

Abstract
Eukaryotic chromatin is a complex and dynamic system in which the DNA double helix is organized and protected by interactions with histone proteins. This system is regulated through, a large network of dynamic post-translational modifications (PTMs) exists to ensure proper gene transcription, DNA repair, and other processes involving DNA. Homogenous protein samples with precisely characterized modification sites are necessary to better understand the functions of modified histone proteins. Here, we discuss sets of chemical and biological tools that have been developed for the preparation of modified histones, with a focus on the appropriate choice of tool for a given target. We start with genetic approaches for the creation of modified histones, including the incorporation of genetic mimics of histone modifications, chemical installation of modification analogs, and the use of the expanded genetic code to incorporate modified amino acids. Additionally, we will cover the chemical ligation techniques that have been invaluable in the generation of complex modified histones that are indistinguishable from the natural counterparts. Finally, we will end with a prospectus on future directions of synthetic chromatin in living systems.
1. Introduction
Eukaryotic chromatin is a complex and dynamic system in which the DNA double helix is organized and protected through interactions with histone proteins to form nucleosomes. These further interact to form higher order chromatin structures. This serves to stabilize and sequester DNA, while regulating interactions with biologically relevant functional partners. At the core of this regulatory system are the dynamic post-translational modifications of histone proteins that help control gene transcription, DNA repair, and a host of other cellular functions. The nucleosome is the unit structure of chromatin (Figure 1). In a canonical nucleosome, there are four primary histone proteins, H3, H4, H2A, and H2B. Two copies of each of H3 and H4 form the H32-H42 tetramer; while one copy each of H2A and H2B form the H2A/H2B dimer. One tetramer and two dimers together form the histone octamer, around which is wrapped ∼147 base pairs of DNA [1]. This structure is elaborated by incorporation of histone variants. For example, there are three major H3 variants (H3.1, H3.2, H3.3) in human chromatin, while the H3 variant CENP-A is found only in centromeric chromatin [2]. Similarly, the dimer may contain variant histones such as H2A variants H2A.X, which plays a role in DNA repair, or H2A.Z, which is implicated in regulation of a variety of cellular functions [3-5]. Specific incorporation of these histone variants is one mechanism by which chromatin function may be dynamically regulated.
Figure 1.

The structure of the nucleosome [141]. Wrapped DNA is depicted in gray. Histone H3 is shown in dark red; histone H4 in purple; histone H2A in dark blue, and histone H2B in light blue. The nucleosome dyad and entry-exit regions are labeled for clarity.
Conceptually, the nucleosome may be subdivided into two distinct functional areas: the highly structured histone core, which forms the primary binding surface for DNA, and the histone tails, which project out from the core and are typically unstructured in the context of a mononucleosome. Histone octamers are deposited to form nucleosomes in arrays along the DNA molecule, which along with linker histone H1 can compact into higher order chromatin structures. Taken as a whole, the nucleosome core packages and protects DNA, counteracting the negative charge of the phosphate backbone with the positive charge of the basic histones, and the act of wrapping physically occludes the DNA from interaction with cellular partners. The histone tails are poised to coordinate interactions between the nucleosomes and to recruit binding partners to regulate biological activity through complex patterns of modifications. All of these functions are dynamically mediated by combinations of histone post-translational modifications which, with a focus on the histone tails, has been described as the “histone code” [6, 7].
Histone proteins are extensively and specifically modified throughout the tail and core regions. Our knowledge of the nature and number of histone modifications is constantly expanding, and an exhaustive list of modifications is an ever-moving target [8, 9]. In part, lysines are commonly mono-, di-, or tri-methylated, acetylated, ubiquitinated, sumoylated, biotinylated, formylated, or crotonylated [10-12]. Arginine can be methylated or converted to citrulline. Ser, Thr, Tyr, and His may be phosphorylated, and Ser & Thr are glycosylated [13]. In vivo, these modifications are coordinated by an elaborate interplay of regulatory enzymes. In vitro, histones have served as high-value targets to develop protein chemistry tools to generate homogenous and precisely-modified histone proteins due to the number and importance of PTMs in human health and disease. These modified proteins can then be refolded back into functional nucleosomes and nucleosome arrays, to determine the molecular mechanisms by which this multitude of modifications functions.
Here, we discuss sets of chemical and biological tools for the creation of modified histones, focusing on the appropriate choice of tool for a given target. There have been several excellent and detailed reviews on the rapid expanding field of chemical ligation chemistry as applied to histone proteins [14-17]. We hope to find a unique niche with a general overview designed for those new to the field, with a focus on practical aspects of design and selected case studies, rather than an exhaustive survey. We will discuss genetic approaches to modified histones, including the incorporation of genetic mimics of histone modifications, chemical installation of residue analogs, and the use of expanded genetic code techniques to incorporate modified amino acids. Next, we will cover the chemical ligation techniques which have been invaluable in the generation of complex modified histones that are indistinguishable from the natural counterparts. We will discuss a variety of ligation approaches that have been developed for the production of these designer histones and chromatin. Finally, we will end with a perspective on future directions of synthetic chromatin in living systems.
2. Genetic Approaches for Modified Histones
2.1 Genetic mimics of histone modifications
Techniques in chemical biology allow unparalleled control over each residue of a protein, leading in the ideal case, to the generation of a chemically modified protein that is otherwise indistinguishable from the native counterpart. However, the simplest approach to studying a histone modification is the introduction of one of the 20 natural amino acids that mimics the features of the modified amino acid of interest (Figure 2). For example, glutamine has been used to substitute for acetylated lysine, and arginine for constitutively unmodified lysine. Glutamate or aspartate is often substituted for phosphorylated serine, threonine, or tyrosine, while alanine is used to replicate the unmodified residue.
Figure 2.

Side chains relevant to genetic mimics of modifications. 1: Unmodified lysine. 2: Acetylated lysine. 3: Arginine, used to mimic constitutively unmodified lysine. 4: Glutamine, commonly used to mimic constitutively acetylated lysine. 5: Threonine. 6: Phosphorylated threonine. 7: Aspartate and 8: Glutamate, commonly used to mimic constitutively phosphorylated residues.
These approaches have two primary advantages. First, using amino acids that are naturally available allows access to incredibly powerful genetic tools to screen for phenotypic effects of a histone modification [18]. Many early leads on functionally significant histone residues emerged out of large-scale mutational screens in yeast, and were later confirmed by mass spectrometry or other studies [19]. Of note, since these mimics introduce a completely different chemical moiety than either the modified or unmodified state, the sites of mimic incorporation are not capable of undergoing dynamic modification by histone modifying enzymes such as histone acetyltransferases, methyltransferases, or histone deacetylases, even in a cellular context. As such, observed changes may be due to a substitution mimicking either the modified or unmodified state, or through restriction of dynamic modification at the static residue. Second, nucleosomes are easily refolded and reconstituted from recombinant histone proteins expressed in and purified from E. coli [20]. Any laboratory with expertise in recombinant protein expression can generate large quantities of histones bearing mimics of acetylation or phosphorylation using standard techniques. This opens up the use of biochemical or biophysical techniques that require milligram quantities of a histone, such as crystallography, to assess the role of a modification [21-23].
When employing genetic substitution to mimic a modified amino acid, it is important to confirm that the mimic alters the same properties of a nucleosome as the modification that is being studied. In some cases, such as the effect of lysine acetylation in the H3 and H4 tails on chromatin compaction, Gln often appears to replicate the effects of lysine acetylation, suggesting that neutralization of the positive charge of an unmodified lysine, coupled with potential hydrogen bonding capabilities of the Gln amide, is sufficient to replicate acetylation[24, 25]. These results are contradicted by other studies suggesting that Gln mimics do not replicate the effect of precisely acetylated histone tails in compaction [26]. The study of histone modifications located in structured histone-DNA interfaces in the core of the nucleosome has offered a more nuanced look at these mimics. In the dyad region, H3-K115ac and H3-K122ac have been shown to destabilize the nucleosome structure [27]. However, H3-K115Q and H3-K122Q do not replicate this effect and, in fact, may stabilize the nucleosome slightly as assessed by competitive nucleosome reconstitution. Phosphorylation of H3-T118, also in the nucleosome dyad region, significantly destabilizes the nucleosome [28] by 40-fold relative to unmodified nucleosome standards and, in fact, can support a stable altered nucleosome structure [29]. H3-T118E as a mimic of phosphorylation does not have these effects, suggesting that negative charge is insufficient to replicate phosphorylation in the context of these functions.
Within the nucleosome entry-exit region, where DNA begins to contact the histone surface, the effectiveness of mimics is also ambiguous. Acetylation of H3-K56 has been demonstrated to increase DNA unwrapping from the histone octamer and to enhance transcription factor binding [30-32]. Incorporation of Gln as H3-K56Q qualitatively enhances DNA unwrapping, but does not quantitatively reproduce the effect of acetylation. However, as Gln does enhance DNA unwrapping, it may be sufficient to mimic this effect, depending on the precision required. These studies suggest that the ease of production of these natural amino acid mimics must be balanced against the elements of the modified amino acid that are essential for function. Further, mimics are a crude tool for some modifications, as there are no good mimics to distinguish, for instance, mono-, di-, and/or tri-methylation of lysine. These considerations are crucial to choose the tool appropriate to the task. Since each of the more elaborate methods developed for more precise replication of a modified residue also requires extra chemical steps or processing, it is reassuring to know that the extra effort and expense of modified histone preparation is justified by the improved accuracy and reliability of results.
2.2 Codon suppression: Expanded genetic code approaches to modified histones
Expanded genetic code approaches have been developed to site specifically insert unnatural amino acids at the amber codon (UAA) using orthogonal aminoacyl-tRNA synthetases (aaRS) and tRNA (Figure 3). These provide enhanced ability to use molecular biology approaches to generate modified histone proteins containing a wide range of modifications from naturally occurring PTMs to PTM mimics, fluorescent amino acids, and amino acids modified to alter their intrinsic chemistry. These subjects have been extensively discussed elsewhere [33, 34]; here, we will not attempt an exhaustive review but will highlight a few topics that span the diversity of applications in histone proteins.
Figure 3.
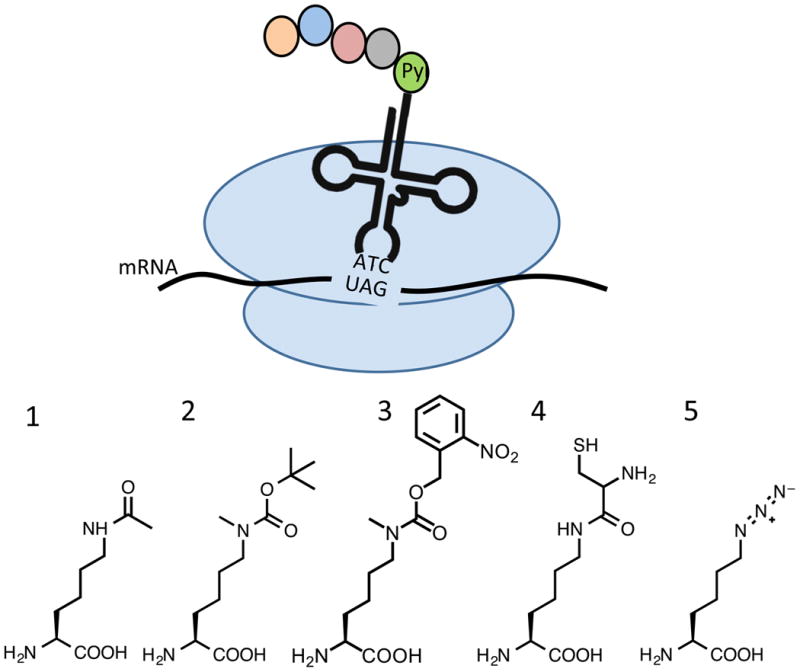
Top: Schematic for expanded genetic code incorporation of lysine mimics by modified pyrrolysine incorporation machinery. Bottom: Representative modified amino acids incorporated using expanded genetic code techniques. 1: Acetylated lysine. 2: Boc-protected Nε-methyllysine. 3: Photocaged Nε-methyllysine. 4: Nε(Cys)-Lysine 5: Azidonorleucine.
2.2.1 Encoded lysine modifications
Given the number of histone modifications that occur on lysine, the discovery of the pyrrolysine incorporation machinery in methanogens and subsequent development of artificial pyrrolysyl-tRNA synthetase (PylRS)/tRNA pairs for efficient incorporation of modified lysine residues has had a tremendous impact on the histone modification field [35, 36]. The synthetase has been effective at incorporating several lysine variants that are chemically similar to pyrrolysine, for example with a modification that includes an amide bond at the ε-amine of the lysine side chain (Figure 3, compounds 1-4). This is exemplified by the classic work of Neumann and coworkers in which they demonstrated the genetic incorporation of acetyllysine into H3-K56ac, located in the entry-exit region of the nucleosome [31]. Milligram quantities of uniformly acetylated histone H3 were generated using this genetic encoding system, which enabled bulk and single molecule FRET experiments that demonstrated an increase of DNA unwrapping and SWI/SNF-dependent chromatin remodeling upon acetylation without destabilizing the nucleosome as assessed by salt-induced dissociation studies, or affecting higher order chromatin structure in nucleosome arrays. Genetically incorporated acetyllysine has since been used to probe the role of lysine acetylation at several positions in the nucleosome core. In one key study, H3 acetylated at K122 was used to elucidate the role of H3-K122ac in transcriptional activation, partially through chromatin assembled in vitro with recombinantly expressed acetylated histone H3 [37]. An additional study of note used genetic incorporation of acetyllysine at H3-K64 to demonstrate that acetylation at this residue increased chromatin remodeling by Chd1, but not RSC, and additionally caused destabilization of the nucleosome as assessed by salt dependence of nucleosome dissociation and competitive nucleosome reconstitution [38]. In each case, the ability to easily generate sufficient quantities of uniformly acetylated histone proteins for nucleosome reconstitution was essential for the success of the study.
The genetic incorporation system used for acetylated lysines has been further developed for site-specific incorporation of methylated lysines. The development of a synthetase with specificity for methylated lysine over unmodified lysine is challenging due to their structural similarity. Therefore, methylated lysines are typically introduced as a protected derivative that is deprotected to reveal the methylated species. For example, Chin and coworkers generated H3 monomethylated at K9 through introduction of a Boc-protected monomethyllysine that could be deprotected with 2% TFA after purification (Figure 3, compound 2) [39]. A particularly elegant example is the genetic encoding of a photocaged Nε-methyl lysine (Figure 3, compound 3) by Liu and coworkers that can be deprotected by photolysis under mild conditions [40]. This opens the intriguing possibility of the dynamic introduction of methyllysine in living cells. More esoteric lysine modifications, including proprionyl, butyryl, or crotonyl-lysine have been found in histone proteins [41] as markers of active regions of chromatin. [10]. PylRS-tRNA pairs have been found to insert each of these modified lysines into histone proteins. [42, 43].
Expanded genetic code approaches in combination with other protein chemistry tools have been used to probe site-specific ubiquitination of histones and other proteins as well. Of note here are two interesting modified lysine variants that can be genetically incorporated as intermediates to ubiquitinated proteins. Lysine in which cysteine has been coupled to the ε-amine (Figure 3, compound 5) can be genetically incorporated, and then used as an avenue for native chemical ligation onto the lysine side chain [44, 45]. Similarly, azidonorleucine (Figure 3, compound 5) can serve as a protected lysine derivative, allowing chemistry specifically at this side chain position to introduce diubiquitin [46]. With the rapid pace of ongoing discovery in this area, many more interesting derivatives of lysine suitable for genetic incorporation may be anticipated in the future.
2.2.2 Combined genetic and chemical approaches: Modifications introduced through dehydroalanine
Several researchers have exploited genetic approaches to install chemical moieties that may be converted to the bioorthogonal, reactive dehydroalanine an entry point for the introduction of a wide variety of modified residues (Figure 4). Schultz and coworkers introduced phenylselenocysteine, which is susceptible to oxidative elimination to yield a reactive dehydroalanine moiety, at H3-K9. Michael addition of an appropriate thiol reagent results in a thioether analog of a post-translational modification [47]. Liu and coworkers evolved the pyrrolysine incorporation machinery to accept Nε-Cbz-lysine for site-specific incorporation at H3-K9, with protein yields reported at 100mg/L Mild oxidation again resulted in conversion to the reactive dehydroalanine, which was further converted into thioether analogs of methylated lysine, acetylated lysine, and phosphocysteine [48].
Figure 4.

Schematic for installation of PTM-mimics through a dehydroalanine intermediate. Alkylated selenocysteine or cysteine is converted to the reactive dehydroalanine with loss of stereochemistry. Michael addition of thiol reagent results in the corresponding post-translational modification mimic.
The site-specific incorporation of dehydroalanine is not restricted to expanded genetic code approaches. Davis and coworkers developed gentle chemical approaches for the conversion of a cysteine residue to dehydroalanine, by treatment with 2,5-dibromohexanediamide [49]. Through this approach, they introduced PTM mimics at single cysteine residues introduced at H3-K4, H3-K9, or H3-K79 [50]. Intriguingly, they also demonstrated that cysteine residues could be introduced simultaneously at H3-K4 and H3-K79, which resulted in two dehydroalanine moieties and therefore two PTM mimics installed at separate locations within the histone. However, it should be noted that conversion of cysteine or selenocysteine to dehydroalanine eliminates the chirality of the α-carbon. The PTM mimics are therefore not fully chirally resolved, although there have been suggestions that the inherent chirality of the protein molecule may lead to enrichment of the L-form PTM mimic.
2.2.3: Encoded phosphoserine
Serine phosphorylation in histones is essential to regulation of several cellular events. Phosphorylation of H3S10, in particular, is thought to be interdependent with acetylation and methylation of surrounding lysine residues in a network of modification switches [51]. Park and coworkers developed an expanded genetic code approach to the introduction of phosphoserine into proteins in E.coli, but with poor expression yields [52]. However, they used H3-S10 as an ideal platform to refine and improve the level of expression for genetically incorporated phosphoserine in the context of a physiologically relevant substrate[53]. They were able to improve yields to 3mg/L of culture, a 3000 fold improvement over previous work, and bringing expression to levels that are useful for production of designer nucleosomes. With the increased expression, they were able to demonstrate the importance of context in histone H3 modification by carrying out histone acetyltransferase (HAT) assays using Gcn5 and the Saga complex on H3 protein alone, octamers, and nucleosome arrays with and without H3-S10ph. They found that H3 alone demonstrated a decrease in acetylation compared to wild-type, histone octamers demonstrated similar levels with phosphorylated and unmodified substrates, while acetylation increased with H3-S10ph in nucleosome arrays. These results clearly demonstrate the importance of precisely characterized and regulated substrates in determining the function of histone modifications.
Expanded genetic code systems clearly are very powerful for the generation of modified proteins, particularly in laboratories that are most comfortable with molecular biology approaches to protein production[54]. They expand the reach of precisely regulated modified histone proteins, particularly with acetylated lysine, to a wider audience of chromatin researchers to determine the interplay between histone modifications and other cellular functions. The primary limitation is that without advanced techniques that are only beginning to be explored, only one modification at a time may be introduced into the histone sequence due to the limitation of the available matched stop codon pairs, and to the reduced yield often observed with each incorporation event. This is likely to change as designer organisms that lack amber codons in their genome become more common [55]. As an example, amber codon (UAG) suppression is one method used in nature by select organisms to expand their own genetic code, for example to include either selenocysteine (Sec) or pyrrolysine (Pyl) [56]. Since the native pyrrolysine tRNA/aaRS pair performs amber codon suppression, this is often used as the starting point for the directed co-evolution of new noncanonical lysine analog incorporation systems for efficient and specific amino-acyl charging of a noncanonical amino acids (reviewed in Liu et al. 2010 [33]). The use of tRNA that read quadruplet codons [57] was originally discovered in Salmonella typhimurium as a suppressor of a +1 frameshift mutation, and since then has been used as a way to make changes to translation without mutations to the ribosomal complex, which are often lethal [58, 59]. This has even given rise to suppression systems in which two modified amino acids can be inserted into a protein.
3: Chemical Installation of PTM Analogs at Single Cysteine Sites
Among the different functional groups presented by the 20 common amino acids, the cysteine sulfhydryl has unique chemical reactivity and is relatively rare in natural protein sequences. With careful selection of a modification reagent, cysteine therefore makes an attractive target for the site-specific incorporation of mimics of post-translational modifications. Cysteine modification can be extremely powerful due to the ease of site-directed mutagenesis for the incorporation of a nonnative cysteine, and the relatively high yields of recombinant histone proteins with these single site substitutions. This same reactivity is commonly used to introduce moieties which allow for direct characterization of a modified biomolecule or complex including spin labels [60, 61], fluorophores [62], cross-linkers [63], and foot printing reagents [64]allowing for biophysical characterization of nucleosomes. In this section, we will discuss methods that have been developed to exploit the reactivity of the cysteine sulfhydryl to introduce PTM mimics that resemble native modifications. We will discuss methods that alkylate cysteine directly to generate modified lysine mimics, or that use the unique properties of cysteine for a disulfide “staple” to reversibly link a modification such as ubiquitin to a histone. Methods that exploit the reactivity of cysteine are particularly appropriate for use in histone proteins, since there are no indispensable cysteine residues in the nucleosome. The only native cysteines in a typical Homo sapiens nucleosome composed of standard histone variants are found in histone H3; at position C110 in histone variants H3.1, H3.2, and H3.3, and at position 96 in histone variant H3.1 [65]. These cysteines are commonly replaced by Ser at position 96 and Ala at C110, with minimal to no perturbation of nucleosome structure, function, or dynamics [66, 67]. This allows introduction of a single cysteine elsewhere in the nucleosome without cross-reactivity with native residues.
3.1: MLAs: Methyllysine analogs
Shokat and coworkers revolutionized the study of methylated lysine residues in histones through the development of simple and elegant cysteine alkylation techniques that are accessible to a wide range of research groups [68]. The aminoethylation of cysteine to generate a lysine analog in which the γ-methylene is replaced with sulfide, with sufficiently similar properties to allow cleavage by trypsin, had been known for decades [69]. Shokat and coworkers made the key recognition that commercially available derivatives of the aminoethylation reagents could be exploited to produce the corresponding mono-, di-, or tri-methylated lysine analogs (MLA) (Figure 5A). Substituting the methylene with a sulfide decreases the pKa of the residue by 1.1, and increases the length of the sidechain by only 0.28 Å. The method is sufficiently high-throughput that in the initial study, specific methylated lysine variants were incorporated at H3-K4, H3-K9, H3-K36, H3-K79, and H4-K20. The proteins readily refolded into histone octamer for reconstitution into nucleosomes, and the MLA-modified nucleosomes were able to recapitulate modification-specific recognition by natural binding partners, such as interaction of H3-K9me2 with HP1α. MLAs were directly tested against the native modification in synthetic peptides to assess activity of the methyltransferase SUV39H1, which targets substrates methylated at H3-K9; the MLAs demonstrated equivalent activity to the native modification. Further, antibodies raised against the natural H3-K9me, H3-K9me2, H3-K9me3, H4-K20me, H3-K4me3, H3-K36me3, and H3-K79me2 also recognized the MLA equivalents of each of these nucleosomes. The one caveat was that the H3-K9me2 antibody showed 5-fold lower affinity, which suggests that the functional equivalency of the MLA is somewhat context-dependent. A survey of the literature suggests that this theme continues; in the majority of cases, MLAs are accessible and cost-efficient mimics of methylated histone function that allow the incorporation of methylated lysine mimics throughout the histone sequence, although concerns are raised in rare cases regarding the impact of the thioether moiety on specific interactions with the MLA.
Figure 5.

Modification of unique cysteine sites A. MLA approach, illustrated for dimethyllysine. B. Generation of the thio-methyl MTCTK acetylation mimic. C. Generation of acetyllysine mimic by thiol-ene chemistry.
The elegance and simplicity of the MLA approach (together with the commercial availability of methylated histones prepared using this approach) has enabled discovery and characterization of important functional histone interactions [70]. To select just a few examples: Crystallography requires large quantities of homogenous modified protein to explore the role of a histone modification in the nucleosome. MLAs were used to prepare nucleosomes that included H3-Kc79me2, H4-K20me3, or unmodified histone cores. Analysis of the crystal structures suggested that H3-K79 methylation alters local sidechain structure to partially reveal a hydrophobic pocket on the nucleosome surface. The ability to generate large quantities of nucleosome arrays with these pure MLA-modified histones also allowed experimental characterization of these modifications by analytical ultracentrifugation, which revealed an influence of the tail modification, but not the core, in chromatin compaction [71]. Similarly, MLAs have enabled structural characterization of methylated histone binding partners by nuclear magnetic resonance, for example low affinity binding of methylated H3-K36 by the histone deacetylase complex Rpd3S [72] or high affinity binding of nucleosomes methylated at H3-K36 to the PWWP domain of LEDGF, which plays an important role in HIV integration [73]. In this study, the ability to prepare homogenous MLA-nucleosomes was essential to the identification of cooperativity between a hydrophobic H3-K36me3 binding site and a basic surface patch that interacts with DNA [72]. While modified peptide pull-down studies were able to identify interactions of H3-K4me3 with the ING4 PHD finger, which is important to tumor suppression, only the ability to prepare MLA-nucleosomes identified that this interaction mediates acetylation on histone H3 within the nucleosome; the ability to prepare these well-defined samples is essential to understanding similar crosstalk between histone modifications [74]. The ability to rapidly generate different modification states of each lysine allows rapid screening across large numbers of differentially modified nucleosomes [75]. In sum, MLAs have proven to be a valuable chemical tool enabling biochemical and biophysical characterization of histone methylation important for biological function.
3.2: Acetyllysine analogs via cysteine alkylation
Alkylation of cysteine to form MLAs is rapid and high yielding using methylated 2-bromoethylamine derivatives. Unfortunately, reaction with corresponding alkylated reagents provide poor kinetics and poor yields [76]. The Cole Laboratory developed the methylthiocarbonyl-aziridine (MTCA) reagent as an alternate approach to introduce methylthiocarbonyl-thiaLys (MTCTK) acetylation mimics at cysteine sites (Fig. 5B), where the side chain includes the γ-sulfide as well as methylthiocarbonyl in place of the acetyl group. While the additional sulfur does add considerable steric bulk to the modification, specifically modified peptides could be recognized by interaction partners, including the Brdt bromodomain and specific antibodies, although with 2-4-fold lower affinity than the precisely modified acetyllysine, and could stimulate Rtt109 HAT activity, although to a lesser extent than acetyllysine. While MTCTK may not fully recapitulate the effect of lysine acetylation, the ease of installation may make it a valuable tool in the nucleosome context.
3.3: Thiol-ene chemistry to introduce modification analogs
An alternate approach to acetyllysine uses free radical induced thiol-ene (or “thiol click” [77]) chemistry to add N-vinyl-acetamide at a single cysteine site [78] to generate the corresponding acetyllysine mimic (Fig. 5C). This mimic replicated lysine deacetylation qualitatively, although not fully quantitatively. The mimic was further reconstituted into nucleosome arrays where it appeared to fully reproduce the impact of acetylation at H4-K16 on chromatin compaction. Fujimori and coworkers used a similar approach to introduce several methylarginine analogs into histone proteins (Figure 5D) [79]. In the methylated arginine mimics, the ε-nitrogen of the guanidyl group is replaced with a methylene, which perturbs both geometry and polarity of the arginine side chain. However, the mimic was functional across a wide range of methylarginine binding partners, indicating that reproduction of the terminal groups of methylarginine may dominate these interactions.
3.4: Disulfide stapling
The unique ability of cysteine to form disulfide bonds has been exploited for chemical ligation in histone proteins through the disulfide stapling approach, exemplified by the Muir Laboratory in preparation of a disulfide-linked histone-ubiquitin library [80]. To enable this approach, ubiquitin was expressed as a fusion with an intein domain (Figure 6). Thiolysis with the 1,2-aminothiol reagent 2-mercaptoethylamine generates a transient intermediate that undergoes rearrangement to form the stable, amide-linked thiol derivative, which can then be activated and incubated with a histone protein with a single cysteine to generate the disulfide-linked ubiquitylated histone. While this does not recapitulate the native isopeptide linkage of a ubiquitylated protein, nucleosomes with ubiquitin at H2B-K120C were able to stimulate the H3-K79 methyltransferase activity of hDot1L, suggesting the mimic was sufficiently similar for recognition. Further, this linkage enables the dynamic removal of a protein modification via reduction of the disulfide bond, mimicking the dynamic behavior of protein modifications under controlled conditions. The Muir Laboratory exploited this approach to determine the position-dependent effect of ubiquitin in histones H2A and H2B while demonstrating that H2B ubiquitylation perturbs chromatin compaction[80, 81]. A similar approach was used to prepare histone H4 with sumoylation at H4-K12 in the N-terminal tail, and demonstrated that SUMO-3 inhibits higher order chromatin structure required for chromatin compaction [12].
Figure 6.

Ubiquitin-histone disulfide stapling. Ubiquitin-intein fusion is thiolized by 2 mercaptoethylamine, which undergoes rearrangement to form the stable thiol derivative. Activation and disulfide bond formation results in the stapled ubiquitin-histone conjugate for study.
Each of the approaches described in this section benefits from the overexpression of a recombinant histone protein with a cysteine substitution at the site or sites of interest, with diverse yet elegant methods to elaborate these cysteines into a modification mimic. The power of these techniques lies in the ability to easily generate milligram quantities of modified histones. Consequently, an important limitation of these techniques are that only a single class of modification may be easily introduced within a single molecule due to the requirement for complete reaction of all cysteine residues to yield a homogenous modified sample. Highly modified histone proteins often contain varying numbers and types of post-translational modifications within a single molecule. Cysteine modification approaches are therefore unlikely to reach this level of diversity.
4. Chemical ligation for the preparation of modified histone proteins
While each method of preparing modified histones has advantages, to date, only chemical ligation has allowed access to histone proteins bearing the complex mixtures of multiple, different modifications representative of native modified proteins. In the general case, chemical ligation is any specific reaction of two peptide or protein segments via a bioorthogonal, chemoselective mechanism to generate a joined product. The most common type of chemical ligation used is native chemical ligation (NCL). In the original form, a peptide with an α-thioester at the C-terminus is reacted with a peptide that contains a 1,2-aminothiol, typically a cysteine residue, at the N-terminus to form a product peptide with a native peptide bond at the juncture [82]. If carried out at a site with a native Cys, the reaction generates a product indistinguishable from the parent sequence. If a peptide segment is generated synthetically, this approach allows full chemical control over each amino acid within the sequence. This allows the introduction of chemical moieties that are challenging to incorporate biologically – for example, amino acids bearing precise post-translational modifications at any desired location in the component peptide sequence. The utility of such an approach to chemically modified histones is immediately apparent.
4.1 Histone semi-synthesis by expressed protein ligation: Modifications near the histone N-terminus
Of course, the preparation of component peptides is limited by the typical parameters of solid phase peptide synthesis, such that peptide segments longer than ∼40-50 amino acids are challenging through standard techniques. The core histone proteins are approximately 100-140 amino acids in length, and a single ligation step is not sufficient to generate a full-length protein from fully synthetic peptide segments. However, many interesting histone modifications occur in the N-terminal histone tails. To access modifications in these regions, only the tails need be prepared synthetically to permit the controlled introduction of histone modifications. The remainder, and largest portion, of the protein sequence may be expressed recombinantly in E. coli, without any modifications along the sequence, as depicted in Figure 7A. In general, native chemical ligation carried out with at least one recombinant protein segment has been termed expressed protein ligation (EPL).
Figure 7.

Expressed protein ligation to generate A) H3 with modifications clustered near the N-terminus or B) H3 with modifications clustered near the C-terminus. Recombinant histone fragments are in blue; synthetic peptide fragments are in red. Similar schemes apply for other histone proteins, and desulfurization is optional depending on selection of ligation site.
A main challenge in employing EPL to generate histones with modifications in the N-terminal tails is the expression of the C-terminal portion of the protein with an N-terminal cysteine. Several methods have been used in histone proteins. The simplest approach is to introduce an initiator Met prior to a Cys, such that removal of the Met by aminopeptidase results in an N-terminal Cys [83]. This expressed protein can then be used directly in a ligation reaction. This approach does not work in all situations; N-terminal Cys is thought to be alkylated by pyruvate in a number of cases, which renders it incapable of acting as a chemoselective ligation site. Another common approach is to follow the initiator Met with a specific proteolysis site such that processing of the expressed protein will result in the active N-terminal Cys. This method also allows for the inclusion of an affinity tag N-terminal to the protease sequence. Proteases including Factor Xa [84], TEV protease [85], or SUMO protease[86] have been successfully used to reveal an N-terminal Cys on recombinant histone fragments with high yield and purity.
Historically, EPL has been used by many groups to study modifications in the N-terminal tails of histones [68, 83, 84, 87-92]. Shogren-Knaak and coworkers were the first to exploit this technique to generate histone H3 containing a phosphorylated serine residue (H3-S10ph, T32C). They demonstrated that the semi-synthetic histone could be refolded into octamer and reconstituted into homogeneous nucleosome arrays, resulting in an increased histone acetyltransferases activity with Gcn5 over WT [84]. However, the physiological Gcn5 complex, SAGA, does not show the same increase in acetyltransferases activity as recombinant Gcn5 and the phosphate modification did not introduce a disruption in the higher-order chromatin structure [87]. Later that same year, He et al 2003 reported the production of semi-synthetic histone H3 containing a methylated lysine residue (H3-K9me3) and histones H3 and H4 with multiple acetylated lysine residues (H3-K4ac,9ac,14ac,18ac,22ac; H4-K5ac,8ac and H4-K5ac,8ac,12ac). This group showed that both semi-synthetic modified histone H3 and H4 were able to form tetramer and serve as active substrate for histone modifying enzymes [83].
In a landmark early study, Shogren-Knaak and coworkers prepared histone H4-K16ac with an R23C substitution by ligating N-terminal peptide H3(1-22)-K16ac to the C-terminal histone fragment H3(23-102, R23C). They found that acetylation of lysine 16 affected higher-order chromatin structure by preventing chromatin compaction in nucleosome arrays, and interrupted protein-histone interaction with adenosine triphosphate-utilizing chromatin remodeling and assembly factor (ACF) [88]. This study clearly demonstrated the potential of semi-synthetic histones to address questions in chromatin structure and function. In another early study, Ferreira and coworkers prepared semi-synthetic, tetra-acetylated H3 (H3-K9ac,14ac,18ac,23ac,S28C) and H4 proteins (H4-K5ac,8ac,12ac,16ac,V21C) to investigate the effects of these modification on chromatin remodeling by Snf2, and found that combinations of acetylation impacted chromatin remodeling rates differentially. H3 tetra-acetylation increased thermal mobility of nucleosomes by two-fold over unmodified nucleosomes, and increased recruitment of chromatin structure remodeling (RSC) complex by 16-fold. H4 tetra-acetylation does not alter thermal mobility of nucleosomes and slightly reduced remodeling activity of both chromodomain helicase DNA binding protein 1 (Chd1) and Isw2 [89]. These results suggested a complex network of unique mechanisms that, although dissimilar, are interrelated to allow for multiple modes of regulation.
Approaches for the semi-synthesis of histones with modified N-terminal tails have reached near-ubiquity in the field, such that semi-synthetic modified histones have even become commercially available (Active Motif). As such, an exhaustive listing of studies with histones prepared using these approaches would be somewhat impractical. However, one recent advance of note is the use of sequential EPL, in which the N-terminal synthetic segment is split into two distinct segments. This allows the preparation of short peptide segments to achieve modifications of histone residues either closer to the core domain (such as H3-R42me2, prepared from recombinant H3(47-135) and synthetic H3(1-28) and H3(29-46)) [93], or somewhat synthetically challenging such as simultaneous modification of H3-K4me3 and H3-K27me3 prepared from recombinant H3(29-135) and synthetic H3(1-20) and H3(21-28)[94].
4.2: Considerations for selection of appropriate ligation sites
One challenge in the design of ligation strategies is the selection of appropriate ligation sites selected for the modification of interest. Many different ligation sites have been successfully used to generate modified histone proteins, and selection of an ideal ligation site depends on several factors. 1) The peptides must be synthetically accessible, of a reasonable length and maintaining desirable solubility properties. 2) The kinetics of the ligation reaction are dependent on the C-terminal residue of the thioester peptide such that β-branched residues should be avoided if possible, and proline is unsuitable[95]. 3) The classical NCL reaction results in a cysteine at the ligation junction. EPL of histone H3 with a synthetic C-terminal peptide carried out by Manohar and coworkers resulted in a cysteine at H3-C110 [27], which is the only native cysteine in nucleosomes in X. laevis. For all other EPL schemes, it is important to select a ligation site that can permit residue substitution by cysteine if no further chemical steps are planned. H3 residues 25 [96], 28 [89], and 31 [84], as well as H4 residues 23 [97], and 20 [89] have been used as ligation sites resulting in cysteine substitutions.
Because cysteine is rare in histone proteins, these cysteine substitutions result in a non-native histone sequence for most ligation schemes. Methods to convert cysteine to alanine through desulfurization dramatically increase the number of potential ligation sites that can result in native sequences, eliminating any potential effects of the substitution on histone function. Yan and Dawson introduced cysteine desulfurization by treatment with Raney nickel [98],. Early histone ligation protocols including those by the McCafferty Laboratory [83]exploited these approaches successfully to generate native histone sequences. Free radical desulfurization was introduced by Wan and Danishefsky in 2007 [99], using mild reagents fully compatible with ligation conditions. This alternate technique rapidly gained wide-spread adoption within the field and is currently the method of choice for most groups carrying out histone ligation chemistry. Free radical desulfurization requires tris(2-carboxy)phosphine (TCEP) and the water soluble radical initiator 2,2′-azobis[2-(2-imaidazolin-2-yl)propane]dihydrochloride (VA-044US), with a thiol proton source [99]. Several different thiols are compatible with both NCL and with free radical desulfurization, including mercaptoethanesulfonic acid (MESNA), which allows desulfurization to be carried out directly on crude ligation mixtures. However, aromatic thiols such as mercaptophenylacetic acid (MPAA) that are often used for improved ligation kinetics appear to quench desulfurization, requiring complete removal from a reaction mixture prior to desulfurization [100].
While the ability to use alanine ligation sites does expand the choices of ligation sites available, not all modifications are conveniently accessible through ligation schemes with these limited sites. The ability to carry out desulfurization, when combined with non-native 1,2-aminothiol derivatives of amino acids, has allowed the expansion of ligation sites to many more residues, reviewed in [101]. For example, penicillamine is a β-thiol derivative of valine that is commercially available in Fmoc- and Boc-protected forms, including as the thiazolidine derivative required for complex ligation schemes discussed in section 4.4. Ligation with an N-terminal penicillamine followed by desulfurization thus generates a valine residue at the ligation site valine [102]. Similar approaches have allowed ligation at an expanding number of amino acid sites including phenylalanine [103], leucine [46], and lysine [44] one of the most common residues in histone proteins, which will likely become more commonly used as the reagents become more accessible. Of course, using these expanded ligation sites requires that the peptide segment bearing the 1,2-aminothiol occur in a synthetic histone fragment; they are not suitable for a one-step EPL reaction scheme to introduce modifications into the N-terminal tail. Another consideration is that any methods for modification of cysteine, described in section 2, can be used to mask a residual ligation site. For example, alkylation of a ligation site Cys by 2-bromoethylamine or derivatives will yield thiolysine analogs [26].
4.3: Histone semi-synthesis by expressed protein ligation: Modifications near the histone C-terminus
While modifications near the N-terminus of histone proteins are easily accessible by the methods described above, several interesting modifications near the C-terminus of histone proteins require a different approach. Modifications of interest within ∼30 residues of the histone C-terminus include PTM sites in the C-terminal tail of histone H2A, as well as modifications at key interfaces in the structured nucleosome core in histones H3 and H4 [104, 105]. Access to these modified residues requires a synthetic C-terminal histone peptide that can incorporate the modified residues, and a recombinant N-terminal histone fragment that remains unmodified (Figure 7B). This has most commonly been accomplished by fusion of the N-terminal protein fragment with an intein domain. Folded intein domains are capable of transferring the fused protein segment, or “extein”, to a thiol side chain to form an intramolecular thioester [106]. This can be intercepted by free thiol to form a reactive thioester in solution. The Mxe GyrA intein has historically been the most commonly used due to its early discovery and commercial availability as the pTXB1 plasmid (New England Biolabs), the capability to refold the functional protein, and robust cleavage conditions including the presence of denaturant and high ionic strength [107].
The behavior of histone proteins fused to an intein is somewhat unpredictable. Muir and coworkers found that histone H2B(1-116) expresses as a soluble protein when fused to a GyrA intein-chitin binding domain construct (GyrA-CBD) [108] and may be purified over chitin columns to easily generate the functional intein derivative, but that histone H2A(1-113)-GyrA-His6 expressed into inclusion bodies, which is typical of most recombinant histone proteins. Similarly, our own laboratory has found that histone H3(1-109)-GyrA-CBD and H4(1-75)-GyrA-CBD express into inclusion bodies and, further, interact nonspecifically with chitin columns such that the chitin binding domain is not appropriate for purification purposes [27, 109]. As such, these proteins must be resolubilized from inclusion bodies and refolded to the active intein prior to thiolysis to generate the functional thioester for ligation. At this point, ligation may proceed with standard considerations.
Several modifications in the structured nucleosome core are accessible only via EPLwith a C-terminal synthetic peptide. This approach was used to explore the impact of acetylation in the histone-DNA interface at the nucleosome dyad region, at H3-K115ac,K122ac. In this approach, a native cysteine (H3-C110) was used as a ligation site, such that ligation generated the native histone sequence with the modification of interest. These studies demonstrated that acetylation in the nucleosome dyad reduces histone-DNA affinity and increases thermal repositioning, suggesting a destabilization of the histone-DNA interface[27]. Interestingly, Gln substitution for acetylated lysine does not replicate these effects. In addition, acetylation at the dyad increases nucleosome disassembly in the context of mechanically unwrapped DNA [110] and in the context of chromatin remodeling by the hMSH2/hMSH6 DNA repair complex [111]. Subsequent work by Tropberger and coworkers using H3-K122ac prepared using an expanded genetic code approach suggested a role for H3-K122ac in transcriptional activation that is consistent with the biophysical work carried out with the semi-synthetic histones [37]. EPL followed by desulfurization was used to determine the impact of acetylation at H4-K77ac,K79ac in the histone-DNA interface in the Loss of Ribosomal Silencing (LRS) region of the nucleosome [109], resulting in the generation of the native H4 sequence with an alanine at the ligation site. Acetylation in this region of the histone-DNA interface was found to increase DNA unwrapping and site exposure for transcription factor binding, with no effect on nucleosome disassembly. Taken together with studies of the nucleosome dyad region described above and for acetylation of H3-K56ac in the entry-exit region [30, 31]. These studies suggest that different regions of the histone-DNA interface play distinct structural and functional roles in the regulation of nucleosome unwrapping and disassembly [110].
Histone phosphorylation remains best accessed by ligation approaches. EPL enabled study of H3-T118ph, which places a phosphate group into the histone-DNA interface at the nucleosome dyad. This site had been highlighted in genetic screens in yeast as likely important for transcriptional regulation and DNA repair [19, 112, 113]. Preparation by EPL and incorporation into mononucleosomes revealed a significant impact. H3-T118ph dramatically decreases histone-DNA affinity by 2 kcal/mol, increases nucleosome mobility in thermal repositioning assays by 28-fold, and increases DNA accessibility near the dyad by 6-fold. Consistent with this picture of a destabilized histone-DNA interface, this modification also dramatically increased nucleosome disassembly by both hMSH2/hMSH6 and by the SWI/SNF chromatin remodeling complex [28]. Reconstitution of nucleosomes with H3-T118ph also resulted in formation of a second defined histone-DNA construct that was structurally distinct from nucleosomes [29]. Extensive characterization of this species suggested that on short segments of DNA, a nucleosome duplex is formed in which two histone octamers are wrapped by two segments of DNA. On long stretches of DNA up to 3 kb in length, structures form in which two histone octamers are wrapped by a single piece of DNA. These structures were reminiscent of the altosomes described during SWI/SNF chromatin remodeling [114, 115]. This study demonstrated an important role for H3-T118 in regulating DNA wrapping, and was the first to demonstrate that a single histone modification could result in large-scale alterations of the nucleosome structure. In both studies, reconstitution of nucleosomes with glutamate substitution for the phosphorylated threonine failed to recapitulate the effects of the modifications, which is a strong argument for the necessity of precise chemical modifications to understand the role of PTMS in chromatin structure and function.
The Muir Laboratory has extensively studied ubiquitylation of histones H2A and H2B using EPL schemes with synthetic C-terminal peptides, and optimizations to their ligation approaches over time have resulted in dramatically improved product yields. Their first chemical strategy to site-specifically introduce ubiquitin into a histone, produced ubiquitylated H2B (uH2B) via a three-piece ligation scheme in which the N-terminal fragment of H2B and the first 75 residues of ubiquitin were each expressed as intein fusions to generate the reactive thioester. H2B(117-125)-A117C was generated synthetically, with a photocleavable ligation auxiliary linked to the ε-amine of the lysine side chain. After ligation, desulfurization of the Cys to Ala (see section 3.3) and photolysis of the ligation auxiliary resulted in a traceless ligation. Significantly, this study found that ubiquitylation of H2B-K120 enhanced methylation of H3-K79 by the methyltransferase Dot1, a clear example of histone modification crosstalk [108]. Subsequent ligation approaches eliminated the synthetically costly photocleavable auxiliary in favor of a cysteine linked to the ε-amine of the lysine through an isopeptide bond, such that after ligation and desulfurization, the product maintained a native histone sequence but with a G76A mutation in the ubiquitin domain [116]. Preparation of ubiquitylated H2A added an additional twist, in that no suitable Cys or Ala ligation sites were available. Instead, H2A(1-113) was expressed as a thioester, and H2A(114-128) was synthesized with the thiazolidine derivative of penicillamine such that after ligation and desulfurization, the histone ligation junction was converted to the native Val residue [100].
While several laboratories have refined EPL approaches for the preparation of modified histone proteins, the study that perhaps best illustrates the full power of this methodology is the development of high-throughput libraries of nucleosomes, prepared with semi-synthetic modified histones, marked by reconstitution onto barcoded DNA for identification [117]. These libraries, currently prepared on the order of 10s of nucleosomes, could be assembled rapidly with different targeted modification marks in the N- or C-terminal histone tails of all four histone proteins, either individually or simultaneously incorporated within the same nucleosome to allow the identification of individual, synergistic, or antagonistic effects of different histone modifications in the physiologically relevant context of a full nucleosome. This study found robust evidence of crosstalk among modification marks on different histone proteins within the same nucleosome across several histone marks “readers” or “writers”.
4.4: Total Synthesis of Histone Proteins by NCL
While EPL grants access to modifications sequestered near the N- or C-terminus of a histone protein, histone proteins do contain modifications throughout the entire sequence. Some of the modifications are centrally located such that a single synthetic peptide does not span the required region. Other modification schemes require the simultaneous incorporation of modifications in the N-terminal tail of a histone and in the folded core domain. These cases require the development of elaborate ligation schemes that enable total synthesis of a histone protein, enabling complete chemical control of the entire histone sequence. This field is rapidly advancing, such that any survey will be outdated almost upon publication. However, some trends are immediately apparent by assessing current progress and directions.
The first total synthesis of a histone protein was carried out by the Ottesen Laboratory to prepare H3-K56ac via a sequential native chemical ligation scheme (Figure 8A). In a typical sequential native chemical ligation scheme, C-terminal peptide is synthesized with a reactive 1,2-aminothiol. The central peptide segment, in this case bearing the acetylated lysine of interest, is prepared with the reactive thioester at the C-terminus as well as a protected 1,2-aminothiol at the N-terminus, often introduced as a thiazolidine derivative that remains masked through the first ligation step. After the first ligation step, the reactive 1,2-aminothiol can be revealed to take part in the final ligation step to generate the full-length target histone protein. Interestingly, the first generation synthesis of H3-K56ac serves as an example of the need to carefully select ligation sites and chemistry to avoid distorting results. The first generation ligation scheme used peptides split with ligation junctions at H3-R40C based on sequence alignment to a divergent histone from Cairina moschata and H3-S96C based on the human histone variant H3.1. However, analysis of the first generation product H3-R40C,S96C with and without acetylated K56 revealed that these semiconservative cysteine substitutions destabilized the histone-DNA interface to a greater extent than the modification of interest. An improved second generation ligation scheme (Figure 8A) identified alanine ligation sites at residues H3-47 and H3-91. Of note, all peptides in this first generation synthesis were prepared by solid phase peptide synthesis using Boc chemistry, which is in use only among synthesis-intensive laboratories. Conversion of the synthesis to Fmoc chemistry, which is more amenable to automation, required development of improved techniques for the preparation of the Fmoc-thioester peptide segments [118].
Figure 8.

Total chemical synthesis of histones. A) Sequential NCL of peptide thioesters with Thz-Cys protection. Purification is carried out after each ligation/deprotection step. B) Sequential NCL of peptide hydrazides. Purification is carried out after each ligation step. C) One-pot NCL of peptide hydrazides with Dobz-Cys protection. A single purification step is carried out after ligation, deprotection, and desulfurization.
Synthesis of these segments followed by sequential ligation and desulfurization resulted in a native-like histone sequence acetylated at H3-K56ac. Importantly, the use of desulfurization in histone H3 requires the use of the H3-C110A substitution, similarto any cysteine-modification approach. This substitution has been widely used in studies requiring cysteine modification, and no effect of this substitution on nucleosome structure, dynamics, or function has been detected. Studies using this synthetic histone revealed that H3-K56ac increased transcription factor binding in the histone-DNA interface by 3-fold, consistent with results from the Chin laboratory using H3-K56ac prepared by codon suppression techniques (section 2.2) [31]. Direct comparison of H3-K56ac prepared synthetically by sequential native chemical ligation and through genetic incorporation of the H3-K56ac species demonstrates the utility of each approach. For a single acetylation site such as H3-K56ac in the center of the histone sequence, exploitation of the pyrrolysine incorporation machinery resulted in multimilligram quantities of protein and is more suitable for repeated expression. Total synthesis by sequential native chemical ligation provided 7% overall yield of the total histone protein, and each repetition of synthesis is as labor-intensive as the first. The true power of the total synthesis approach, then, lies in the ability to prepare histones with combinations of modifications throughout the sequence, such as H3-Y41ph,K56ac (unpublished data from the Ottesen Laboratory).
Sequential native chemical ligation using masked Cys protection is limited to ligation in a C-to-N direction. In an exciting advance, Liu and coworkers introduced peptide hydrazides as a stable masked thioester that could be converted to a reactive thioester in situ for use in native chemical ligation, allowing sequential ligation in the reverse direction (Figure 8B) [119]. Peptide hydrazides are stable under ligation conditions, but can be converted to a reactive thioester by treatment with NaNO2 and an external ligation-compatible thiol, typically MPAA. They applied this methodology to carry out the sequential ligation of histone H3 with the same split sites as the Ottesen scheme, but in reverse order, such that the N-terminal and central segments were ligated to generate an initial ligation product, followed by conversion of the central hydrazide to a reactive thioester for ligation to the C-terminal peptide segment. Selective sidechain protection of H3-C110 allowed desulfurization followed by ligation to yield Ala at the ligation sites, while maintaining the native H3-C110 residue. However, similar to the C to N synthesis above, they found that yields were limited by the challenges of purification of the final product from the component segments.
To eliminate these handling problems, they sought to develop a one-pot chemical ligation approach (Figure 8C). The key advance in this study was the identification of an N-terminal cysteine protection strategy compatible with the chemistry required for conversion of the peptide hydrazide, since the Thz group commonly used for N-terminal cysteine protection is labile to the NaNO2 conversion step. These requirements were satisfied by p-boronobenzyloxycarbonyl (Dobz) protection of the central segment, which could then be reversed by reaction with H2O2to regenerate the active cysteine. Overall, this one pot approach resulted in 20% isolated yield of histone H3-K4me3, a substantial improvement over the Ottesen approach. The Liu group extended the one-pot chemical ligation approach with peptide hydrazides and Dobz protection to the total synthesis of histone H4 [120]. Using the same approaches, they achieved an average 18% overall isolated yield of H4-K16ac. While these percentage yields are not commensurate with an EPL strategy for N-terminal tail modification, they are highly respectable for a total synthesis that can be applied in the future to combinations of modifications throughout the histone sequence, and generated multimilligram quantities of each modified histone. These amounts are sufficient for nearly all biochemical and biophysical assays for characterization of modified nucleosomes.
The Brik Laboratory has introduced two alternate solutions to the challenges posed by sequential native chemical ligation in the context of histone H2B. In 2013, Siman and coworkers developed a convergent ligation strategy that exploits the regulated reactivity of peptide hydrazides used by Liu and coworkers (Figure 9) [121]. In this strategy, H2B is split into four synthetically accessible peptide fragments. In the first ligation step, H2B(1-20) is ligated to H2B(22-56)-hydrazide and purified, while Thz-H2B(59-96) is ligated to H2B(98-125), the N-terminal Cys is revealed by treatment with methoxylamine, and the segment is purified. The ligated H2B(1-56)-hydrazide segment is then converted to reactive thioester by treatment with NaNO2, and ligated to the H2B(59-125) segment to generate the full length histone sequence. Desulfurization and purification yields the final product. While the synthesis of H2B alone is a significant achievement, the potential of total histone synthesis was further demonstrated by modification of the reaction scheme to incorporate ubiquitin at H2B-K34 [122]. Brik synthetically introduced a δ-mercaptolysine residue at position H2B-K34 in the H2B(22-56) peptide hydrazide, which enabled orthogonal ligation to the ubiquitin thioester to generate the H2B(22-56, K34Ub)-hydrazide fragment. This peptide could then be plugged into the convergent ligation scheme to generate the full-length H2B-K34Ub in low yields, but sufficient for incorporation into 12-mer nucleosome arrays suitable for biochemical characterization.
Figure 9.

Total chemical synthesis of histone H2A by convergent ligation. Purification is carried out after synthesis of each two-fragment ligation product.
Solid phase synthesis chemistry has had profound implications for the preparation of complex molecules, including individual peptide segments. Some elements of solid phase chemistry would appear ideal for use with the assembly of histone proteins via sequential solid phase ligation reaction. The Brik Laboratory explored this concept for an improved total synthesis of histone H2B (Figure 10) [122]. A key element of any solid phase ligation approach is the use of a chemical linker to connect the growing protein chain to a solid support that is stable for all ligation conditions, but labile to orthogonal cleavage conditions after synthesis. Here, researchers selected the acid-labile Rink linker that is commonly used for synthesis of peptide amides. They first assembled a solid support connected to a Rink linker with an N-terminal cysteine suitable for ligation. The prepared four peptide segments using similar split sites to those exploited in the convergent synthesis to assemble the final product: H2B(1-20)-thioester, Thz-H2B(22-57)-thioester, Thz-H2B(59-96)-thioester, and Thz-H2B(98-124)-thioester. Each round of solid phase ligation then consisted of a repeated cycle of ligation, wash, Cys deprotection, and wash steps. After chain assembly, free radical desulfurization was used to convert all Cys residues to Ala on the solid phase. Treatment with trifluoroacetic acid then revealed the full-length product in 10% isolated yield, which is significantly improved over the convergent approach for this histone. Of note, the final protein product did result in a H2B-K125A substitution, and generation of the C-terminal amide rather than acid derivative. Since the C-terminal tail of histone H2B is not folded into the nucleosome core, these substitutions are likely permitted. However, care would be required to consider alternate resin attachment strategies for proteins such as H3 and H4 in which the C-terminal tail forms interactions within the structured nucleosome core.
Figure 10.

Total chemical synthesis of histone H2B by solid phase native chemical ligation on PEGA resin with a Rink linker. A single purification step is carried out after on-resin desulfurization and cleavage.
Total protein synthesis by NCL offers the greatest potential level of chemical control over every residue within a histone sequence. These methods offer the possibility of complex combinations of chemically precise modifications installed throughout the nucleosome. However, due to the challenges inherent to these synthetics routes, this comes as a trade-off with effort and yield in synthesis. Continual rapid advances in the field are likely to reduce the barriers to preparation of fully synthetic histones for researchers who are not specialists in chemical protein synthesis.
5. Prospects: Synthetic histone proteins in the eukaryotic cell
The previous sections have considered different ways to prepare precisely modified histone proteins with single or multiple PTMs. We have available extremely powerful tools that cover the full range from fast, simple, and large scale preparation of modification mimics, to careful, precise chemical control over the full histone sequence. These amazing toolkits have enabled biochemical and biophysical investigation of the structural, dynamic, and functional properties of modified chromatin. However, carefully modified histones, for the most part, been restricted to in vitro studies, isolated from the full biological complexity of the cellular environment. While major advances have been made in understanding histone modification cross-talk [123] and the local chromatin environment through preparation of complex synthetic chromatin arrays [15, 124], the next frontier is the introduction of synthetic histones directly into a live eukaryotic cell for incorporation into functional chromatin. If this could be accomplished, the effects of specific sets of modifications could be probed directly.
The most promising lead for this work is the slime mold Physarum polycephalum. This fascinating myxomycete has several different growth stages that range from free-swimming amoeba to micro- and macro-plasmodia, single-celled states that can grow up to 30 cm in diameter, in which each cell contains tens to millions of nuclei synchronized across the cell cycle [125]. In the microplasmodial and macroplasmodial stages, this organism has been shown to spontaneously uptake exogenous histone proteins from media, transport these histones into the nucleus, and incorporate them into active regions of transcription within their chromatin [126, 127]. This myxomycete is easy to grow, and is widely used in the field of bio-computing [128, 129].
Work carried out primarily by Thiriet and Hayes over the past decade has demonstrated that properly folded histone H2A/H2B dimers or H3/H4 tetramers added exogenously to P. polycephalum microplasmodia and macroplasmodia for spontaneous uptake, as demonstrated by following the localization of fluorescein- or FLAG-tagged histones. Uptake is most rapid during the end of the G2 phase of the cell cycle, which occurs simultaneously for all nuclei within a single micro- or macroplasmodium [126, 130, 131]. Both H2A/H2B dimers and H3/H4 tetramers are incorporated into chromatin, although H2A/H2B dimers are deposited at a higher rate than H3/H4 tetramers [132, 133]. Thiriet and coworkers have explored a range of questions in histone transport and chromatin assembly using these techniques. In a recent study, they introduced recombinant histone proteins with Gln mimics of acetylation in the H4 tails at combinations of positions 5, 8, 12, and 16. In general, these acetylation mimics appeared to increase nucleosome exchange, although glutamine substitution at solely H4-K8 and/or H4-K16 abolished uptake into the nuclei. There would appear to be no reason why synthetic and semi-synthetic histones could not be used similarly, to directly probe histone modification cross-talk in the context of the cellular environment.
While the spontaneous uptake of exogenous histone complexes has been described as unique to P. polycephalum, the true target for designer chromatin would be a mammalian or human cell line that could incorporate chemically modified histones into chromatin. The ultimate target for designer chromatin would be a human cell line that could take up histones and incorporate them into chromatin. The N-terminal tails of histones do have stretches of highly positive charge that resemble cell penetrating peptides [134, 135] and, in fact, researchers have demonstrated that histone-derived CPPs can be used to carry protein cargo such as bovine serum albumin into Leishmania tarentolae and into protoplasts of petunia cells [136, 137]. Initial reports suggested that unfolded histones might penetrate HeLa and Colo-205 cells to enter the nucleus, possibly through direct translocation across the plasma membrane rather than through endocytosis, although several questions remain unresolved [138]. Histones have also been proposed to increase uptake of plasmids into human cell lines in a process denoted “histonefection” [139] through a poorly-understood, non-endosome mediated pathway, but is poorly understood [140]. If any of these methods are validated, they could provide the ability to take the power of chemistry to prepare precisely modified histone proteins into the complexity of the biological test tube that is the cell.
References
- 1.Luger K, et al. Crystal structure of the nucleosome core particle at 2.8Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Lochmann B, Ivanov D. Histone H3 Localizes to the Centromeric DNA in Budding Yeast. PLoS Genetics. 2012;8 doi: 10.1371/journal.pgen.1002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redon C, et al. Histone H2A variant H2AX and H2AZ. Current Opinion in Genetics & Development. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 4.Ward IM, et al. Accumulation of Checkpoint Protein 53BP1 at DNA Breaks Involves Its Binding to Phosphorylated Histone H2AX. Journal of Biological Chemistry. 2003;278:19579–19582. doi: 10.1074/jbc.C300117200. [DOI] [PubMed] [Google Scholar]
- 5.Park YJ, et al. A New Fluorescence Resonance Energy Transfer Approach Demonstrates That the Histone Variant H2AZ Stabilizes the Histone Octamer within the Nucleosome. Journal of Biological Chemistry. 2004;279:24274–24282. doi: 10.1074/jbc.M313152200. [DOI] [PubMed] [Google Scholar]
- 6.Jenuwein T. Translating the Histone Code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 8.Schwammle V, et al. Large Scale Analysis of Co-existing Post-translational Modifications in Histone Tails Reveals Global Fine Structure of Cross-talk. Molecular & Cellular Proteomics. 2014;13:1855–1865. doi: 10.1074/mcp.O113.036335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin S, Garcia BA. Examining Histone Posttranslational Modification Patterns by High-Resolution Mass Spectrometry. 2012;512:3–28. doi: 10.1016/B978-0-12-391940-3.00001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan M, et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh MP, Wijeratne SSK, Zempleni J. Biotinylation of lysine 16 in histone H4 contributes toward nucleosome condensation. Archives of Biochemistry and Biophysics. 2013;529:105–111. doi: 10.1016/j.abb.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhall A, et al. Sumoylated Human Histone H4 Prevents Chromatin Compaction by Inhibiting Long-range Internucleosomal Interactions. J Biol Chem. 2014;289:33827–37. doi: 10.1074/jbc.M114.591644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakabe K, Wang Z, Hart GW. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci USA. 2010;107:19915–20. doi: 10.1073/pnas.1009023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fierz B, Muir TW. Chromatin as an expansive canvas for chemical biology. Nature Chemical Biology. 2012;8:417–427. doi: 10.1038/nchembio.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pick H, Kilic S, Fierz B. Engineering chromatin states: Chemical and synthetic biology approaches to investigate histone modification function. Biochimica et Biophysica Acta (BBA) -Gene Regulatory Mechanisms. 2014;1839:644–656. doi: 10.1016/j.bbagrm.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 16.Fierz B. Synthetic Chromatin Approaches To Probe the Writing and Erasing of Histone Modifications. ChemMedChem. 2014;9:495–504. doi: 10.1002/cmdc.201300487. [DOI] [PubMed] [Google Scholar]
- 17.Frederiks F, et al. A Modified Epigenetics Toolbox to Study Histone Modifications on the Nucleosome Core. ChemBioChem. 2011;12:308–313. doi: 10.1002/cbic.201000617. [DOI] [PubMed] [Google Scholar]
- 18.Matsubara K, et al. Global analysis of functional surfaces of core histones with comprehensive point mutants. Genes Cells. 2007;12:13–33. doi: 10.1111/j.1365-2443.2007.01031.x. [DOI] [PubMed] [Google Scholar]
- 19.Hyland EM, et al. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:10060–70. doi: 10.1128/MCB.25.22.10060-10070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of Nucleosome Core Particle from Recombinant Histones. Methods in Enzymology. 1999;304:1–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe S, et al. Structural characterization of H3K56Q nucleosomes and nucleosomal arrays. Biochim Biophys Acta. 2010;1799:480–6. doi: 10.1016/j.bbagrm.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muthurajan UM, et al. Crystal structures of histone Sin mutant nucleosomes reveal altered protein-DNA interactions. EMBO. 2004;23:260–270. doi: 10.1038/sj.emboj.7600046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwasaki W, et al. Comprehensive Structural Analysis of Mutant Nucleosomes Containing Lysine to Glutamine (KQ) Substitutions in the H3 and H4 Histone-Fold Domains. Biochemistry. 2011;50:7822–7832. doi: 10.1021/bi201021h. [DOI] [PubMed] [Google Scholar]
- 24.Yu Q, et al. Differential contributions of histone H3 and H4 residues to heterochromatin structure. Genetics. 2011;188:291–308. doi: 10.1534/genetics.111.127886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Hayes JJ. Acetylation Mimics within Individual Core Histone Tail Domains Indicate Distinct Roles in Regulating the Stability of Higher-Order Chromatin Structure. Molecular and Cellular Biology. 2008;28:227–236. doi: 10.1128/MCB.01245-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allahverdi A, et al. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Research. 2010;39:1680–1691. doi: 10.1093/nar/gkq900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manohar M, et al. Acetylation of Histone H3 at the Nucleosome Dyad Alters DNA-Histone Binding. Journal of Biological Chemistry. 2009;284:23312–23321. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.North JA, et al. Phosphorylation of histone H3(T118) alters nucleosome dynamics and remodeling. Nucleic Acids Research. 2011;39:6465–6474. doi: 10.1093/nar/gkr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.North JA, et al. Histone H3 phosphorylation near the nucleosome dyad alters chromatin structure. Nucleic Acids Research. 2014;42:4922–4933. doi: 10.1093/nar/gku150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimko JC, et al. Preparation of Fully Synthetic Histone H3 Reveals That Acetyl-Lysine 56 Facilitates Protein Binding Within Nucleosomes. Journal of Molecular Biology. 2011;408:187–204. doi: 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neumann H, et al. A Method for Genetically Installing Site-Specific Acetylation in Recombinant Histones Defines the Effects of H3 K56 Acetylation. Molecular Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masumoto H, et al. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 33.Liu CC, Schultz PG. Adding New Chemistries to the Genetic Code. Annual Review of Biochemistry. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 34.Chin JW. Expanding and Reprogramming the Genetic Code of Cells and Animals. Annual Review of Biochemistry. 2014;83:379–408. doi: 10.1146/annurev-biochem-060713-035737. [DOI] [PubMed] [Google Scholar]
- 35.Hao B, et al. A New UAG-Encoded Residue in the Structure of a Methanogen Methyltransferase. Science. 2002;296:1462–1466. doi: 10.1126/science.1069556. [DOI] [PubMed] [Google Scholar]
- 36.Tarrant MK, Cole PA. The Chemical Biology of Protein Phosphorylation. Annual Review of Biochemistry. 2009;78:797–825. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tropberger P, Schneider R. Scratching the (lateral) surface of chromatin regulation by histone modifications. Nature Structural & Molecular Biology. 2013;20:657–661. doi: 10.1038/nsmb.2581. [DOI] [PubMed] [Google Scholar]
- 38.Di Cerbo V, et al. Acetylation of histone H3 at lysine 64 regulates nucleosome dynamics and facilitates transcription. Elife. 2014;3 doi: 10.7554/eLife.01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen DP, et al. Genetically Directing ε-N, N-Dimethyl-l-Lysine in Recombinant Histones. Chemistry & Biology. 2010;17:1072–1076. doi: 10.1016/j.chembiol.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 40.Wang YS, et al. A genetically encoded photocaged Nε-methyl-l-lysine. Molecular BioSystems. 2010;6:1557. doi: 10.1039/c002155e. [DOI] [PubMed] [Google Scholar]
- 41.Xie Z, et al. Lysine Succinylation and Lysine Malonylation in Histones. Molecular & Cellular Proteomics. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gattner MJ, Vrabel M, Carell T. Synthesis of ε-N-propionyl-, ε-N-butyryl-, and ε-n-crotonyl-lysine containing histone H3 using the pyrrolysine system. Chemical Communications. 2013;49:379. doi: 10.1039/c2cc37836a. [DOI] [PubMed] [Google Scholar]
- 43.Kim CH, et al. Site-Specific Incorporation of ε-N-Crotonyllysine into Histones. Angewandte Chemie International Edition. 2012;51:7246–7249. doi: 10.1002/anie.201203349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang R, et al. Dual Native Chemical Ligation at Lysine. JACS Communications. 2009;131:13592–13593. doi: 10.1021/ja905491p. [DOI] [PubMed] [Google Scholar]
- 45.Li X, et al. A Pyrrolysine Analogue for Site-Specific Protein Ubiquitination. Angewandte Chemie International Edition. 2009;48:9184–9187. doi: 10.1002/anie.200904472. [DOI] [PubMed] [Google Scholar]
- 46.Yang R, et al. Native chemical ubiquitination using a genetically incorporated azidonorleucine. Chemical Communications. 2014;50:7971. doi: 10.1039/c4cc03721a. [DOI] [PubMed] [Google Scholar]
- 47.Guo J, et al. Site-Specific Incorporation of Methyl- and Acetyl-Lysine Analogues into Recombinant Proteins. Angewandte Chemie International Edition. 2008;47:6399–6401. doi: 10.1002/anie.200802336. [DOI] [PubMed] [Google Scholar]
- 48.Wang ZU, et al. A Facile Method to Synthesize Histones with Posttranslational Modification Mimics. Biochemistry. 2012;51:5232–5234. doi: 10.1021/bi300535a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chalker JM, et al. Methods for converting cysteine to dehydroalanine on peptides and proteins. Chemical Science. 2011;2:1666. [Google Scholar]
- 50.Chalker JM, et al. Conversion of cysteine into dehydroalanine enables access to synthetic histones bearing diverse post-translational modifications. Angew Chem Int Ed Engl. 2012;51:1835–9. doi: 10.1002/anie.201106432. [DOI] [PubMed] [Google Scholar]
- 51.Sawicka A, Seiser C. Sensing core histone phosphorylation — A matter of perfect timing. Biochimica et Biophysica Acta (BBA) -Gene Regulatory Mechanisms. 2014;1839:711–718. doi: 10.1016/j.bbagrm.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park HS, et al. Expanding the Genetic Code of Escherichia coli with Phosphoserine. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee S, et al. A Facile Strategy for Selective Incorporation of Phosphoserine into Histones. Angewandte Chemie International Edition. 2013;52:5771–5775. doi: 10.1002/anie.201300531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chin JW, et al. Progress Toward an Expanded Eukaryotic Genetic Code. Chemistry & Biology. 2003;10:511–519. doi: 10.1016/s1074-5521(03)00123-6. [DOI] [PubMed] [Google Scholar]
- 55.Lajoie MJ, et al. Genomically Recoded Organisms Expand Biological Functions. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ambrogelly A, Palioura S, Söll D. Natural expansion of the genetic code. Nature Chemical Biology. 2007;3:29–35. doi: 10.1038/nchembio847. [DOI] [PubMed] [Google Scholar]
- 57.Anderson JC, et al. An expanded genetic code with a functional quadruplet codon. Proceedings of the National Academy of Sciences. 2004;101:7566–7571. doi: 10.1073/pnas.0401517101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang K, Schmied WH, Chin JW. Reprogramming the Genetic Code: From Triplet to Quadruplet Codes. Angewandte Chemie International Edition. 2012;51:2288–2297. doi: 10.1002/anie.201105016. [DOI] [PubMed] [Google Scholar]
- 59.Riddle DL, Carbon J. Frameshift Suppression: a Nucleotide Addition in the Anticodon of a Glycine Transfer RNA. Nature New Biology. 1973;242:230–234. doi: 10.1038/newbio242230a0. [DOI] [PubMed] [Google Scholar]
- 60.Bowman A, et al. Probing the (H3-H4)2 histone tetramer structure using pulsed EPR spectroscopy combined with site-directed spin labelling. Nucleic Acids Research. 2010;38:695–707. doi: 10.1093/nar/gkp1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ward R, et al. Long Distance PELDOR Measurements on the Histone Core Particle. Journal of the American Chemical Society. 2009;131:1348–1349. doi: 10.1021/ja807918f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tims HS, Widom J. Methods. Vol. 41. San Diego, Calif: 2007. Stopped-flow Fluorescence Resonance Energy Transfer for Analysis of Nucleosome Dynamics; pp. 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dechassa ML, et al. Architecture of the SWI/SNF-Nucleosome Complex. Molecular and Cellular Biology. 2008;28:6010–6021. doi: 10.1128/MCB.00693-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferreira H, et al. Histone Tails and the H3 αN Helix Regulate Nucleosome Mobility and Stability. Molecular and Cellular Biology. 2007;27:4037–4048. doi: 10.1128/MCB.02229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kurumizaka H, et al. Current progress on structural studies of nucleosomes containing histone H3 variants. Current Opinion in Structural Biology. 2013;23:109–115. doi: 10.1016/j.sbi.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 66.Flaus A, et al. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc Natl Acad Sci USA. 1996;93:1370–1375. doi: 10.1073/pnas.93.4.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poirier MG, et al. Dynamics and function of compact nucleosome arrays; Nature Structural &. Molecular Biology. 2009;16:938–944. doi: 10.1038/nsmb.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simon MD, et al. The Site-Specific Installation of Methyl-Lysine Analogs into Recombinant Histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kenyon G, Bruice TW. Novel Sulfhydryl Reagents. Methods in Enzymology. 1977;47:407–430. doi: 10.1016/0076-6879(77)47042-3. [DOI] [PubMed] [Google Scholar]
- 70.Lauberth SM, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–36. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu X, et al. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat Struct Mol Biol. 2008;15:1122–4. doi: 10.1038/nsmb.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu C, et al. Structural basis for the recognition of methylated histone H3K36 by the Eaf3 subunit of histone deacetylase complex Rpd3S. Structure. 2008;16:1740–50. doi: 10.1016/j.str.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eidahl JO, et al. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Research. 2013;41:3924–3936. doi: 10.1093/nar/gkt074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hung T, et al. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol Cell. 2009;33:248–56. doi: 10.1016/j.molcel.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Margueron R, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–7. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang R, et al. Site-Specific Introduction of an Acetyl-Lysine Mimic into Peptides and Proteins by Cysteine Alkylation. Journal of the American Chemical Society. 2010;132:9986–9987. doi: 10.1021/ja103954u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoyle CE, Bowman CN. Thiol-Ene Click Chemistry. Angewandte Chemie-International Edition. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 78.Li F, et al. A Direct Method for Site-Specific Protein Acetylation. Angewandte Chemie-International Edition. 2011;50:9611–9614. doi: 10.1002/anie.201103754. [DOI] [PubMed] [Google Scholar]
- 79.Shiau C, et al. Reconstitution of Nucleosome Demethylation and Catalytic Properties of a Jumonji Histone Demethylase. Chemistry & Biology. 2013;20:494–499. doi: 10.1016/j.chembiol.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chatterjee A, et al. Disulfide-directed histone ubiquitylation reveals plasticity in hDot1L activation. Nature Chemical Biology. 2010;6:267–269. doi: 10.1038/nchembio.315. [DOI] [PubMed] [Google Scholar]
- 81.Whitcomb SJ, et al. Histone Monoubiquitylation Position Determines Specificity and Direction of Enzymatic Cross-talk with Histone Methyltransferases Dot1L and PRC2. Journal of Biological Chemistry. 2012;287:23718–23725. doi: 10.1074/jbc.M112.361824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dawson PE, et al. Synthesis of Proteins by Native Chemical Ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 83.He S, et al. Facile synthesis of site-specifically acetylated and methylated histone proteins: Reagents for evaluation of the histone code hypothesis. Proceedings of the National Academy of Sciences. 2003;100:12033–12038. doi: 10.1073/pnas.2035256100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shogren-Knaak MA, Fry CJ, Peterson CL. A Native Peptide Ligation Strategy for Deciphering Nucleosomal Histone Modifications. Journal of Biological Chemistry. 2003;278:15744–15748. doi: 10.1074/jbc.M301445200. [DOI] [PubMed] [Google Scholar]
- 85.Fierz B, et al. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nature Chemical Biology. 2011;7:113–119. doi: 10.1038/nchembio.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nguyen DP, et al. Genetic Encoding of Photocaged Cysteine Allows Photoactivation of TEV Protease in Live Mammalian Cells. Journal of the American Chemical Society. 2014;136:2240–2243. doi: 10.1021/ja412191m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fry CJ, Shogren-Knaak MA, Peterson CL. Histone H3 Amino-Terminal Tail Phosphorylation and Acetylation: Synergistic or Independent Transcriptional Regulatory Marks? Cold Spring Harbor Symposia on Quantitative Biology. 2004;69:219–226. doi: 10.1101/sqb.2004.69.219. [DOI] [PubMed] [Google Scholar]
- 88.Shogren-Knaak M. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 89.Ferreira H, Flaus A, Owen-Hughes T. Histone modifications influence the action of Snf2 family remodelling enzymes by different mechanisms. J Mol Biol. 2007;374:563–79. doi: 10.1016/j.jmb.2007.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu Y, et al. Influence of histone tails and H4 tail acetylations on nucleosome-nucleosome interactions. J Mol Biol. 2011;414:749–64. doi: 10.1016/j.jmb.2011.10.031. [DOI] [PubMed] [Google Scholar]
- 91.Chiang KP, et al. A semisynthetic strategy to generate phosphorylated and acetylated histone H2B. Chembiochem. 2009;10:2182–7. doi: 10.1002/cbic.200900238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Casadio F, et al. H3R42me2a is a histone modification with positive transcriptional effects. PNAS. 2013;110:14894–14899. doi: 10.1073/pnas.1312925110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim J, et al. The n-SET domain of Set1 regulates H2B ubiquitylation-dependent H3K4 methylation. Mol Cell. 2013;49:1121–33. doi: 10.1016/j.molcel.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen Z, Gryzbowski AT, Ruthenburg AJ. Traceless Semisynthesis of a Set of Histone 3 Species Bearing Specific Lysine Methylation Marks. ChembioChem. 2014;15:2071–2075. doi: 10.1002/cbic.201402313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hackeng TM, Dawson PE. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc Natl Acad Sci USA. 1999;96:10069–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li S, Shogren-Knaak MA. Cross-talk between histone H3 tails produces cooperative nucleosome acetylation. Proceedings of the National Academy of Sciences. 2008;105:18243–18248. doi: 10.1073/pnas.0804530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fry CJ, et al. The LRS and SIN Domains: Two Structurally Equivalent but Functionally Distinct Nucleosomal Surfaces Required for Transcriptional Silencing. Molecular and Cellular Biology. 2006;26:9045–9059. doi: 10.1128/MCB.00248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu Y, van der Donk W. Convergent Synthesis of Peptide Conjugates Using Dehydroalaninesfor Chemoselective Ligations. Organic Letters. 2001;3:1189–1192. doi: 10.1021/ol015648a. [DOI] [PubMed] [Google Scholar]
- 99.Wan Q, Danishefsky SJ. Free-Radical-Based, Specific Desulfurization of Cysteine: A Powerful Advance in the Synthesis of Polypeptides and Glycopolypeptides. Angewandte Chemie International Edition. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 100.Fierz B, et al. Stability of Nucleosomes Containing Homogenously Ubiquitylated H2A and H2B Prepared Using Semisynthesis. Journal of the American Chemical Society. 2012;134:19548–19551. doi: 10.1021/ja308908p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wong CTT, et al. Realizing serine/threonine ligation: scope and limitations and mechanistic implication thereof. Frontiers in Chemistry. 2014;2:28. doi: 10.3389/fchem.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Haase C, Rohde H, Seitz O. Native Chemical Ligation at Valine. Angewandte Chemie International Edition. 2008;47:6807–6810. doi: 10.1002/anie.200801590. [DOI] [PubMed] [Google Scholar]
- 103.Crich D, Banerjee A. Native Chemical Ligation at Phenylalanine. JACS Communications. 2007;129:10064–10065. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]
- 104.Mersfelder EL, Parthun MR. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Research. 2006;34:2653–2662. doi: 10.1093/nar/gkl338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jack AM, Hake S. Getting down to the core of histone modifications. Chromosoma. 2014;123:355–371. doi: 10.1007/s00412-014-0465-x. [DOI] [PubMed] [Google Scholar]
- 106.Wood DW, Camarero JA. Intein applications: from protein purification and labeling to metabolic control methods. J Biol Chem. 2014;289:14512–9. doi: 10.1074/jbc.R114.552653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ayers B, et al. Introduction of unnatural amino acids into proteins using expressed protein ligation. Peptide Science. 1999;51:343–354. doi: 10.1002/(SICI)1097-0282(1999)51:5<343::AID-BIP4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 108.McGinty RK, et al. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature. 2008;453:812–6. doi: 10.1038/nature06906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shimko JC, et al. Preparing Semisynthetic and Fully Synthetic Histones H3 and H4 to Modify the Nucleosome Core. 2013;981:177–192. doi: 10.1007/978-1-62703-305-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Simon M, et al. Histone fold modifications control nucleosome unwrapping and disassembly. Proc Natl Acad Sci USA. 2011;108:12711–6. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Javaid S, et al. Nucleosome remodeling by hMSH2-hMSH6. Mol Cell. 2009;36:1086–94. doi: 10.1016/j.molcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kruger W, et al. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes & Development. 1995;9:2770–2779. doi: 10.1101/gad.9.22.2770. [DOI] [PubMed] [Google Scholar]
- 113.Hurd PJ, et al. Phosphorylation of Histone H3 Thr-45 Is Linked to Apoptosis. Journal of Biological Chemistry. 2009;284:16675–16683. doi: 10.1074/jbc.M109.005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ulyanova NP, Schnitzler GR. Human SWI/SNF generates abundant, structurally altered dinucleosomes on polynucleosomal templates. Mol Cell Biol. 2005;25:11156–70. doi: 10.1128/MCB.25.24.11156-11170.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schnitzler GR, Sif S, Kingston RE. Human SWI/SNF Interconverts a Nucleosome between Its Base State and a Stable Remodeled State. Cell. 1998;94:17–27. doi: 10.1016/s0092-8674(00)81217-9. [DOI] [PubMed] [Google Scholar]
- 116.McGinty RK, et al. Structure-Activity Analysis of Semisynthetic Nucleosomes: Mechanistic Insights into the Stimulation of Dot1L by Ubiquitylated Histone H2B. ACS Chemical Biology. 2009;4:958–968. doi: 10.1021/cb9002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nguyen UTT, et al. Accelerated chromatin biochemistry using DNA-barcoded nucleosome libraries. Nature Methods. 2014;11:834–840. doi: 10.1038/nmeth.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mahto SK, et al. A Reversible Protection Strategy To Improve Fmoc-SPPS of Peptide Thioesters by the N-Acylurea Approach. ChemBioChem. 2011;12:2488–2494. doi: 10.1002/cbic.201100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fang GM, Wang JX, Liu L. Convergent chemical synthesis of proteins by ligation of peptide hydrazides. Angew Chem Int Ed Engl. 2012;51:10347–50. doi: 10.1002/anie.201203843. [DOI] [PubMed] [Google Scholar]
- 120.Li J, et al. One-pot native chemical ligation of peptide hydrazides enables total synthesis of modified histones. Organic & Biomolecular Chemistry. 2014;12:5435. doi: 10.1039/c4ob00715h. [DOI] [PubMed] [Google Scholar]
- 121.Siman P, et al. Convergent Chemical Synthesis of Histone H2B Protein for the Site-Specific Ubiquitination at Lys34. Angewandte Chemie International Edition. 2013;52:8059–8063. doi: 10.1002/anie.201303844. [DOI] [PubMed] [Google Scholar]
- 122.Jbara M, Seenaiah M, Brik A. Solid phase chemical ligation employing a rink amide linker for the synthesis of histone H2B protein. Chem Communications. 2014;50:12534–12537. doi: 10.1039/c4cc06499b. [DOI] [PubMed] [Google Scholar]
- 123.Linghu C, et al. Discovering common combinatorial histone modification patterns in the human genome. Gene. 2013;518:171–178. doi: 10.1016/j.gene.2012.11.038. [DOI] [PubMed] [Google Scholar]
- 124.Zheng C, Hayes JJ. Intra- and Inter-nucleosomal Protein-DNA Interactions of the Core Histone Tail Domains in a Model System. Journal of Biological Chemistry. 2003;278:24217–24224. doi: 10.1074/jbc.M302817200. [DOI] [PubMed] [Google Scholar]
- 125.Mohberg J, Rusch HP. Isolation of the nuclear histones from the myxomycete, Physarum polycephalum. Archives of Biochemistry and Biophysics. 1969;134:577–589. doi: 10.1016/0003-9861(69)90320-8. [DOI] [PubMed] [Google Scholar]
- 126.Thiriet C, Hayes JJ. Histone Proteins in Vivo: Cell-Cycle-Dependent Physiological Effects of Exogenous Linker Histones Incorporated into Physarum polycephalum. Methods: A Companion to Methods in Enzymology. 1999;17:140–150. doi: 10.1006/meth.1998.0725. [DOI] [PubMed] [Google Scholar]
- 127.Prior CP, et al. Incorporation of exogenous pyrene-labeled histone into Physarum chromatin: a system for studying changes in nucleosomes assembled in vivo. Cell. 1980;20:597–608. doi: 10.1016/0092-8674(80)90306-2. [DOI] [PubMed] [Google Scholar]
- 128.Adamatzky A. Slimeware: engineering devices with slime mold. Artificial Life. 2013;19:317–30. doi: 10.1162/ARTL_a_00110. [DOI] [PubMed] [Google Scholar]
- 129.Taylor B, et al. Physarum polycephalum: Towards a biological controller. Biosystems. 2014;127C:42–46. doi: 10.1016/j.biosystems.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 130.Ejlassi-Lassallette A, et al. H4 replication-dependent diacetylation and Hat1 promote S-phase chromatin assembly in vivo. Molecular Biology of the Cell. 2010;22:245–255. doi: 10.1091/mbc.E10-07-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thiriet C, Hayes JJ. A novel labeling technique reveals a function for histone H2A/H2B dimertail domains in chromatin assembly in vivo. Genes & Development. 2001;15:2048–2053. doi: 10.1101/gad.910201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Studitsky VM, Clark DJ, Felsenfeld G. A Histone Octamer Can Step around a Transcribing Polymerase without Leaving the Template. Cell. 1994;76:371–382. doi: 10.1016/0092-8674(94)90343-3. [DOI] [PubMed] [Google Scholar]
- 133.Kireeva ML, et al. Nucleosome Remodeling Induced by RNA Polymerase II: Loss of the H2A-H2B Dimer during Transcription. Molecular Cell. 2002;9:541–552. doi: 10.1016/s1097-2765(02)00472-0. [DOI] [PubMed] [Google Scholar]
- 134.Nishiyama A, et al. Intracellular delivery of acetyl-histone peptides inhibits native bromodomain-chromatin interactions and impairs mitotic progression. FEBS letters. 2008;582:1501–1507. doi: 10.1016/j.febslet.2008.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Heo K, et al. Cell-penetrating H4 tail peptides potentiate p53-mediated transactivation via inhibition of G9a and HDAC1. Oncogene. 2013;32:2510–20. doi: 10.1038/onc.2012.273. [DOI] [PubMed] [Google Scholar]
- 136.Keller AA, et al. Transduction of proteins into leishmania tarentolae by formation of non-covalent complexes with cell-penetrating peptides. J Cell Biochem. 2014;115:243–52. doi: 10.1002/jcb.24654. [DOI] [PubMed] [Google Scholar]
- 137.Rosenbluh J, et al. Non-endocytic penetration of core histones into petunia protoplasts and cultured cells: a novel mechanism for the introduction of macromolecules into plant cells. Biochim BiophysActa. 2004;1664:230–40. doi: 10.1016/j.bbamem.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 138.Hariton-Gazal E, et al. Direct translocation of histone molecules across cell membranes. Journal of Cell Science. 2003;116:4577–4586. doi: 10.1242/jcs.00757. [DOI] [PubMed] [Google Scholar]
- 139.Balicki D, et al. Structure and function correlation in histone H2A peptide-mediated gene transfer. Proceedings of the National Academy of Sciences. 2002;99:7467–7471. doi: 10.1073/pnas.102168299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kaouass M, Beaulieu R, Balicki D. Histonefection: Novel and potent non-viral gene delivery. Journal of Controlled Release. 2006;113:245–254. doi: 10.1016/j.jconrel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 141.Davey CA, et al. Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9Å Resolution. Journal of Molecular Biology. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]