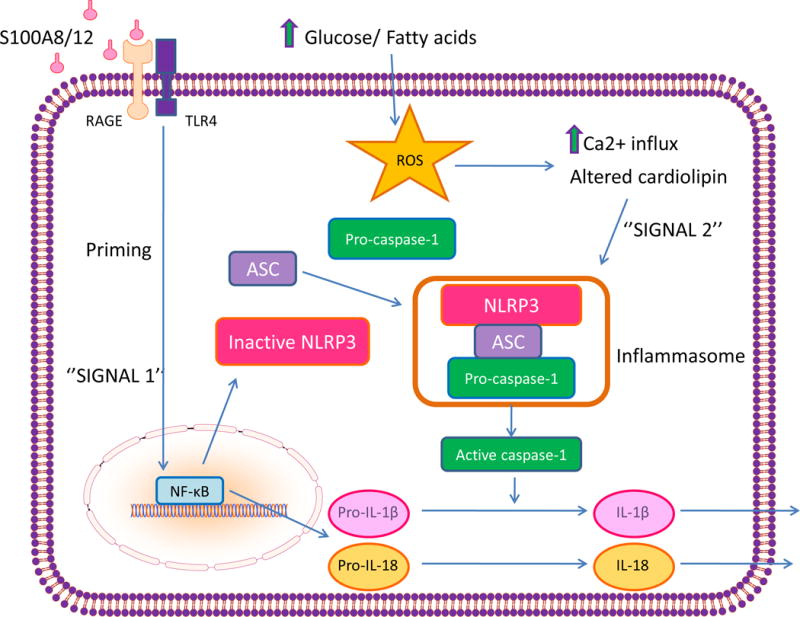
Intracellular hyperglycemia-induced ROS in monocytes and vascular endothelial cells increases RAGE expression, which heterodimerizes with TLR4. Signaling from this complex causes NFκB-mediated transcription of inactive NOD-like receptor family, pyrin domain-containing 3 (NRLP3), pro-IL-1β, and pro-IL-18. Increased intracellular Ca++ triggers oligomerization of inactive NLRP3, associated with apoptosis-associated speck-like protein (ASC), and pro-caspase-1. This activated inflammsome complex catalyzes the conversion of procaspase-1 to caspase-1, and of pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18. S100A8/12, calgranulin A/B heterodimer ligand for RAGE.
Consequences of oxidative miRNA modification and ROS-induced downreguation of specific miRNAs
ROS oxidatively modify certain microRNAs. ROS can hydroxylate guanine to produce 8-oxo-7,8,-dihydroguanosine (8OHG)113. Oxidized miR-184 was shown to mismatch with the 3′ UTRs of the anti-apoptotic proteins Bcl-xL and Bcl-w, thereby sensitizing cardiomyocytes to apoptosis. Administration of oxidatively modified miR-184 increased infarct size in an ischemia/reperfusion model. Increased expression of several miRNAs which may contribute to diabetic atherosclerosis is correlated with insulin resistance, but virtually nothing is known about the mechanisms regulating expression of these miRNAs. A recent study of miRNA-QTL using liver tissue from 424 morbidly obese, insulin-resistant subjects identified an association of miR-128-1 and miR-148a expression with SNPs linked to abnormal human blood lipid levels. In vivo studies in the Apoe−/− mouse verified that miR-128-1 and miR-148a do regulate cholesterol/lipid and energy homeostasis114. MiR-128-1 also regulates expression of SIRT1, the NAD+-dependent lysine deacetylase, and these miRNAs also affect expression of several components of the heterotrimeric AMP-activated protein kinase (AMPK). Diabetes-induced ROS have been implicated in the downregulation of miR-499, -133a, and -373 in diabetic cardiomyocytes115. Although most individual miRNAs target hundreds of specific mRNAs, thereby coordinately regulating complex gene networks, transgenic mice overexpressing miR-499 displayed pathological cardiac remodeling accompanied by cardiac dysfunction116. Downregulation of MiR-133a in a normal adult genetic background was sufficient to induce cardiac hypertrophy, and its downregulation is a prerequisite for the development of apoptosis, fibrosis, and prolongation of the QT interval in animal models117.
Insulin-resistance associated down-regulation of miR-128-1 and perhaps also miR-34a may also play a critical role in diabetic atherosclerosis. MiR-34a directly targets and decreases SIRT1 expression. SIRT1 activity would be reduced further by miR34a targeting of the rate-limiting enzyme in the salvage pathway for NAD+ biosynthesis, NAMPT118. In a well-established porcine model of diabetic atherosclerosis which develops complex atherosclerotic plaques resembling human complex plaques, the expression and activity of the NAD+-dependent deacetylase SIRT1 were markedly reduced119. As mentioned earlier, ROS-activated PARP depletes NAD+ in the process of synthesizing ADP-ribose by cleaving NAD+ into ADP-ribose and nicotinamide. This depletion of NAD+ would also inhibit SIRT1’s enzymatic activity. In the porcine diabetic atherosclerosis model, the activity of AMPK was also reduced by 40%, due to decreased phosphorylation at Thr-172. Since SIRT1-mediated deacetylation is necessary for full function of the AMPK-activating kinase LKB1, AMPK-dependent inhibition of fatty acid synthesis would be reduced by diabetes-induced increased production of mitochondrial ROS120. In addition, overexpression of miR-451 in diabetes reduces AMPK activating phosphorylation of LKB1 by reducing levels of the LKB1 scaffold protein Calcium-binding protein 39 (Cab39)120. Normally, SIRT1 activity and AMPK activity suppress the activating cleavage processing of sterol regulatory element binding proteins (SREBP-1 and -2).
In the porcine model of diabetic atherosclerosis, the cleavage processing of SREBP-1 and -2 and expression of their target genes were increased in endothelial cells and infiltrating macrophages of both fatty streaks and of advanced lesions with fibrous caps, necrotic cores, and cholesterol cores. SIRT1 and AMPK activity were also reduced119. Increased SREBP-1a in macrophages also directly upregulates transcription of NLRP3 inflammasomes121, and in porcine diabetic atherosclerosis, increased NLRP3 was found both in macrophages of advanced lesions, and in endothelial cells and smooth muscle cells. The changes found in porcine diabetic atherosclerosis were also present in coronary atherosclerosis samples from diabetic humans.
Mechanisms of Insulin Resistance-Induced Cardiovascular Damage
Insulin resistance is operationally defined as an impaired ability of different cell types to respond normally to insulin. In people with type 2 diabetes, insulin resistance is caused by underlying heritable factors and is exacerbated by environmental factors such as obesity.
Insulin signaling is initiated by its binding to tyrosine kinase insulin receptors (IR). Insulin receptor activation of the phosphatidylinositol 3-kinase- (PI3K)–Akt pathway is responsible for most of the metabolic actions of insulin. The IR has two splice isoforms, both of which can phosphorylate at least six known insulin receptor substrate proteins (IRS). These IRSs are capable of interacting with eight known forms of the PI3K regulatory subunit. PI3K regulatory subunits in turn can associate with three forms of the PI3K catalytic subunit, and the product of PI3K activity can then activate three isoforms of Akt. The combinatorial possibilities of the IR-IRS-PI3K-Akt part of the insulin signaling pathway alone exceed 1,000. When differential compartmentalization, stoichiometry, and kinetics of the various downstream signaling components are included, this number increases dramatically122. Many of these steps are negatively regulated by action of phosphatases or inhibitory proteins. The complexity of this signaling system is essential to selectively mediate the large variety of known responses to insulin123. Because of this complexity, the molecular pathogenesis of insulin resistance is still incompletely understood.
It has been suggested that insulin resistance is a physiologic mechanism that protects the cardiovascular system from nutrient-induced injury, and that therapies attempting to override it with intensive insulin therapy in an effort to lower plasma glucose levels could therefore be harmful124. Since insulin resistance in critical tissues appears to be pathway-specific for glucose metabolism, attempts to treat insulin resistance with intensive insulin therapy may well increase deleterious effects of insulin on lipid and lipoprotein metabolism125. However, physiologic hyperinsulinemia in response to pathway-specific insulin resistance in liver without exogenous insulin is responsible for the flooding of the heart with triglyceride-derived fatty acids125. Insulin resistance itself also appears to be harmful due to associated metabolic inflexibility, in which nutrient overload and heightened substrate competition result in mitochondrial dysfunction, impaired fuel switching, and energy dysregulation. Metabolic inflexibility occurs early in the course of glucose intolerance, and obesity-induced perturbations in substrate switching persist in isolated muscle mitochondria. Current evidence suggests that myocardial cells (and those of striated muscle and liver) function optimally when they retain their capacity to switch freely between oxidative substrates in response to nutritional physiologic cues126.
Increased endothelial fatty acid oxidation, increased mitochondrial ROS, and diabetic atherosclerosis
Insulin resistance increases fatty acid oxidation in arterial endothelial cells. In two insulin- resistant nondiabetic animal models, inhibition of either free fatty acid (FFA) release from adipocytes or FFA oxidation in arterial endothelium prevented the increased production of ROS and its damaging effects127. In arterial endothelial cells, this FFA-induced increase in ROS activates the same damaging pathways seen with high glucose: AGEs, PKC, the hexosamine pathway (GlcNAc), and NFκB. FFA-induced overproduction of superoxide also activates a variety of proinflammatory signals previously implicated in hyperglycemia-induced vascular damage, and inactivates two important antiatherogenic enzymes, prostacyclin synthase and eNOS127.
King first proposed that in diabetes, a pathway-specific insulin resistance in diabetic vascular cells reduces the antiatherogenic actions of insulin, while leaving insulin’s proatherogenic actions unaffected128. Subsequently, this group showed in apoE null mice that specific overexpression of PKCβ2 in endothelial cells caused pathway-specific insulin resistance by inhibiting downstream PI3K-Akt signaling, thereby inhibiting eNOS activation36. NO released from endothelial cells is a potent inhibitor of platelet aggregation and adhesion to the vascular wall. Endothelial NO also controls the expression of genes involved in atherogenesis. It decreases expression of MCP-1, and of surface adhesion molecules such as CD11/CD18, P-selectin, VCAM-1 and ICAM-1. Since PI3K-Akt signaling also causes the sequestration of FOXO transcription factors in the cytoplasm, thereby preventing transcription of a variety of pro-atherogenic genes129, inhibition of PI3K-Akt signaling by PKCβ2 would be expected to increase transcription of these pro-atherogenic genes.
Surprisingly, in human arterial endothelial cells, cells from diabetics showed higher levels of the activating Ser 1177 phosphorylation of eNOS, despite increased PKCβ expression, reduced PI3K-Akt signaling, and increased levels of oxidative stress. PKCβ activity was associated with lower flow-mediated dilation which was reversible by PKC inhibition130. These results are consistent with hyperglycemia- and fatty acid-induced ROS overproduction. Increased ROS activates PKC, which like PI3K-Akt, phosphorylates eNOS at Ser 1177. However, increased ROS also reduce eNOS activity by two mechanisms: oxidation of tetrahydrobiopterin (BH4), the essential cofactor of endothelial nitric oxide synthase (eNOS), and uncoupling of dimeric eNOS to monomeric eNOS. These changes convert the nitric oxide-producing dimeric eNOS to superoxide-producing monomeric eNOS131. In contrast to IR inhibition of PI3K-Akt signaling, insulin signaling through MAPK and ERK is unaffected by insulin resistance. In arterial endothelial cells, this resulted in increased expression of pro-atherogenic proteins like endothelin-1. In apoE null mice with specific overexpression of PKCβ2 in endothelial cells, the severity of atherosclerotic lesions was increased by 70%36.
Increased myocardial fatty acid oxidation, increased mitochondrial ROS, and diabetic cardiomyopathy
The normal adult heart consumes a large amount of energy and uses a variety of substrates to produce ATP. 70% of total energy is derived from mitochondrial oxidative phosphorylation of fatty acids, with most of the rest derived from glucose oxidation132. Fatty acid uptake in the heart is mediated by the cluster of differentiation 36 (CD36), a scavenger receptor class B type I, and fatty acid translocase (FAT), while glucose uptake is mediated by insulin-stimulated translocation of glucose transporter 4 (GLUT4). In diabetes, myocardial insulin resistance with impaired insulin signaling decreases GLUT4 translocation to the cell surface, while increasing cell-surface CD36133. At the same time, insulin resistance in adipose tissue and liver causes an increased delivery of triglycerides and triglyceride-rich VLDL to the heart, where heart-specific LpL releases fatty acids. In diabetes, heart-specific LPL activity is upregulated134, 135. Thus, insulin resistance causes a major increase in fatty acid flux into the myocardium. Similarly, murine models of insulin-dependent and non-insulin-dependent diabetes had serum triglyceride levels 2.6- and 4.2-fold higher, respectively, than normal mice, and 7- and 3.5-fold higher levels of heart microsomal CD36, respectively, than control mice136. Thus, there is a dramatic shift away from glucose utilization and an overreliance on fatty acids as the energy source in the diabetic heart. Even in TIDM, where substrate-induced insulin resistance is much less severe than the intrinsic insulin resistance of T2D, studies have shown a dramatic impairment in glucose uptake in cardiomyocytes because of diminished insulin-induced transcription and translocation of GLUT-4137, 138. In streptozotocin-induced TIDM animals, levels of GLUT-4 were significantly reduced, forcing the cardiomyocytes to rely on fatty acids139.
Both increased fatty acid uptake and increased fatty acid oxidation in the diabetic heart are mediated in part by increased PPARα activity140, 141. Like the other two members of this nuclear receptor family, PPARβ/δ and PPAR γ, PPARα forms heterodimers with the retinoid X receptors (RXRs), which bind to PPAR-responsive elements (PPREs). PPARα activation by binding of its endogenous ligand (1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC)142 causes bound transcriptional repressors (such as SMRT and N-CoR) to be exchanged for transcriptional co-activators—primarily PGC-1α and PGC-1β—which form active transcriptional complexes with CBP/p300, SRC-1, and a number of other proteins. PPARα transcriptional activity is further increased by PKCβ2-mediated Ser phosphorylation143. Since increased ROS activate PKCβ2 and several other PKC isoforms (discussed previously), this may contribute to the upregulation of PPARα-target genes in diabetic heart. PPARα (−/−) mice were protected from the development of diabetes-induced cardiac hypertrophy, while the combination of diabetes and the MHC-PPARα genotype resulted in a more severe cardiomyopathic phenotype than either did alone. Cardiomyopathy in diabetic MHC-PPARα mice was accompanied by myocardial long-chain triglyceride accumulation. Reactive oxygen intermediates were identified as candidate mediators of cardiomyopathic effects in MHC-PPARα mice140. Recently, the muscle ring finger-3 (MuRF3) ubiquitin ligase was shown to stabilize PPARα activity in vivo144. Both diabetes and treatment with the cardiotoxic ROS-producing chemotherapeutic agent doxorubicin increase MuRF3 expression. MuRF3 expression was increased in hearts from insulin resistant high fat fed mice, and was shown to mono-ubiquitinate cardiac PPARα.
Cardiomyopathy resulting from this increased fatty acid flux is called “lipotoxicity”. Two general mechanisms have been proposed to explain lipotoxicity. One mechanism proposes that in diabetic cardiomyocytes, increased fatty acid flux results in increased synthesis of diacylglycerols, diglycerides, and ceramide145. Aberrant accumulation of these signaling intermediates, particularly ceramide, appears to be cardiotoxic146. In transgenic mice with heart-specific LpL overexpression, more lipid accumulated in hearts expressing the transgene, and myocytes were enlarged and exhibited abnormal architecture. Hearts of transgenic mice were dilated, and left ventricular systolic function was impaired135. Inhibition of de novo ceramide biosynthesis reduced fatty acid oxidation and increased glucose oxidation in isolated perfused LpL hearts, and improved systolic function and prolonged survival rates of cardiac specific LpL overexpressing mice146.
The second general mechanism proposes that increased fatty acid oxidation results in increased ROS production, which causes cardiomyopathy by a variety of downstream actions. Although the oxidation of fatty acids normally yields significantly more energy per carbon atom than does the oxidation of glucose, oxidative phosphorylation capacity is impaired in hearts from insulin-resistant db/db mice. However, H2O2 production is increased147. In human myocardium from insulin resistant diabetic patients, mitochondrial H2O2 production was also increased during oxidation of lipid-based substrates compared with carbohydrate-based substrates148. The explanation for this apparent paradox is that the major site of electron leakage from increased fatty acid oxidation is electron transfer flavoprotein (ETF), which receives electrons from the FADH2 formed during the first oxidation step of β-oxidation54 (Figure 7). Increased myocardial ROS production in the diabetic heart occurs very early, before accumulation of TGs are evident, due to increased fatty acid oxidation and resultant oxidative damage to mitochondrial cardiolipin (CL)149, the specific phospholipid of mitochondrial membranes. Cardiolipin is important for efficient electron flux, ATP synthesis, and reduced ROS formation. In addition, cardiolipin is involved in mitochondrial-mediated apoptosis, and it plays a critical role in regulating mitochondrial fission and fusion150–152. In non-diabetic heart, the major species of cardiolipin contains four linoleic acids (tetra 18:2 cardiolipin). This unique acyl composition is not derived from de novo synthesis of CL, but rather from a remodeling process that involves phospholipases and acyltransferase-transacylases. In diabetic myocardium from murine models with insulin resistance (ob/ob, db/db, high-fat diet) and in models of severe insulin-deficient type 1 diabetes (STZ), the fatty acyl content of the more saturated 18:2 cardiolipin is dramatically reduced, while the content of longer chain, more unsaturated fatty acyl cardiolipin is substantially increased112. Because of this increase in highly unsaturated side chains, diabetic heart cardiolipin is more vulnerable to oxidative damage. Cardiac overexpression of cardiolipin synthase increases tetra 18:2 cardiolipin in diabetic mice and prevents diabetes-induced changes in cardiolipin lipid remodeling. The cardiolipin deficiency and profound remodeling caused by diabetes and by diet-induced obesity is caused by ROS-induced transcription of Acyl-CoA:lysocardiolipin acyltransferase 1 (ALCAT1)(Figure 7). ALCAT1 catalyzes the transfer of linoleoyl-CoA onto mono- or dilysocardiolipin. Overexpression of ALCAT1 caused cardiolipin deficiency and fatty acid compositional changes similar to diabetes and obesity, with increased production of ROS, while ALCAT1 deficiency increased levels of tetra 18:2 cardiolipin in mouse heart and reduced ROS production111.
Figure 7. Increased cardiac fatty acid oxidation, ROS formation and cardiolipin remodeling.
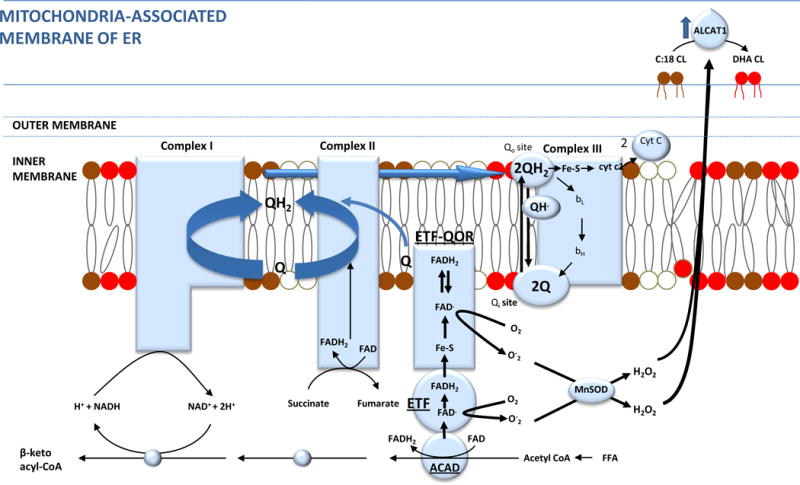
Insulin resistance-induced increased cardiac β oxidation of free fatty acids (FFA) causes greater H2O2 production than increased glucose oxidation because of increased electron leakage from the electron transfer flavoprotein (ETF) complex. These ROS activate Acyl-CoA:lysocardiolipin acyltransferase 1 (ALCAT1). ALCAT1, located in the mitochondrial associated membrane (MAM) of the ER, which causes pathologic remodeling of cardiolipin from tetra 18:2 cardiolipin to cardiolipin with highly unsaturated fatty acid side chains and cardiolipin deficiency due to oxidative damage. This reduces ETC electron flux, ATP synthesis, and further increases ROS.
High levels of ROS, through JNK signaling, are also a major proximal activator of the forkhead box O (FOXO) family of transcription factors. Increased ROS stimulate FOXO translocation from the cytosol into the nucleus by increasing FOXO GlcNAcylation, direct cysteine oxidation, and CaMKII activation153, 154. Numerous other signals and post-translational modifications can also regulate FOXOs, and FOXOs regulate a multitude of diverse processes by interacting with many different transcription factors and other nuclear proteins. In the hearts of diabetic mice and of mice with high fat diet-induced insulin resistance, FOXO proteins were persistently activated155. This persistent activation was associated with downregulation of insulin receptor substrate 1 (IRS1), reduced activity of IRS1 and its downstream target Akt, and the development of cardiomyopathy. In cardiomyocyte-specific FOXO1 knockout mice fed a high fat diet, neither insulin resistance nor cardiomyopathy occurred.
Altered mitochondrial dynamics
Mitochondria dynamics are the continuous processes of mitochondrial fusion, fission, biogenesis and mitophagy which maintain optimal cellular bioenergetics and ROS homeostasis. The topic of altered mitochondrial dynamics and cardiovascular disease has been reviewed quite recently156, and the reader is referred to that comprehensive review for more extensive discussion. In the context of insulin resistance and diabetic cardiomyopathy, the balance between mitochondrial fusion and fission is altered in favor of increased fission and decreased fusion. This increased ratio of fission to fusion causes increased production of ROS and decreased mtDNA, reducing the metabolic capacity and efficiency of the mitochondrial electron transport chain and ATP synthesis. The primary regulators of mitochondrial fusion are dynamin-related GTPases termed mitofusins 1 and 2 (MFN1 and MFN2) and optic atrophy protein 1 (OPA1) located in the inner mitochondrial membrane. In humans with Type 2 diabetes, expression of the fusion protein MFN1 is decreased in myocardium157.
The primary regulator of mammalian mitochondrial fission is the GTPase dynamin-related protein 1 (DRP1), which is recruited from the cytosol to the mitochondrial outer membrane where it binds to four DRP1 receptors- — mitochondrial fission factor (MFF), Mitochondrial dynamics protein of 49 kDa (MID49) and 51kDa (MID51), and FIS1158, 159. Oligomerization of Drp1 is believed to provide the mechanical force to constrict mitochondrial membranes and to fragment the organelle. Drp1 activity is regulated by the Ca2+/calmodulin-dependent serine/threonine phosphatase calcineurin. Dephosphorylation of Drp1 at Ser 656 by calcineurin activates DRP1 and increases mitochondrial fission. Calcineurin itself is activated by the calcium-activated neutral cysteine protease calpain. Since increased ROS activate calpain and calcineurin in cardiomyocytes160, 161, the increased mitochondrial ROS production from increased fatty acid oxidation described earlier could explain, in part, the observed increased fission in hearts of insulin resistant diabetics (Figure 8). Increased mitochondrial fragmentation also occurs in mitochondria from mouse coronary endothelial cells (MCECs), where the level of the fusion protein OPA1 is decreased, and the level of DRP1 is increased162. Insulin resistance itself may also contribute to the increased ratio of fission to fusion. In non-diabetic non-insulin resistant cardiomyocytes, insulin increased OPA-1 levels and mitochondrial fusion, and reduction of either OPA-1 or MFN2 with siRNA prevented this163. 52
Figure 8. Diabetes-induces increased mitochondrial fission.
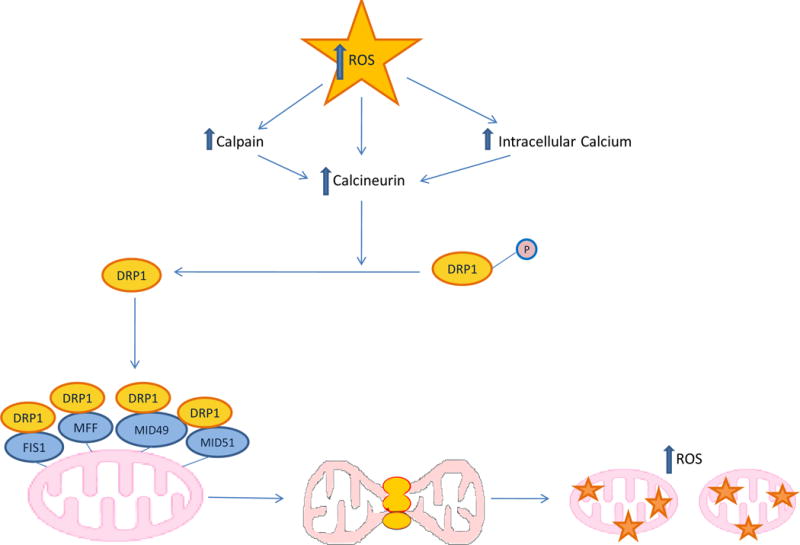
Increased ROS from either excess glucose in vascular cells or fatty acids in cardiomyocytes increases intracellular Ca++. Increased Ca++ activates the calcium-activated neutral cysteine protease calpain, which activates the Ca2+/calmodulin-dependent/CaM) (Ca2+)serine/threonine phosphatase calcineurin. Calcineurin dephosphorylates the GTPase dynamin-related protein 1 (DRP1), which is then recruited from the cytosol to the mitochondrial outer membrane where it binds to four DRP1 receptors- — mitochondrial fission factor (MFF), Mitochondrial dynamics protein of 49 kDa (MID49) and 51 kDa (MID51), and FIS1. Drp1 oligimerization provides the mechanical force to constrict mitochondrial membranes and fragment the organelle (mitochondrial fission). Increased fission causes further increases in ROS production and mitochondrial dysfunction. FIS1, Mitochondrial fission 1 protein.
Circadian clocks, Metabolic Networks, and Diabetic Cardiovascular Disease
Cell autonomous circadian clocks encoded by a transcription-translation feedback loop exist in most peripheral tissues including liver, fat, muscle, and pancreatic β cells. In the liver, they promote lipid catabolism, gluconeogenesis, and mitochondrial biogenesis during sleep/fasting, and lipogenesis, glycogen synthesis, and cholesterol and bile acid synthesis in the awake/feeding state164. In adipose tissue, they promote lipid catabolism during sleep/fasting, and lipogenesis when awake and feeding. In muscle, they promote oxidative metabolism during sleep/fasting, and fatty acid update during feeding. Different tissues exhibit distinct clock-controlled properties164. The core transcriptional components of the mammalian circadian clock are the transcriptional activators CLOCK and BMAL1, which co-activate transcription at E-box-containing gene promoters, including the period (Per) and cryptochrome (Cry) genes. PER and CRY proteins form a complex that is imported into the nucleus and inhibits their own transcription. During the night/fasting, these proteins are degraded, resulting in reactivation of their transcription in the early morning. A short feedback loop consisting of the orphan nuclear receptor REV-ERBα and the retinoic acid orphan receptors (ROR) α and β activate and repress, respectively, Bmal1 transcription. REV-ERBα controls oscillation, abundance and activation of SREBPs through modulation of INSIG2. PGC-1alpha, a transcriptional coactivator that regulates mitochondrial biogenesis and energy metabolism, is rhythmically expressed in the liver and skeletal muscle of mice. PGC-1alpha stimulates the expression of clock genes including Bmal1 and Rev-Erbα through co-activation of the retinoic acid orphan receptors (ROR) family of nuclear receptors. Mice lacking PGC-1alpha show abnormal diurnal rhythms metabolic rate. The disruption of physiological rhythms in these animals is correlated with aberrant expression of clock genes and those involved in energy metabolism. Analyses of PGC-1α-deficient fibroblasts and mice with liver-specific knockdown of PGC-1α indicate that it is required for cell-autonomous clock function165.
Metabolic networks influenced by oscillation of clock genes and downstream transcription factors also reciprocally influence clock function164, 166. Major metabolic coupling signals include NAD+, SIRT1, AMPK, and ROS164, 166, 167. NAD+ activates SIRT1, which deacetylates and thereby inhibits the CLOCK:BMAL1 complex. PARP-1 also ADP-ribosylates the CLOCK protein. AMPK controls proteolytic degradation of PER and CRY. CLOCK mutant mice develop hypertriglyceridemia, and hepatic steatosis168, while ablation of Bmal1 increases arterial superoxide, uncoupling of eNOS169, and arterial wall lesion extent after endothelial injury170. Circadian transcription factors also regulate levels of the pro-thrombotic plasminogen activator inhibitor type 1 (PAI-1)171, 172. Circadian rhythmicity of the clock-dependent oscillator krupple-like factor 15 (Klf15) controls cardiac expression of Kv channel-interacting protein 2 (KChIP2), a critical subunit required for generating the transient outward potassium current and QT-interval duration. Deficiency or excess of Klf15 causes loss of rhythmic QT variation, abnormal repolarization and enhanced susceptibility to ventricular arrhythmias173.
In the diabetic cardiovascular system, however, continuously increased ROS production caused by hyperglycemia and insulin resistance depletes NAD+ by activating PARP1, which cleaves NAD+ into ADP-ribose and nicotinamide in the process of synthesizing ADP-ribose. This depletion of NAD+ inhibits SIRT1’s enzymatic activity, which normally deacetylates and activates both PGC1α and LKB1, the kinase that activates AMPKα2. Decreased SIRT1 activity would decrease activity of LKB1, PGC1α, and AMPK174, causing decreased mitochondrial biogenesis, increased ROS production, and a profoundly disturbed clock synchronization of glucose and lipid metabolism. Recently, the flavone nobiletin was discovered to improve the amplitude of the clock repressor PERIOD 2, acting through RORs. Nobiletin treatment of wild type mice on a high fat diet prevented weight gain and visceral adiposity, while treatment had no effect in Clock homozygous mutant mice. Nobiletin had potent insulin-sensitizing actions, consistent with the observed reduction in hepatic and serum triglycerides, and reversed high fat diet-induced alterations in clock gene expression in liver and fat164, 175.
Conclusion
Knowledge about the biochemical, molecular, and cellular mechanisms responsible for cardiovascular disorders in diabetes has increased enormously in recent years. As a consequence, the clinical correlations linking hyperglycemia and insulin resistance with accelerated atherosclerosis, heart failure (both HFpEF and HFrEF), and increased post-MI fatality rates are increasingly understood in mechanistic terms. For each of these clinical entities, the multiple mechanisms discussed in this review seem to share a common element: prolonged increases in ROS production in diabetic cardiovascular cells. In normal cardiovascular physiology, ROS production is coupled to circadian clocks and metabolic networks, and ROS species (H2O2) function as signaling molecules essential for normal cellular homeostasis67, 68. In contrast, ROS production at too high a level, for too long, or at an inappropriate location, leads to impaired cellular function and cardiovascular pathology74. Intracellular hyperglycemia causes excessive ROS production by increasing electron leak from the mitochondrial electron transport chain. This can be amplified by ROS-induced uncoupling of nitric oxide synthase (eNOS) and activation of NADPH oxidases. Increased mitochondrial ROS cause DNA double strand breaks damage by releasing free iron and H2O2, which diffuse into the nucleus. These DNA strand breaks activate latent nuclear poly(ADP ribose) polymerase (PARP). PolyADP-ribosylation of glyceraldehyde-3-dehydrogenase (GAPDH) by PARP leads to partial inhibition of this key glycolytic enzyme. As a result, early glycolytic intermediates accumulate and are then diverted into pathogenic signaling pathways. These pathways include increased substrate conversion by the enzyme AKR1B1, increased formation of methylglyoxal, the major advanced glycation product precursor, activation of protein kinase C isoforms β,δ, and θ, and increased protein modification by O-GlcNAc. Activation of GS3Kβ by excessive ROS production reduces intracellular Nrf2, the transcription factor that normally increases expression of ROS-degrading enzymes. Nrf2 also regulates expression of the rate-limiting enzyme of the glyoxalase system, GLO1, which degrades the major AGE precursor methylglyoxal.
PARP activation also degrades NAD+, reducing activity of the NAD+-dependent sirtuin deacetylases. SIRT1 normally deacetylates and activates both PGC1α and LKB1, the kinase that activates AMPKα2. Reduced SIRT1 deacetylation decreases activities of LKB1, PGC1α, and AMPKα2174, causing decreased mitochondrial biogenesis, increased ROS production, and a profoundly disturbed circadian clock synchronization of glucose and lipid metabolism. Excessive ROS production also facilitates nuclear transport of NFAT transcription factors by activating the Ca2+/calmodulin-dependent serine/threonine phosphatase calcineurin. Excessive ROS also increase transcription of Pad4, thee neutrophil enzyme initiating NETosis, which primes macrophages for pro-inflammatory responses in atherosclerotic plaques. Many reported activators of the NRLP3 inflammasome also converge on excessive production of ROS.
Insulin resistance causes excessive ROS production by cardiomyocytes because it increases fatty acid uptake and oxidation. Thus, there is a dramatic shift away from glucose utilization and an overreliance on fatty acids as the energy source in the diabetic heart. Although oxidation of fatty acids normally yields significantly more energy per carbon atom than does the oxidation of glucose, oxidative phosphorylation capacity is impaired in hearts from insulin-resistant animal models and human diabetics. Despite a reduced oxidative phosphorylation capacity mitochondrial H2O2 production is increased during oxidation of lipid-based substrates compared with carbohydrate-based substrates. This reflects the fact that the major site of electron leakage from increased fatty acid oxidation is electron transfer flavoprotein (ETF), which receives electrons from β oxidation-generated FADH2, independent of the electron transport chain complexes. Increased myocardial ROS production in the diabetic heart occurs very early, before accumulation of TGs are evident, due to increased fatty acid oxidation and resultant ROS-induced cardiolipin remodeling by ALCAT1. Finally, in the context of insulin resistance and diabetic cardiomyopathy, the balance between mitochondrial fusion and fission is altered in favor of increased fission and decreased fusion. This increased ratio of fission to fusion causes increased production of ROS and decreased mtDNA, reducing the metabolic capacity and efficiency of the mitochondrial electron transport chain and ATP synthesis.
Reactive oxygen intermediates have also been identified as candidate mediators of the cardiomyopathic effects of cardiac overexpression of the nuclear receptor PPARα140. High levels of ROS are also a major proximal activator of the forkhead box O (FOXO) family of transcription factors. The increased ROS associated with insulin resistance stimulate translocation of FOXO from the cytosol into the nucleus, and persistent myocardial FOXO1 activation in insulin resistant diabetic mice causes cardiomyopathy.
In contrast to the mechanisms underlying diabetic accelerated atherosclerosis and diabetic cardiomyopathy, much less is known about the mechanisms responsible for diabetes-associated increases in both early and post-MI fatality rates. A major cause of post-MI mortality is ventricular arrhythmia. Mitochondrial ROS play a central role in gap junction remodeling and fatal arrhythmia generation176. ROS also increase myocardial protein modification by O-GlcNAc, which downregulates SERCA2a transcription and increases phosphorylation of the ryanodine receptor by autonomously activating CaMKII6, 7. This contributes to potentially fatal arrhythmias such as premature ventricular complexes and delayed afterdepolarizations. Overexpression of GlcNAcase normalized CaMKII activity, restored SERCA2a transcription, and reduced arrhythmic events. In addition, ROS can activate CaMKII directly through a mitochondrial/oxidized-CaMKII pathway8. Activation of this mitochondrial ROS-oxidized CaMKII pathway increased mortality after myocardial infarction in diabetic mouse models8. Deficiency or excess of the clock-dependent oscillator krupple-like factor 15(Klf15) causes loss of rhythmic QT variation, abnormal repolarization and enhanced susceptibility to ventricular arrhythmias in mice173. While human repolarization occurs through a complex interaction of multiple repolarizing ionic currents, rather than the outward potassium current which is central in mice, altered klf15 expression may also contribute to human arrhythmogenesis by the ROS-dependent mechanisms described above. klf15 controls rhythmic expression of key enzymes involved in normal glucose, lipid, and nitrogen homeostasis. Disruption of branched chain amino acid (BCAA) catabolism caused by reduced klf15 suppresses cardiac mitochondrial respiration and induces superoxide production177.
In human diabetic hearts, regional myocardial autonomic denervation may also predispose patients to malignant arrhythmias178. Much work remains to be done in this area. However, increased ROS have been shown to depress sympathetic ganglion synaptic transmission by oxidizing residue Cys 239 of the nAch receptor α3 subunit179. This subunit is also present in parasympathetic neurons. These were not examined, but the same mechanism likely depresses diabetic parasympathetic ganglia synaptic transmission as well.
Supplementary Material
Acknowledgments
Owing to space limitations, a comprehensive list of reference citations could not be included. We apologize to those colleagues whose work is not specifically referenced, and gratefully acknowledge their contributions to the field.
Sources of funding
This work was supported by grants from the NIH, the Juvenile Diabetes Research Foundation and the American Diabetes Association.
Footnotes
Nonstandard Abbreviations and Acronyms
None
Disclosures
None.
REFERECES
- 1.Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care. 1998;21:1138–45. doi: 10.2337/diacare.21.7.1138. [DOI] [PubMed] [Google Scholar]
- 2.Virmani R, Burke AP, Kolodgie F. Morphological characteristics of coronary atherosclerosis in diabetes mellitus. The Canadian journal of cardiology. 2006;22(Suppl B):81b–84b. doi: 10.1016/s0828-282x(06)70991-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gu K, Cowie CC, Harris MI. Diabetes and decline in heart disease mortality in US adults. Jama. 1999;281:1291–7. doi: 10.1001/jama.281.14.1291. [DOI] [PubMed] [Google Scholar]
- 4.Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donahoe SM, Stewart GC, McCabe CH, Mohanavelu S, Murphy SA, Cannon CP, Antman EM. Diabetes and mortality following acute coronary syndromes. Jama. 2007;298:765–75. doi: 10.1001/jama.298.7.765. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee PS, Ma J, Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci U S A. 2015;112:6050–5. doi: 10.1073/pnas.1424017112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–6. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. The Journal of Clinical Investigation. 2013;123:1262–1274. doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinik AI, Ziegler D. Diabetic cardiovascular autonomic neuropathy. Circulation. 2007;115:387–97. doi: 10.1161/CIRCULATIONAHA.106.634949. [DOI] [PubMed] [Google Scholar]
- 10.Battiprolu P, Wnag Zhao V, Hill Joseph A. Diabetic Cardiomyopathy Mediators and Mechanisms. In: McGuire DK, Marx N, editors. Diabetes in cardiovascular disease : a companion to Braunwald’s heart disease. pp. 290–230. [Google Scholar]
- 11.Ramasamy R, Yan SF, Schmidt AM. Vascular Biology of Atherosclerosis in Patients with Diabetes. In: McGuire DK, Marx N, editors. Diabetes in cardiovascular disease : a companion to Braunwald’s heart disease. 2015. 1 online resource. [Google Scholar]
- 12.Plutzky J, Zafir B, Brown JD. Vascular Biology of Atherosclerosis in Patients with Diabetes. In: McGuire DK, Marx N, editors. Diabetes in cardiovascular disease : a companion to Braunwald’s heart disease. Elsvier; 2015. 1 online resource. [Google Scholar]
- 13.Ramasamy R, Goldberg IJ. Aldose reductase and cardiovascular diseases, creating human-like diabetic complications in an experimental model. Circ Res. 2010;106:1449–58. doi: 10.1161/CIRCRESAHA.109.213447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vedantham S, Noh H, Ananthakrishnan R, Son N, Hallam K, Hu Y, Yu S, Shen X, Rosario R, Lu Y, Ravindranath T, Drosatos K, Huggins LA, Schmidt AM, Goldberg IJ, Ramasamy R. Human aldose reductase expression accelerates atherosclerosis in diabetic apolipoprotein E−/− mice. Arterioscler Thromb Vasc Biol. 2011;31:1805–13. doi: 10.1161/ATVBAHA.111.226902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vedantham S, Thiagarajan D, Ananthakrishnan R, Wang L, Rosario R, Zou YS, Goldberg I, Yan SF, Schmidt AM, Ramasamy R. Aldose reductase drives hyperacetylation of Egr-1 in hyperglycemia and consequent upregulation of proinflammatory and prothrombotic signals. Diabetes. 2014;63:761–74. doi: 10.2337/db13-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srivastava S, Vladykovskaya E, Barski OA, Spite M, Kaiserova K, Petrash JM, Chung SS, Hunt G, Dawn B, Bhatnagar A. Aldose reductase protects against early atherosclerotic lesion formation in apolipoprotein E-null mice. Circ Res. 2009;105:793–802. doi: 10.1161/CIRCRESAHA.109.200568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J. 2003;375:581–92. doi: 10.1042/BJ20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thornalley PJ. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. The Biochemical journal. 1990;269:1–11. doi: 10.1042/bj2690001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao D, Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes. 2010;59:249–55. doi: 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rong LL, Gooch C, Szabolcs M, Herold KC, Lalla E, Hays AP, Yan SF, Yan SSD, Schmidt AM. RAGE: a journey from the complications of diabetes to disorders of the nervous system – striking a fine balance between injury and repair. Restorative neurology and neuroscience. 2005;23:355–65. [PubMed] [Google Scholar]
- 21.Tikellis C, Pickering RJ, Tsorotes D, Huet O, Cooper ME, Jandeleit-Dahm K, Thomas MC. Dicarbonyl stress in the absence of hyperglycemia increases endothelial inflammation and atherogenesis similar to that observed in diabetes. Diabetes. 2014;63:3915–25. doi: 10.2337/db13-0932. [DOI] [PubMed] [Google Scholar]
- 22.Hanssen NM, Wouters K, Huijberts MS, Gijbels MJ, Sluimer JC, Scheijen JL, Heeneman S, Biessen EA, Daemen MJ, Brownlee M, de Kleijn DP, Stehouwer CD, Pasterkamp G, Schalkwijk CG. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur Heart J. 2014;35:1137–46. doi: 10.1093/eurheartj/eht402. [DOI] [PubMed] [Google Scholar]
- 23.Tian C, Alomar F, Moore CJ, Shao CH, Kutty S, Singh J, Bidasee KR. Reactive carbonyl species and their roles in sarcoplasmic reticulum Ca2+ cycling defect in the diabetic heart. Heart failure reviews. 2014;19:101–12. doi: 10.1007/s10741-013-9384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molgat AS, Tilokee EL, Rafatian G, Vulesevic B, Ruel M, Milne R, Suuronen EJ, Davis DR. Hyperglycemia inhibits cardiac stem cell-mediated cardiac repair and angiogenic capacity. Circulation. 2014;130:S70–6. doi: 10.1161/CIRCULATIONAHA.113.007908. [DOI] [PubMed] [Google Scholar]
- 25.Vulesevic B, McNeill B, Geoffrion M, Kuraitis D, McBane JE, Lochhead M, Vanderhyden BC, Korbutt GS, Milne RW, Suuronen EJ. Glyoxalase-1 overexpression in bone marrow cells reverses defective neovascularization in STZ-induced diabetic mice. Cardiovasc Res. 2014;101:306–16. doi: 10.1093/cvr/cvt259. [DOI] [PubMed] [Google Scholar]
- 26.Nam DH, Han JH, Lee TJ, Shishido T, Lim JH, Kim GY, Woo CH. CHOP deficiency prevents methylglyoxal-induced myocyte apoptosis and cardiac dysfunction. Journal of molecular and cellular cardiology. 2015;85:168–77. doi: 10.1016/j.yjmcc.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 27.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell metabolism. 2009;9:474–81. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parodi EM, Kuhn B. Signalling between microvascular endothelium and cardiomyocytes through neuregulin. Cardiovasc Res. 2014;102:194–204. doi: 10.1093/cvr/cvu021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craven PA, Davidson CM, DeRubertis FR. Increase in diacylglycerol mass in isolated glomeruli by glucose from de novo synthesis of glycerolipids. Diabetes. 1990;39:667–74. doi: 10.2337/diab.39.6.667. [DOI] [PubMed] [Google Scholar]
- 30.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:11059–63. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiba T, Inoguchi T, Sportsman JR, Heath WF, Bursell S, King GL. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. The American journal of physiology. 1993;265:E783–93. doi: 10.1152/ajpendo.1993.265.5.E783. [DOI] [PubMed] [Google Scholar]
- 32.Cosentino-Gomes D, Rocco-Machado N, Meyer-Fernandes JR. Cell signaling through protein kinase C oxidation and activation. International journal of molecular sciences. 2012;13:10697–721. doi: 10.3390/ijms130910697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–31. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong L, Shen X, Lin L, Leitges M, Rosario R, Zou YS, Yan SF. PKCbeta promotes vascular inflammation and acceleration of atherosclerosis in diabetic ApoE null mice. Arterioscler Thromb Vasc Biol. 2013;33:1779–87. doi: 10.1161/ATVBAHA.112.301113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durpes MC, Morin C, Paquin-Veillet J, Beland R, Pare M, Guimond MO, Rekhter M, King GL, Geraldes P. PKC-beta activation inhibits IL-18-binding protein causing endothelial dysfunction and diabetic atherosclerosis. Cardiovasc Res. 2015;106:303–13. doi: 10.1093/cvr/cvv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q, Park K, Li C, Rask-Madsen C, Mima A, Qi W, Mizutani K, Huang P, King GL. Induction of vascular insulin resistance and endothelin-1 expression and acceleration of atherosclerosis by the overexpression of protein kinase C-beta isoform in the endothelium. Circ Res. 2013;113:418–27. doi: 10.1161/CIRCRESAHA.113.301074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Way KJ, Isshiki K, Suzuma K, Yokota T, Zvagelsky D, Schoen FJ, Sandusky GE, Pechous PA, Vlahos CJ, Wakasaki H, King GL. Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C beta2 activation and diabetes. Diabetes. 2002;51:2709–18. doi: 10.2337/diabetes.51.9.2709. [DOI] [PubMed] [Google Scholar]
- 38.Verma SK, Deshmukh V, Liu P, Nutter CA, Espejo R, Hung ML, Wang GS, Yeo GW, Kuyumcu-Martinez MN. Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. The Journal of biological chemistry. 2013;288:35372–86. doi: 10.1074/jbc.M113.507426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell metabolism. 2013;17:20–33. doi: 10.1016/j.cmet.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardiville S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell metabolism. 2014;20:208–13. doi: 10.1016/j.cmet.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. The Journal of cell biology. 2015;208:869–80. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marsh SA, Collins HE, Chatham JC. Protein O-GlcNAcylation and cardiovascular (patho)physiology. The Journal of biological chemistry. 2014;289:34449–56. doi: 10.1074/jbc.R114.585984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Makino A, Dai A, Han Y, Youssef KD, Wang W, Donthamsetty R, Scott BT, Wang H, Dillmann WH. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am J Physiol Cell Physiol. 2015;309:C593–9. doi: 10.1152/ajpcell.00069.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–6. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 45.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. The Journal of clinical investigation. 2001;108:1341–8. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qian J, Fulton D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front Physiol. 2013;4:347. doi: 10.3389/fphys.2013.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G, Lauro R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. 2002;106:466–72. doi: 10.1161/01.cir.0000023043.02648.51. [DOI] [PubMed] [Google Scholar]
- 48.Shrikhande GV, Scali ST, da Silva CG, Damrauer SM, Csizmadia E, Putheti P, Matthey M, Arjoon R, Patel R, Siracuse JJ, Maccariello ER, Andersen ND, Monahan T, Peterson C, Essayagh S, Studer P, Guedes RP, Kocher O, Usheva A, Veves A, Kaczmarek E, Ferran C. O-glycosylation regulates ubiquitination and degradation of the anti-inflammatory protein A20 to accelerate atherosclerosis in diabetic ApoE-null mice. PLoS One. 2010;5:e14240. doi: 10.1371/journal.pone.0014240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. The Journal of biological chemistry. 2003;278:44230–7. doi: 10.1074/jbc.M303810200. [DOI] [PubMed] [Google Scholar]
- 50.Daniels L, Bell JR, Delbridge LM, McDonald FJ, Lamberts RR, Erickson JR. The role of CaMKII in diabetic heart dysfunction. Heart failure reviews. 2015;20:589–600. doi: 10.1007/s10741-015-9498-3. [DOI] [PubMed] [Google Scholar]
- 51.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 52.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 53.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–25. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 54.Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, Hoppel CL. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes. 2012;61:2074–83. doi: 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crabtree MJ, Tatham AL, Al-Wakeel Y, Warrick N, Hale AB, Cai S, Channon KM, Alp NJ. Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: insights from cells with tet-regulated GTP cyclohydrolase I expression. The Journal of biological chemistry. 2009;284:1136–44. doi: 10.1074/jbc.M805403200. [DOI] [PubMed] [Google Scholar]
- 56.Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell metabolism. 2012;15:186–200. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giacco F, Du X, Carratu A, Gerfen GJ, D’Apolito M, Giardino I, Rasola A, Marin O, Divakaruni AS, Murphy AN, Shah MS, Brownlee M. GLP-1 Cleavage Product Reverses Persistent ROS Generation After Transient Hyperglycemia by Disrupting an ROS-Generating Feedback Loop. Diabetes. 2015;64:3273–84. doi: 10.2337/db15-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schaffer SW, Jong CJ, Mozaffari M. Role of oxidative stress in diabetes-mediated vascular dysfunction: unifying hypothesis of diabetes revisited. Vascul Pharmacol. 2012;57:139–49. doi: 10.1016/j.vph.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 60.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–62. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 61.Zou M-H, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes. 2002;51:198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ Res. 2014;114:421–33. doi: 10.1161/CIRCRESAHA.114.302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maalouf RM, Eid AA, Gorin YC, Block K, Escobar GP, Bailey S, Abboud HE. Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am J Physiol Cell Physiol. 2012;302:C597–604. doi: 10.1152/ajpcell.00331.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang M, Kho AL, Anilkumar N, Chibber R, Pagano PJ, Shah AM, Cave AC. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. 2006;113:1235–43. doi: 10.1161/CIRCULATIONAHA.105.581397. [DOI] [PubMed] [Google Scholar]
- 65.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- 66.Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes. 2004;53:1336–43. doi: 10.2337/diabetes.53.5.1336. [DOI] [PubMed] [Google Scholar]
- 67.Finkel T. Signal transduction by reactive oxygen species. The Journal of cell biology. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–80. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 70.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol. 2011;31:1368–76. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 71.Schmelter M, Ateghang B, Helmig S, Wartenberg M, Sauer H. Embryonic stem cells utilize reactive oxygen species as transducers of mechanical strain-induced cardiovascular differentiation. FASEB J. 2006;20:1182–4. doi: 10.1096/fj.05-4723fje. [DOI] [PubMed] [Google Scholar]
- 72.Heo JS, Lee JC. beta-Catenin mediates cyclic strain-stimulated cardiomyogenesis in mouse embryonic stem cells through ROS-dependent and integrin-mediated PI3K/Akt pathways. J Cell Biochem. 2011;112:1880–9. doi: 10.1002/jcb.23108. [DOI] [PubMed] [Google Scholar]
- 73.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–5. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 74.Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nature reviews Immunology. 2013;13:349–61. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham N, Park BK, Souma T, Moriguchi T, Yamamoto M, Thornalley PJ. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem J. 2012;443:213–22. doi: 10.1042/BJ20111648. [DOI] [PubMed] [Google Scholar]
- 77.Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, Lin J, Bierhaus A, Nawroth P, Hannak D, Neumaier M, Bergfeld R, Giardino I, Brownlee M. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. 2003;9:294–9. doi: 10.1038/nm834. [DOI] [PubMed] [Google Scholar]
- 78.Du Y, Kowluru A, Kern TS. PP2A contributes to endothelial death in high glucose: inhibition by benfotiamine. American journal of physiology Regulatory, integrative and comparative physiology. 2010;299:R1610–7. doi: 10.1152/ajpregu.00676.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Katare RG, Caporali A, Oikawa A, Meloni M, Emanueli C, Madeddu P. Vitamin B1 analog benfotiamine prevents diabetes-induced diastolic dysfunction and heart failure through Akt/Pim-1-mediated survival pathway. Circulation Heart failure. 2010;3:294–305. doi: 10.1161/CIRCHEARTFAILURE.109.903450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2-an update. Free radical biology & medicine. 2014;66:36–44. doi: 10.1016/j.freeradbiomed.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–33. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xue M, Qian Q, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R, Thornalley PJ. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes. 2008;57:2809–17. doi: 10.2337/db06-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Zhang Z, Sun W, Tan Y, Liu Y, Zheng Y, Liu Q, Cai L, Sun J. Sulforaphane attenuation of type 2 diabetes-induced aortic damage was associated with the upregulation of Nrf2 expression and function. Oxid Med Cell Longev. 2014;2014:123963. doi: 10.1155/2014/123963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Z, Wang S, Zhou S, Yan X, Wang Y, Chen J, Mellen N, Kong M, Gu J, Tan Y, Zheng Y, Cai L. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. Journal of molecular and cellular cardiology. 2014;77:42–52. doi: 10.1016/j.yjmcc.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 85.Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X, Goldblatt CS, Meyer CJ, Li X, Cai L, Cui T. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625–33. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–32. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 87.Olabisi OA, Soto-Nieves N, Nieves E, Yang TT, Yang X, Yu RY, Suk HY, Macian F, Chow CW. Regulation of transcription factor NFAT by ADP-ribosylation. Mol Cell Biol. 2008;28:2860–71. doi: 10.1128/MCB.01746-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nilsson-Berglund LM, Zetterqvist AV, Nilsson-Ohman J, Sigvardsson M, Gonzalez Bosc LV, Smith ML, Salehi A, Agardh E, Fredrikson GN, Agardh CD, Nilsson J, Wamhoff BR, Hultgardh-Nilsson A, Gomez MF. Nuclear factor of activated T cells regulates osteopontin expression in arterial smooth muscle in response to diabetes-induced hyperglycemia. Arterioscler Thromb Vasc Biol. 2010;30:218–24. doi: 10.1161/ATVBAHA.109.199299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zetterqvist AV, Berglund LM, Blanco F, Garcia-Vaz E, Wigren M, Duner P, Andersson AM, To F, Spegel P, Nilsson J, Bengtsson E, Gomez MF. Inhibition of nuclear factor of activated T-cells (NFAT) suppresses accelerated atherosclerosis in diabetic mice. PLoS One. 2014;8:e65020. doi: 10.1371/journal.pone.0065020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Friedman JK, Nitta CH, Henderson KM, Codianni SJ, Sanchez L, Ramiro-Diaz JM, Howard TA, Giermakowska W, Kanagy NL, Gonzalez Bosc LV. Intermittent hypoxia-induced increases in reactive oxygen species activate NFATc3 increasing endothelin-1 vasoconstrictor reactivity. Vascul Pharmacol. 2014;60:17–24. doi: 10.1016/j.vph.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Letavernier E, Zafrani L, Perez J, Letavernier B, Haymann JP, Baud L. The role of calpains in myocardial remodelling and heart failure. Cardiovasc Res. 2012;96:38–45. doi: 10.1093/cvr/cvs099. [DOI] [PubMed] [Google Scholar]
- 92.Bourajjaj M, Armand AS, da Costa Martins PA, Weijts B, van der Nagel R, Heeneman S, Wehrens XH, De Windt LJ. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. The Journal of biological chemistry. 2008;283:22295–303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- 93.Li Y, Ma J, Zhu H, Singh M, Hill D, Greer PA, Arnold JM, Abel ED, Peng T. Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes. 2011;60:2985–94. doi: 10.2337/db10-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang JC, Zhao Y, Li XD, Zhou NN, Sun H, Sun YY. Proteolysis by endogenous calpain I leads to the activation of calcineurin in human heart. Clinical laboratory. 2012;58:1145–52. [PubMed] [Google Scholar]
- 95.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–22. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 96.Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–20. doi: 10.1126/science.aaa8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 98.Fadini GP, Menegazzo L, Scattolini V, Gintoli M, Albiero M, Avogaro A. A perspective on NETosis in diabetes and cardiometabolic disorders. Nutr Metab Cardiovasc Dis. 2016;26:1–8. doi: 10.1016/j.numecd.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 99.Nahrendorf M, Swirski FK. Immunology. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science. 2015;349:237–8. doi: 10.1126/science.aac7801. [DOI] [PubMed] [Google Scholar]
- 100.Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Frontiers in immunology. 2012;3:360. doi: 10.3389/fimmu.2012.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–9. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Abbas AK, Le K, Pimmett VL, Bell DA, Cairns E, Dekoter RP. Negative regulation of the peptidylarginine deiminase type IV promoter by NF-kappaB in human myeloid cells. Gene. 2014;533:123–31. doi: 10.1016/j.gene.2013.09.108. [DOI] [PubMed] [Google Scholar]
- 103.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 104.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nature immunology. 2005;6:1191–7. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 105.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, Zhang M, Zhang Y, An F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One. 2014;9:e104771. doi: 10.1371/journal.pone.0104771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Frontiers in pharmacology. 2015;6:262. doi: 10.3389/fphar.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature immunology. 2013;14:454–60. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 110.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–23. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, Sun C, Liu X, Jefferson LS, Xiong J, Lanoue KF, Chang Z, Lynch CJ, Wang H, Shi Y. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell metabolism. 2010;12:154–65. doi: 10.1016/j.cmet.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.He Q, Han X. Cardiolipin remodeling in diabetic heart. Chemistry and physics of lipids. 2014;179:75–81. doi: 10.1016/j.chemphyslip.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 113.Wang JX, Gao J, Ding SL, Wang K, Jiao JQ, Wang Y, Sun T, Zhou LY, Long B, Zhang XJ, Li Q, Liu JP, Feng C, Liu J, Gong Y, Zhou Z, Li PF. Oxidative Modification of miR-184 Enables It to Target Bcl-xL and Bcl-w. Mol Cell. 2015;59:50–61. doi: 10.1016/j.molcel.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 114.Wagschal A, Najafi-Shoushtari SH, Wang L, Goedeke L, Sinha S, deLemos AS, Black JC, Ramirez CM, Li Y, Tewhey R, Hatoum I, Shah N, Lu Y, Kristo F, Psychogios N, Vrbanac V, Lu YC, Hla T, de Cabo R, Tsang JS, Schadt E, Sabeti PC, Kathiresan S, Cohen DE, Whetstine J, Chung RT, Fernandez-Hernando C, Kaplan LM, Bernards A, Gerszten RE, Naar AM. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat Med. 2015;21:1290–7. doi: 10.1038/nm.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou Q, Lv D, Chen P, Xu T, Fu S, Li J, Bei Y. MicroRNAs in diabetic cardiomyopathy and clinical perspectives. Front Genet. 2014;5:185. doi: 10.3389/fgene.2014.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Matkovich SJ, Hu Y, Eschenbacher WH, Dorn LE, Dorn GW., 2nd Direct and indirect involvement of microRNA-499 in clinical and experimental cardiomyopathy. Circ Res. 2012;111:521–31. doi: 10.1161/CIRCRESAHA.112.265736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW., 2nd MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106:166–75. doi: 10.1161/CIRCRESAHA.109.202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, Kang Y, Li X, Kemper B, Kemper JK. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging cell. 2013;12:1062–72. doi: 10.1111/acel.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li Y, Xu S, Jiang B, Cohen RA, Zang M. Activation of sterol regulatory element binding protein and NLRP3 inflammasome in atherosclerotic lesion development in diabetic pigs. PLoS One. 2013;8:e67532. doi: 10.1371/journal.pone.0067532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, Izuhara M, Nakao T, Nishino T, Otsu K, Kita T, Kimura T, Ono K. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res. 2015;116:279–88. doi: 10.1161/CIRCRESAHA.116.304707. [DOI] [PubMed] [Google Scholar]
- 121.Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, Raffatellu M, Osborne TF. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell metabolism. 2011;13:540–9. doi: 10.1016/j.cmet.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 123.Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nolan CJ, Ruderman NB, Kahn SE, Pedersen O, Prentki M. Insulin resistance as a physiological defense against metabolic stress: implications for the management of subsets of type 2 diabetes. Diabetes. 2015;64:673–86. doi: 10.2337/db14-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell metabolism. 2008;7:95–6. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 126.Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159:1253–62. doi: 10.1016/j.cell.2014.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Du X, Edelstein D, Obici S, Higham N, Zou M-H, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. The Journal of clinical investigation. 2006;116:1071–80. doi: 10.1172/JCI23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jiang ZY, Lin YW, Clemont A, Feener EP, Hein KD, Igarashi M, Yamauchi T, White MF, King GL. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. The Journal of clinical investigation. 1999;104:447–57. doi: 10.1172/JCI5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–88. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 130.Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N, Farb MG, Rosenzweig J, Ruderman N, Vita JA, Hamburg NM. Protein kinase C-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li H, Forstermann U. Pharmacological prevention of eNOS uncoupling. Current pharmaceutical design. 2014;20:3595–606. doi: 10.2174/13816128113196660749. [DOI] [PubMed] [Google Scholar]
- 132.Bayeva M, Sawicki KT, Ardehali H. Taking diabetes to heart–deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc. 2013;2:e000433. doi: 10.1161/JAHA.113.000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nature reviews Endocrinology. 2015 doi: 10.1038/nrendo.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pulinilkunnil T, Rodrigues B. Cardiac lipoprotein lipase: metabolic basis for diabetic heart disease. Cardiovasc Res. 2006;69:329–40. doi: 10.1016/j.cardiores.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 135.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111:419–26. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Greenwalt DE, Scheck SH, Rhinehart-Jones T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J Clin Invest. 1995;96:1382–8. doi: 10.1172/JCI118173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tremblay F, Dubois MJ, Marette A. Regulation of GLUT4 traffic and function by insulin and contraction in skeletal muscle. Front Biosci. 2003;8:d1072–84. doi: 10.2741/1137. [DOI] [PubMed] [Google Scholar]
- 138.Doria A, Nosadini R, Avogaro A, Fioretto P, Crepaldi G. Myocardial metabolism in type 1 diabetic patients without coronary artery disease. Diabet Med. 1991;8(Spec No):S104–7. doi: 10.1111/j.1464-5491.1991.tb02168.x. [DOI] [PubMed] [Google Scholar]
- 139.Camps M, Castello A, Munoz P, Monfar M, Testar X, Palacin M, Zorzano A. Effect of diabetes and fasting on GLUT-4 (muscle/fat) glucose-transporter expression in insulin-sensitive tissues. Heterogeneous response in heart, red and white muscle. Biochem J. 1992;282(Pt 3):765–72. doi: 10.1042/bj2820765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A. 2003;100:1226–31. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. Journal of molecular and cellular cardiology. 2008;44:968–75. doi: 10.1016/j.yjmcc.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 142.Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, Semenkovich CF. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138:476–88. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C. The protein kinase C signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol Endocrinol. 2004;18:1906–18. doi: 10.1210/me.2003-0327. [DOI] [PubMed] [Google Scholar]
- 144.Quintana MT, He J, Sullivan J, Grevengoed T, Schisler J, Han Y, Hill JA, Yates CC, Stansfield WE, Mapanga RF, Essop MF, Muehlbauer MJ, Newgard CB, Bain JR, Willis MS. Muscle ring finger-3 protects against diabetic cardiomyopathy induced by a high fat diet. BMC Endocr Disord. 2015;15:36. doi: 10.1186/s12902-015-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chaurasia B, Summers SA. Ceramides – Lipotoxic Inducers of Metabolic Disorders. Trends in endocrinology and metabolism: TEM. 2015;26:538–50. doi: 10.1016/j.tem.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 146.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. Journal of lipid research. 2008;49:2101–12. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–66. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 148.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–8. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Han X, Yang J, Yang K, Zhao Z, Abendschein DR, Gross RW. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry. 2007;46:6417–28. doi: 10.1021/bi7004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ren M, Phoon CK, Schlame M. Metabolism and function of mitochondrial cardiolipin. Progress in lipid research. 2014;55:1–16. doi: 10.1016/j.plipres.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 151.Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochimica et biophysica acta. 2014;1837:408–17. doi: 10.1016/j.bbabio.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 152.Frohman MA. Role of mitochondrial lipids in guiding fission and fusion. J Mol Med (Berl) 2015;93:263–9. doi: 10.1007/s00109-014-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 154.Ozcan L, Wong CC, Li G, Xu T, Pajvani U, Park SK, Wronska A, Chen BX, Marks AR, Fukamizu A, Backs J, Singer HA, Yates JR, 3rd, Accili D, Tabas I. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell metabolism. 2012;15:739–51. doi: 10.1016/j.cmet.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, Lavandero S, Hill JA. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest. 2012;122:1109–18. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, Lavandero S. Mitochondrial dynamics, mitophagy and cardiovascular disease. The Journal of physiology. 2016;594:509–25. doi: 10.1113/JP271301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130:554–64. doi: 10.1161/CIRCULATIONAHA.113.008476. [DOI] [PubMed] [Google Scholar]
- 158.Parra V, Verdejo H, del Campo A, Pennanen C, Kuzmicic J, Iglewski M, Hill JA, Rothermel BA, Lavandero S. The complex interplay between mitochondrial dynamics and cardiac metabolism. J Bioenerg Biomembr. 2011;43:47–51. doi: 10.1007/s10863-011-9332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15:634–46. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Min K, Kwon OS, Smuder AJ, Wiggs MP, Sollanek KJ, Christou DD, Yoo JK, Hwang MH, Szeto HH, Kavazis AN, Powers SK. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. The Journal of physiology. 2015;593:2017–36. doi: 10.1113/jphysiol.2014.286518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Burkard N, Becher J, Heindl C, Neyses L, Schuh K, Ritter O. Targeted proteolysis sustains calcineurin activation. Circulation. 2005;111:1045–53. doi: 10.1161/01.CIR.0000156458.80515.F7. [DOI] [PubMed] [Google Scholar]
- 162.Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010;53:1783–94. doi: 10.1007/s00125-010-1770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, Zhu Y, Kuzmicic J, Pennanen C, Lopez-Crisosto C, Jana F, Ferreira J, Noguera E, Chiong M, Bernlohr DA, Klip A, Hill JA, Rothermel BA, Abel ED, Zorzano A, Lavandero S. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes. 2014;63:75–88. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–54. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liu C, Li S, Liu T, Borjigin J, Lin JD. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature. 2007;447:477–81. doi: 10.1038/nature05767. [DOI] [PubMed] [Google Scholar]
- 166.Bass J. Circadian topology of metabolism. Nature. 2012;491:348–56. doi: 10.1038/nature11704. [DOI] [PubMed] [Google Scholar]
- 167.Hoyle NP, O’Neill JS. Oxidation-reduction cycles of peroxiredoxin proteins and nontranscriptional aspects of timekeeping. Biochemistry. 2015;54:184–93. doi: 10.1021/bi5008386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Baggs JE, Price TS, DiTacchio L, Panda S, Fitzgerald GA, Hogenesch JB. Network features of the mammalian circadian clock. PLoS biology. 2009;7:e52. doi: 10.1371/journal.pbio.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Anea CB, Cheng B, Sharma S, Kumar S, Caldwell RW, Yao L, Ali MI, Merloiu AM, Stepp DW, Black SM, Fulton DJ, Rudic RD. Increased superoxide and endothelial NO synthase uncoupling in blood vessels of Bmal1-knockout mice. Circ Res. 2012;111:1157–65. doi: 10.1161/CIRCRESAHA.111.261750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Anea CB, Zhang M, Stepp DW, Simkins GB, Reed G, Fulton DJ, Rudic RD. Vascular disease in mice with a dysfunctional circadian clock. Circulation. 2009;119:1510–7. doi: 10.1161/CIRCULATIONAHA.108.827477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Schoenhard JA, Smith LH, Painter CA, Eren M, Johnson CH, Vaughan DE. Regulation of the PAI-1 promoter by circadian clock components: differential activation by BMAL1 and BMAL2. Journal of molecular and cellular cardiology. 2003;35:473–81. doi: 10.1016/s0022-2828(03)00051-8. [DOI] [PubMed] [Google Scholar]
- 172.Wang J, Yin L, Lazar MA. The orphan nuclear receptor Rev-erb alpha regulates circadian expression of plasminogen activator inhibitor type 1. The Journal of biological chemistry. 2006;281:33842–8. doi: 10.1074/jbc.M607873200. [DOI] [PubMed] [Google Scholar]
- 173.Jeyaraj D, Haldar SM, Wan X, McCauley MD, Ripperger JA, Hu K, Lu Y, Eapen BL, Sharma N, Ficker E, Cutler MJ, Gulick J, Sanbe A, Robbins J, Demolombe S, Kondratov RV, Shea SA, Albrecht U, Wehrens XH, Rosenbaum DS, Jain MK. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature. 2012;483:96–9. doi: 10.1038/nature10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nishikawa T, Brownlee M, Araki E. Mitochondrial reactive oxygen species in the pathogenesis of early diabetic nephropathy. Journal of diabetes investigation. 2015;6:137–9. doi: 10.1111/jdi.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.He B, Nohara K, Park N, Park YS, Guillory B, Zhao Z, Garcia JM, Koike N, Lee CC, Takahashi JS, Yoo SH, Chen Z. The Small Molecule Nobiletin Targets the Molecular Oscillator to Enhance Circadian Rhythms and Protect against Metabolic Syndrome. Cell metabolism. 2016;23:610–21. doi: 10.1016/j.cmet.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sovari AA, Rutledge CA, Jeong EM, Dolmatova E, Arasu D, Liu H, Vahdani N, Gu L, Zandieh S, Xiao L, Bonini MG, Duffy HS, Dudley SC., Jr Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circulation Arrhythmia and electrophysiology. 2013;6:623–31. doi: 10.1161/CIRCEP.112.976787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, Williams NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation. 2016 doi: 10.1161/CIRCULATIONAHA.115.020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Stevens MJ, Dayanikli F, Raffel DM, Allman KC, Sandford T, Feldman EL, Wieland DM, Corbett J, Schwaiger M. Scintigraphic assessment of regionalized defects in myocardial sympathetic innervation and blood flow regulation in diabetic patients with autonomic neuropathy. J Am Coll Cardiol. 1998;31:1575–84. doi: 10.1016/s0735-1097(98)00128-4. [DOI] [PubMed] [Google Scholar]
- 179.Campanucci V, Krishnaswamy A, Cooper E. Diabetes depresses synaptic transmission in sympathetic ganglia by inactivating nAChRs through a conserved intracellular cysteine residue. Neuron. 2010;66:827–34. doi: 10.1016/j.neuron.2010.06.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.