

Abstract
Background
Transient receptor potential melastatin member 4 (TRPM4) is a nonselective cation channel. TRPM4 mutations have been linked to cardiac conduction disease and Brugada syndrome. The mechanisms underlying TRPM4‐dependent conduction slowing are not fully understood. The aim of this study was to characterize TRPM4 genetic variants found in patients with congenital or childhood atrioventricular block.
Methods and Results
Ninety‐one patients with congenital or childhood atrioventricular block were screened for candidate genes. Five rare TRPM4 genetic variants were identified and investigated. The variants were expressed heterologously in HEK293 cells. Two of the variants, A432T and A432T/G582S, showed decreased expression of the protein at the cell membrane; inversely, the G582S variant showed increased expression. Further functional characterization of these variants using whole‐cell patch‐clamp configuration showed a loss of function and a gain of function, respectively. We hypothesized that the observed decrease in expression was caused by a folding and trafficking defect. This was supported by the observation that incubation of these variants at lower temperature partially rescued their expression and function. Previous studies have suggested that altered SUMOylation of TRPM4 may cause a gain of function; however, we did not find any evidence that supports SUMOylation as being directly involved for the gain‐of‐function variant.
Conclusions
This study underpins the role of TRPM4 in the cardiac conduction system. The loss‐of‐function variants A432T/G582S found in 2 unrelated patients with atrioventricular block are most likely caused by misfolding‐dependent altered trafficking. The ability to rescue this variant with lower temperature may provide a novel use of pharmacological chaperones in treatment strategies.
Keywords: atrioventricular block, mutations, temperature‐dependent rescue, transient receptor potential melastatin member 4
Subject Categories: Arrhythmias, Electrophysiology
Introduction
Congenital and childhood atrioventricular block (AVB)1 is characterized by a delay or interruption in the impulse transmission from the atria to the ventricles caused by either anatomical or functional impairment in the conduction system that is observed at birth or at a young age. Conduction can be delayed, intermittent, or absent, and the conduction disturbances can be transient or permanent. Implantation of a pacemaker is the main therapeutic option for congenital AVB. In young patients, congenital AVB could be caused by either (1) acquired autoimmune disease in which maternal autoantibodies against the intracellular ribonucleoproteins Ro/SSA and La/SSB cross the placenta and inhibit l‐type calcium channels2, 3 or (2) genetic variants in NKX2.5, 4, 5 SCN5A, and SCN1B genes coding for the homeobox protein Nkx‐2.5, the voltage‐gated sodium channel Nav1.5, and the auxiliary subunit β1 of the voltage‐gated sodium channel, respectively.5, 6, 7 Recently, at least 10 genetic variants in the transient receptor potential channel melastatin 4 (TRPM4) gene have been linked to different forms of cardiac conduction defect and Brugada syndrome (BrS).8, 9, 10, 11 The ion channel TRPM4 is a member of the transient receptor potential channel family, which comprises at least 28 genes in the human genome. TRPM4 is an intracellular Ca2+‐activated nonspecific cation channel that is not permeable to Ca2+ and is expressed in different cells of the cardiovascular system, such as arterial and venous smooth muscle cells and cardiac cells of the conduction pathways.8, 12, 13, 14 As with all transient receptor potential channels, each monomer contains 6 transmembrane‐spanning segments, whereas the channel pore is formed by a tetramer. The cellular response on TRPM4 activation or inactivation depends on the cell type and the coexpression of membrane transporters. Its activation enables Na+ entry into the cell, leading to cellular membrane depolarization. TRPM4 also participates in intracellular Ca2+ sensing and affects the driving force for Ca2+ and other ions by altering the cellular membrane electrical potential.
In a seminal study,8 the E7K TRPM4 variant was found to be linked to progressive familial type 1 heart block in a large South African pedigree. Characterization of this variant demonstrated that it led to a gain of expression at the cell membrane as well as a gain of function. Further experiments suggested that increased SUMOylation of this variant might be the cause of the observed gain of function. The molecular details of these alterations, however, remain poorly understood. Other TRPM4 variants causing either gain or loss of function have been found in patients with autosomal‐dominant cardiac conduction disease,9 different forms of cardiac conduction alterations,10 and BrS.11 Although the study by Kruse et al8 clearly linked TRPM4 to conduction block based on genetic linkage, more recent studies have failed to demonstrate a direct relationship between the sporadic presence of the variants and the electric phenotypes.10 It has been proposed that these TRPM4 variants may act as modifiers in the context of a complex genetic background.10 Furthermore, how both gain‐ and loss‐of‐function variants may lead to conduction slowing has yet to be determined.15
In the present study, we identified 5 rare variants in the TRPM4 gene of patients with either congenital or childhood AVB. Additional functional and biochemical characterization of these variants identified 2 with loss of function and 1 with gain of function. We further examined the possible mechanisms by which these variants could have altered plasma membrane expression. Moreover, this work also assessed the possibility of rescuing the loss‐of‐function variants and restoring their membrane expression and function.
Material and Methods
The study involving humans conforms to the guiding principles of the Declaration of Helsinki. Human subjects gave informed consent for a study that was approved by the Institutional Committee on Human Research at the authors’ institution.
Samples
A total of 91 unrelated patients and their parents were identified from a multicenter retrospective study from 1980 to 2009 carried out in 13 French tertiary hospitals.16 Patients were included in the study if they had AV conduction disturbance with negative maternal anti‐Ro/SSA and anti‐La/SSB antibodies and without associated structural cardiac malformation. Patients born before 1980 were excluded because of the major progress in prenatal diagnosis and neonatal management of AVB and in pacing technologies. Both incomplete and complete AVB were considered. Patients with traumatic or postoperative heart block were excluded, as were those with myocarditis, neuromuscular disorders, metabolic diseases, congenital structural heart disease likely to account for AVB, and long QT syndrome. Mothers of included patients were all screened for maternal antibodies to soluble nuclear antigens 48‐kDa SSB/La, 52‐kDa SSA/Ro, and 60‐kDa SSA/Ro using quantitative radioligand assays. In case of missing data or negative maternal antibodies diagnosed with a less sensitive technique, parents were contacted and rescreened with radioligand assays. Written informed consent was obtained from all participants or their guardians. For each patient, standard 12‐lead ECGs were collected at time of diagnosis and at time of pacemaker implantation or at last follow‐up if not implanted. ECGs were analyzed by 2 blinded medical investigators, and interval durations were measured using dedicated Datinf Measure software (Datinf). The earliest documentation of a conduction abnormality of a patient was used as the time of diagnosis of AVB. Type of block, heart rate, PR intervals, QRS complexes, and QT interval durations were determined. QT interval measurement was adjusted to the heart rate using the Bazett formula.17 Among these 91 children, 15 showed congenital AVB (6 were diagnosed in utero) and 76 had childhood AVB. The median age at diagnosis was 24 months. Different types of conduction block were diagnosed: (1) 3 cases of AVB type 1, (2) 21 cases of AVB type 2 (23%) (8 Mobitz type 1 and 13 Mobitz type 2), and (3) 67 cases of complete AVB (73.6%). The majority (84.6%) of QRS complex durations were not increased, whereas intraventricular conduction abnormalities were observed in 14 children (15.4%), distributed in 7 blocks of right bundle branch (3 complete and 4 incomplete), 5 left bundle branch block (3 complete and 2 left anterior hemiblock), 1 alternation between right bundle branch and left bundle branch block, and 1 case of undetermined complete block. Implantation of a pacemaker was required in 73 children (80.2%) during a median follow‐up of 9.5 months.
Mutation Screening
Genomic DNA was extracted from peripheral blood by using the NucleoSpin Blood XL kit (Macherey‐Nagel), as described by the manufacturer. NKX2.5, SCN5A, SCN1B, and 10 coding exons of TRPM4 (ENSG00000130529, ENST00000252826) were screened by direct DNA sequencing, and 15 coding exons of TRPM4 were screened by high‐resolution melting curve analysis in all patients. The primers were designed to flank the coding regions. Polymerase chain reaction (PCR) and high‐resolution melting were performed in a single run on a LightCycler 480 instrument (Roche Diagnostics). The high‐resolution melting analysis was performed using Gene Scanning software version 1.5.0 (Roche Instrument Centre), which allows for clustering of the samples into groups on the basis of difference plots obtained by analyzing the differences in melting curve shapes. All samples studied were clustered at the default sensitivity setting (0.5). Samples with an aberrant melting profile were assigned for validation sequencing to identify or exclude sequence variants. Forward‐ and reverse‐sequence reactions were performed with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) using the same primers. The sequence products were analyzed on a 3730 DNA Analyzer (Applied Biosystems).
Molecular Cloning
The following TRPM4 variants were selected for molecular cloning: p.D198G, p.A432T, p.G582S, p.T677I, and p.V921I. Constructions were called, respectively, D198G, A432T, G582S, T677I, and V921I, and the double variant (p.A432T/p.G582S) was called A432T/G582S. The cDNA of TRPM4 was obtained in 2 overlapping fragments from human kidney RNA (Stratagene, Amsterdam, Netherlands) by reverse transcription with Mu‐MLV reverse transcriptase (Eurogentec France SASU). The complete TRPM4 cDNA was cloned in pcDNA4/TO vector (Invitrogen). It was provided by A. Guse (University Hospital Hamburg‐Eppendorf, Hamburg‐Eppendorf, Germany) and P. Bouvagnet (Laboratoire Cardiogenetique, Hospices Civils de Lyon, Groupe Hospitalier Est, Bron, France). TRPM4 p.E7K mutant (called TRPM4 E7K) was provided by P. Bouvagnet.
Primers for reverse transcription and PCR are available on request. Mutant TRPM4 cDNAs were obtained by in vitro mutagenesis using the QuickChange XLII site‐directed mutagenesis kit (Stratagene). Mutagenesis primer sequences are available on request. Mutant cDNA clones were systematically sequenced before use in further experiments.
Cell Culture and Transfection
Human embryonic kidney (HEK293) cells were cultured with DMEM supplemented with 4 mmol/L glutamine, 10% FBS, and 20 μg/mL gentamycin. They were transiently transfected with 240 ng of HA‐TRPM4 wild type (WT), variants (D198G, A432T, G582S, A432T/G582S, T677I and V921I), or empty vector (pcDNA4TO) in a P100 dish (BD Falcon), mixed with 30 μL of JetPEI (Polyplus‐transfection) and 250 μL of 150 mmol/L NaCl. The cells were incubated for 48 hours at 37°C with 5% CO2. The amount of cDNA used was proportionately increased to the amount used for electrophysiological studies. For electrophysiological studies, T25 25‐cm2 flasks of HEK293 cells were transiently cotransfected using X‐tremeGENE 9 DNA transfection mix reagent (Roche Diagnostics) with 80 ng WT or variant TRPM4 channels. All transfections included 200 ng cDNA encoding CD8 antigen as a reporter gene. Anti‐CD8 beads (Dynal) were used to identify transfected cells, and only CD8‐displaying cells were analyzed. Cells were used 48 hours after transfection.
Real‐Time Quantitative PCR
RNA isolation was performed with the NucleoSpin RNA II kit (Macherey‐Nagel), as described by the manufacturer. Using cDNAs of transfected HEK293 cells, we performed quantitative expression analysis with the 7900HT Fast Real‐Time PCR System (Applied Biosystems). The PCR amplification was performed with TaqMan gene expression assay Hs01026070_m1 for human TRPM4. For control, we studied expression of a cotransfected human CD8 gene with TaqMan gene expression assay Hs00233520_m1. For each sample, triplicate determinations were performed in a 96‐well optical plate using 80 ng of cDNA, 1 μL of 20× TaqMan Gene Expression Assay, 10 μL of 2× TaqMan Fast Universal PCR Master Mix No AmpErase UNG, and 5 μL of water in each 20 μL reaction. Plates were heated for 20 seconds at 95°C, and then 40 cycles of 1 second at 95°C and 20 seconds at 60°C were applied. Relative TRPM4 expression (ΔCt) was calculated by subtracting the signal threshold cycle (Ct) of the control (CD8) from the Ct value of TRPM4. Subsequently, ΔΔCt values were obtained for each mutant by subtracting the ΔCt of the TRPM4 WT from the ΔCt of each mutant. Results were then linearized by calculating 2−expΔΔCt.
Cell Surface Biotinylation Assay
Following 48 hours of incubation, transiently transfected HEK293 cells were treated with EZ‐Link Sulfo‐NHS‐SS‐Biotin (Thermo Scientific) 0.5 mg/mL in cold 1× PBS for 15 minutes at 4°C. Subsequently, the cells were washed twice with 200 mmol/L glycine in cold 1× PBS and twice with cold 1× PBS to inactivate and remove excess biotin, respectively. The cells were then lysed with 1× lysis buffer (50 mmol/L HEPES, pH 7.4; 150 mmol/L NaCl; 1.5 mmol/L MgCl2; 1 mmol/L EGTA, pH 8.0; 10% glycerol; 1% Triton X‐100; 1× Complete Protease Inhibitor Cocktail [Roche]) for 1 hour at 4°C. Cell lysates were centrifuged at 16 000g at 4°C for 15 minutes. Next, 2 mg of the supernatant were incubated with 50 μL Streptavidin Sepharose High Performance beads (GE Healthcare) for 2 hours at 4°C, and the remaining supernatant was kept as input. The beads were subsequently washed 5 times with 1× lysis buffer before elution with 50 μL of 2× NuPAGE sample buffer (Invitrogen) plus 100 mmol/L DTT at 37°C for 30 minutes. These biotinylated fractions were analyzed as TRPM4 expressed at the cell surface. The input fractions, analyzed as total expression of TRPM4, were resuspended with 4× NuPAGE Sample Buffer plus 100 mmol/L DTT to give a concentration of 1 mg/mL and incubated at 37°C for 30 minutes.
Immunoprecipitation
Transiently transfected HEK293 cells in P100 plates were harvested after 48 hours of incubation and lysed with 1× cold Ubi lysis buffer (50 mmol/L HEPES, pH 7.4; 150 mmol/L NaCl; 1 mmol/L EGTA, pH 8.0; 10% glycerol; 1× EDTA‐free Complete Protease Inhibitor Cocktail [Roche]; 2 mmol/L N‐ethylmaleimide/NEM [Sigma‐Aldrich], 10 mmol/L iodoacetamide/IAA [Sigma‐Aldrich]) containing 1% Triton X‐100 for 1 hour at 4°C. Cell lysates were then centrifuged at 16 000g at 4°C for 15 minutes. Next, 2 mg of the supernatant (lysate) were incubated at 4°C for 24 hours with 10 μg anti‐RanGAP1 A302‐027A (Bethyl Laboratories) or 20 μg anti‐HA MMS‐101R (Covance) to immunoprecipitate RanGAP1 and HA‐tagged TRPM4, respectively. One volume of 1× cold Ubi lysis buffer without Triton X‐100 (to obtain a final concentration of 0.5% Triton X‐100) was also added in the mix. On the next day, the lysate–antibody mix was transferred to a microcentifuge tube containing 50 μL (1:1 bead:lysis buffer ratio) of Protein G Sepharose beads (GE Healthcare), which were previously washed 3 times with 1× cold Ubi lysis buffer containing 0.5% Triton X‐100. After adding fresh 1× EDTA‐free Complete Protease Inhibitor Cocktail, the mix was incubated overnight at 4°C. The beads were subsequently washed 5 times (4°C; 0.8 g) with 1× cold Ubi buffer containing 0.3% Triton X‐100 before elution with 50 μL of 2× NuPAGE sample buffer plus 100 mmol/L DTT at 37°C for 30 minutes. These samples are designated as immuniprecipitation fractions. The input fractions were resuspended with 4× NuPAGE Sample Buffer plus 100 mmol/L DTT to give a concentration of 1 mg/mL and incubated at 37°C for 30 minutes. All lysis and incubation steps, except elution in sample buffer, were performed in absence of light.
Low‐Temperature Rescue Experiments
HEK293 cells were transiently transfected individual TRPM4 WT or TRPM4 variants (A432T, A432T/G582S). After the first 24 hours of incubation at 37°C (95% O2 and 5% CO2), 1 set of transfected HEK293 cells was kept in the same incubator, and another set was shifted to a 28°C incubator supplied with 95% O2 and 5% CO2 for 24 hours. The following day, cell surface biotinylation assay (as described earlier in Cell Surface Biotinylation Assay) was performed on the 2 sets of HEK293 cells.
Generation of glutathione‐S‐transferase (GST) and GST‐S5a and Pull‐Down Experiments
Glycerol stocks of Escherichia coli DH5α transformed with pGEX‐4T3 (GST alone) and pGEX‐S5a were added to 1 L Luria‐Bertan medium containing ampicillin (0.1 μg/μL) and grown to an OD600 of 0.600. The bacterial cultures were then induced with 1 mmol/L IPTG for 2.5 hours at 29°C. Cells were harvested by centrifugation (800 g, 4°C, 15 minutes in a Sorvall Legend RT+ benchtop centrifuge), resuspended in 25 mL 1× bacterial lysis buffer (0.5 mol/L Tris, pH 7.5; 2.5 mol/L NaCl; 10% Igepal; and 1× Complete Protease Inhibitor Cocktail Roche, IN, USA), added with 0.2 mg/mL lysozyme and then mixed by gently inverting the tube several times until it appeared viscous. The mix was then neutralized by adding 10 mmol/L MgSO4 and sonicated 3 times for 10 seconds each at 100% power (with 10‐second breaks in between) to disrupt DNA. To obtain protein lysate, the mix was transferred to a Sorvall tube and centrifuged in an ss‐43 rotor for 10 minutes at 11 200 g at 4°C. Protein lysate was then transferred to a falcon tube and combined with 3 mL of glutathione‐sepharose beads (GE Healthcare Biosciences) previously washed with distilled water and 1× bacterial lysis buffer and rotated on a wheel at 4°C for 2 hours to facilitate binding. The mix was then washed 5 times with 15 mL 1× bacterial lysis buffer (4°C, 10 minutes at 0.1 g in a Sorvall Legend RT+ benchtop centrifuge). After the concentration of GST fusion protein/glutathione–sepharose complex was determined, it was stored in glycerol buffer at −80°C. Transiently transfected HEK293 cells in P100 plates were harvested after 48 hours of incubation and lysed with 1× cold Ubi lysis buffer (50 mmol/L HEPES, pH 7.4; 150 mmol/L NaCl; 1 mmol/L EGTA, pH 8.0; 10% glycerol; 1× EDTA‐free Complete Protease Inhibitor Cocktail; 2 mmol/L NEM) containing 1% Triton X‐100 for 1 hour at 4°C. Cell lysates were then centrifuged at 16 000g at 4°C for 15 minutes. Next, 1 mg of the supernatant (lysate) was incubated with 100 μg GST or GST‐S5a coupled on glutathione beads at 4°C for 2 hours. The beads were subsequently washed 5 times (4°C; 0.8 g in a tabletop centrifuge) with the same 1× cold Ubi buffer described above except containing 0.5% Triton X‐100 before elution with 50 μL 2× NuPAGE sample buffer plus 100 mmol/L DTT at 37°C for 30 minutes. These samples are designated as pull‐down fractions. The input fractions were resuspended with 4× NuPAGE Sample Buffer plus 100 mmol/L DTT to give a concentration of 1 mg/mL and incubated at 37°C for 30 minutes.
Western Blotting Experiments
Protein samples were analyzed on 9% polyacrylamide gels, transferred with the TurboBlot dry blot system (Bio‐Rad), and detected with anti‐TRPM4 (generated by Pineda), anti‐RanGAP1 A302‐026A (Bethyl Laboratories), anti‐SUMO1 S8070 (Sigma‐Aldrich), anti‐SUMO2/3 ab3742 (Abcam), anti‐Na+/K+ ATPase α1 ab7671 (Abcam), and anti–α‐actin A2066 (Sigma‐Aldrich) antibodies using SNAP id (Millipore). The anti‐TRPM4 antibody was generated by Pineda using the following peptide sequence: NH2‐CRDKRESDSERLKRTSQKV‐CONH2. A fraction of the antisera, which was subsequently used in this study, was then affinity purified.
Cellular Electrophysiology
For patch‐clamp experiments in whole‐cell configuration, intracellular solution contained (in mmol/L) 100 CsAsp, 20 CsCl, 4 Na2ATP, 1 MgCl2, 10 EGTA, and 10 HEPES. The pH was adjusted to 7.20 with CsOH and the free Ca2+ concentration at 100 μmol/L with CaCl2, using the WEBMAXCLITE program (http://www.stanford.edu/cpatton/downloads.htm). Na2ATP has been added to the intracellular solution to be as close as possible to physiological cytosolic solution and to provide phosphate residue for phosphorylation processes. Extracellular solution contained (in mmol/L) 156 NaCl, 1.5 CaCl2, 1 MgCl2, 6 CsCl, 10 glucose, and 10 HEPES. The pH was adjusted to 7.40 with NaOH. Patch‐clamp recordings were carried out in the whole‐cell configuration at room temperature. TRPM4 currents were investigated using a ramp protocol. The holding potential was −60 mV. The 400‐ms increasing ramp from −100 to +100 mV ended with a 300‐ms step at +100 mV and then 300 ms at −100 mV. A new ramp was performed every 2 seconds. Current densities were obtained by dividing the peak current recorded at −100 mV by the cell capacitance.
Statistical Analysis
Data are presented as the mean±SEM. The unpaired 2‐tailed Student t test was used to compare the means, with P<0.05 considered significant. For the experiments, the WT condition was used as the reference for normalization and quantification. The P values were not correct for multiple testing because each variant was compared with the WT allele only and not between variants.
Results
Genetic Screening in Patients With Congenital AVB
All exons of the NKX2.5, SCN5A, SCN1B, and TRPM4 genes were directly sequenced or screened using high‐resolution melting in a cohort of 91 pediatric patients (Table 1) with congenital (n=15) or childhood (n=76) AVB (Figure 1). The genes already identified in cardiac conduction diseases were screened initially (ie, NKX2.5, SCN5A, and SCN1B) (Table 2). A nonsynonymous variant was identified in NKX2.5 (A119S) (Table 2). This variant was detected in a boy with complete AVB that was diagnosed when he was aged 5 years, and a pacemaker was implanted when he was aged 11 years. This A119S mutation was described previously in a patient with severe hypothyroidism.18 A nonsynonymous variant was identified in SCN1B (C211Y) in a young boy with complete AVB diagnosed at age 18 months, and a pacemaker was implanted at age 30 months. A frame shift variant was identified in SCN5A (T1806SfsX27) in a girl with first‐degree AVB diagnosed at age 7 years. This child, with a follow‐up of 4 years, was never implanted with a pacemaker (Table 2).
Table 1.
Clinical and ECG Characterizations of Congenital and Childhood Atrioventricular Block Patients
| Total | Without TRPM4 Variants | With TRPM4 Variants | |
|---|---|---|---|
| General characterizations | |||
| Number of children | 91 | 77 | 14 |
| Mean age at diagnosis, months | 43.9±48.0 | 45.0±48.9 | 37.3±44.3 |
| Median age at diagnosis, months | 24 (in utero 228) | 24 (in utero 228) | 12 (in utero 126) |
| Congenital atrioventricular block, % | 16.5 | 13.0 | 35.7 |
| Childhood atrioventricular block, % | 83.5 | 87.0 | 64.3 |
| ECG characterizations | |||
| Complete atrioventricular block, % | 73.6 | 67.8 | 78.6 |
| Intraventricular conduction disorder, % | 15.4 | 11.7 | 35.7 |
| Heart beat, bpm | 56.0±16.4 | 56.0±16.0 | 54.0±19.5 |
| QRS duration, ms | 80.0±28.8 | 79.0±28.5 | 88.0±31.1 |
| QT interval corrected, ms | 433.1±67.1 | 427.0±45.3 | 469.0±143.7 |
| Pacemaker implant | |||
| Pacemaker implant, % | 80.2 | 77.9 | 92.8 |
| Mean age at implant, months | 69.2±61.6 | 72.6±62.0 | 53.9±59.6 |
| Median age at implant, months | 54 (0–264) | 54 (0–264) | 24 (0–338) |
| Median time between diagnosis and implantation, months | 9.5 (0–144) | 12.0 (0–144) | 0.0 (0–102) |
Figure 1.
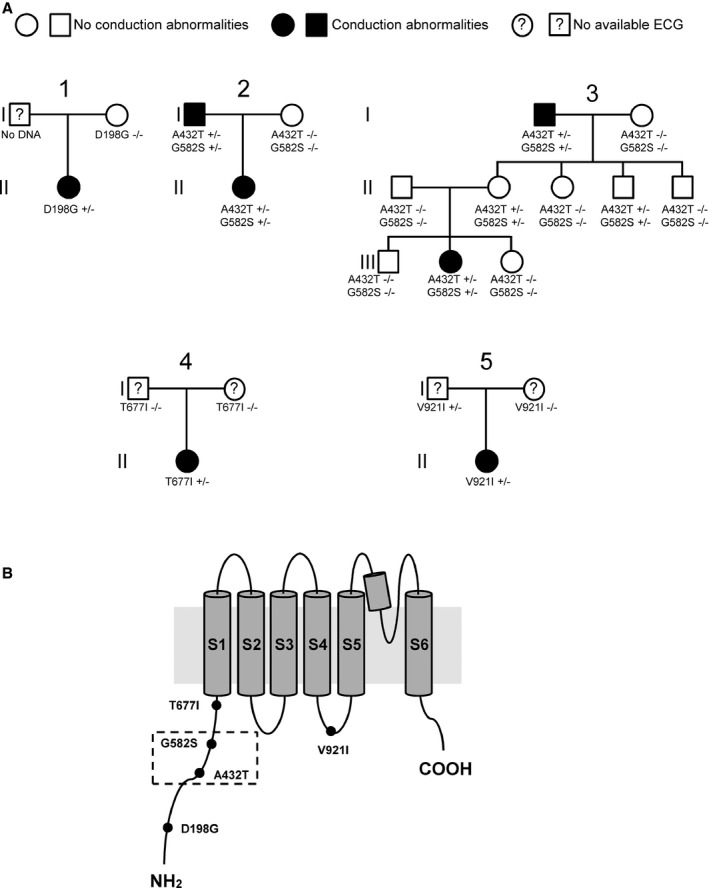
Families with AVB. A, Pedigrees of families with members harboring TRPM4 variants. B, Illustration of the TRPM4 channel showing the location of the 5 congenital atrioventricular block variants described in this study. The dashed box highlights the presence of 2 variants in the same patient. AVB indicates atrioventricular block.
Table 2.
Clinical and Genetic Features From Patients Carrying Rare TRPM4 Variants
| Patients | TRPM4 Variants | Variant Frequency in ExAc (Controls) | ||||||
|---|---|---|---|---|---|---|---|---|
| No. | Age and Rhythm at Diagnosis | Age and Rhythm at Implantation | Exon | Nucleotide | Amino Acid | European | ||
| 1 | 10.5 years | Complete AVB | 13 years | Complete AVB | 05 | c.593A>G | p.Asp198Gly | 0 |
| 2 | 6 months |
AVB1 AVB2 Type 1 |
9 years | Complete AVB | 06 | c.755G>A | p.Arg252His | 0.0082 |
| 3a | 5 years | Complete AVB | 11 years | Complete AVB |
11 13 |
c.1294G>A c.1744G>A |
p.Ala432Thr p.Gly582Ser |
0.0005 0.0006 |
| 4a | 7 years | First‐degree AVB | No pacemaker | — |
11 13 |
c.1294G>A c.1744G>A |
p.Ala432Thr p.Gly582Ser |
0.0005 0.0006 |
| 5 | 6 years | Complete AVB | 9 years | Complete AVB | 11 | c.1458_1493del36 | p.Lys487_Leu498del | 0 |
| 6 | 4 months | Complete AVB | 5 months | Complete AVB | 15 | c.2030C>T | p.Thr677Ile | 0 |
| 7 |
In utero 33 weeks of gestation |
Complete AVB | 3 days | Complete AVB | 16 | c.2209G>A | p.Gly737Arg | 0.0017 |
| 8 |
In utero 36 weeks of gestation |
Complete AVB | 1 day | Complete AVB | 17 | c.2531G>A | p.Gly844Asp | 0.0044 |
| 9 | At birth | Complete AVB | 3 days | Complete AVB | 17 | c.2531G>A | p.Gly844Asp | 0.0044 |
| 10 | 8.5 years | Complete AVB | 8.5 years | Complete AVB | 17 | c.2531G>A | p.Gly844Asp | 0.0044 |
| 11 | At birth | Complete AVB | 2 days | Complete AVB | 17 | c.2531G>A | p.Gly844Asp | 0.0044 |
| 12 |
In utero 38 weeks of gestation |
Complete AVB | 1 day | Complete AVB | 17 | c.2531G>A | p.Gly844Asp | 0.0044 |
| 13 | 18 months |
AVB1 AVB 2/1 |
2 years | Complete AVB | 18 | c.2761G>A | p.Val921Ile | 0.0001 |
| 14 | 5 years | Complete AVB | 5.5 years | Complete AVB | 24 | c.3611C>T | p.Pro1204Leu | 0.0051 |
TRPM4 genotypes and their respective allele frequencies in controls are presented for 14 congenital AVB cases. Age and rhythm at diagnosis and pacemaker implantation are presented for each patient. (1) c.355G>T, p.A119S. (2) c.5415_5418del415 p.Thr1806SerfsX27. AVB indicates atrioventricular block; ExAc, Exome Aggregation Consortium.
Patients 3 and 4 also harbor NKX2.5 and SCN5A point mutations, respectively.
TRPM4 was screened in this cohort (Table 2) because 4 recent studies have reported variants in TRPM4 in patients with cardiac conduction disease and BrS.8, 9, 10, 11 We identified 10 TRPM4 variants in 14 patients, and 3 of those variants were already reported in familial cases of progressive cardiac conduction disease (A432T, G582S, and G844D)9, 10 (Table 2). The G582S variant was found to be isolated only with A432T variants in 2 patients belonging to 2 unrelated families (Table 2). Two of the 10 TRPM4 variants were previously classified as polymorphisms (P1204L and K487L498del)8 because of their frequencies in control populations, whereas 5 nonsynonymous variants were never previously described (D198G, R252Q, T677I, G737R, V921I) (Table 2).
Pathogenicity Assessment of TRPM4 Variants
Allele frequency was evaluated in a cohort of controls. The results are shown in Table 2. Five variants with an allele frequency ≥0.1% were classified as polymorphisms and were excluded from functional analysis (R252Q, K487L498del, G737R, G844D and P1204L) (Table 2). Five rare variants with a frequency ≤0.1% (D198G, A432T, G582S, T677I, and V921I), found in conserved regions of TRPM4 (Figure 2), were subjected to biochemical and functional investigations.
Figure 2.

Human, mouse, and rat TRPM4 sequence alignments and localization of variants described in this study.
The Double Variant: A432T/G582S
The TRPM4 mutant A432T was shown previously to generate a gain of function.9 In the present study, we identified 2 unrelated patients with this variant (Figures 1 and 3). In contrast to the former study,9 these patients also had a second variant, G582S (Figure 1), on the same haplotype. Functional studies were simultaneously performed on A432T, G582S, and A432T/G582S to evaluate the impact of the individual variants on TRPM4 function. These 2 patients also carried NKX2.5 and SCN5A variants in their genomes, as described earlier (Table 2).
Figure 3.
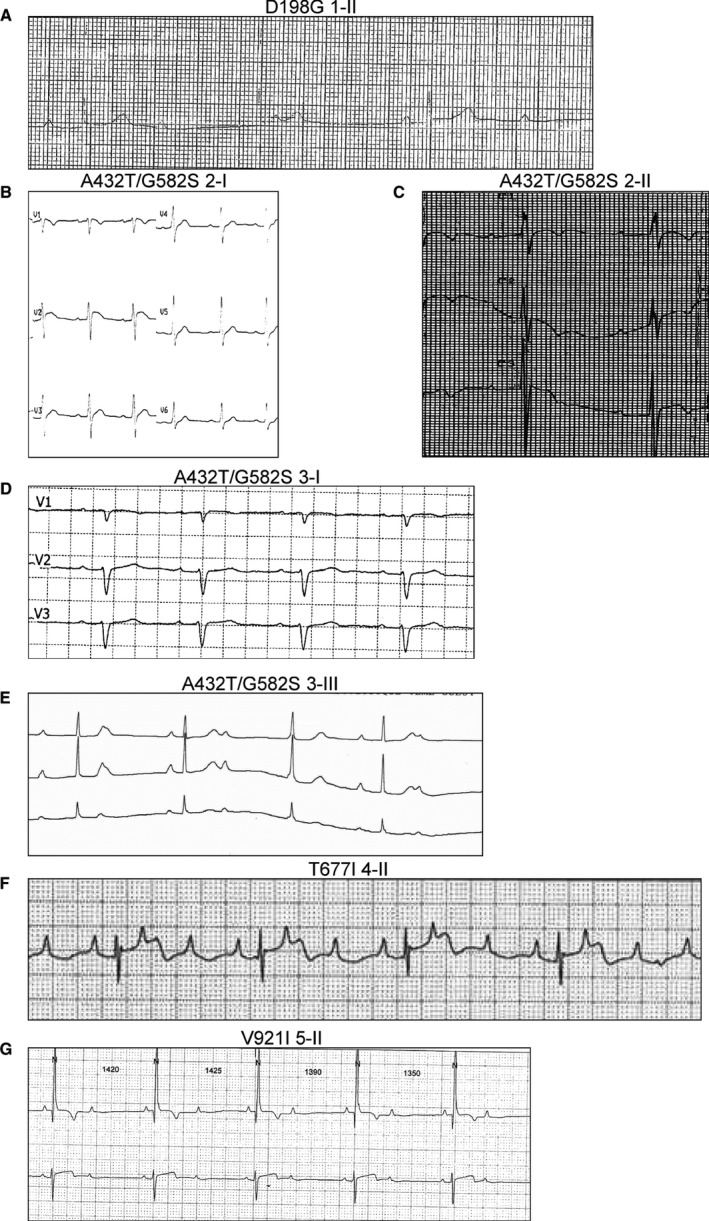
ECGs of patients harboring TRPM4 variants. A, ECG of a girl aged 14 years showing severe bradycardia caused by complete atrioventricular block, with narrow QRS complexes (heart rate 37 beats/min, QRS complex 69 ms). B, ECG in a man aged 45 years showing atrioventricular block type 1 (PR interval 240 ms) associated with an incomplete left bundle branch block (RS pattern in right precordial leads, RS pattern in left precordial leads, QRS complex 110 ms, QRS axis −15°). C, ECG in a girl aged 7 years showing atrioventricular block type 1 (PR interval 280 ms) associated with incomplete right bundle branch block (RSR′ pattern in right precordial leads, QRS complex 94 ms, QRS axis 135°). D, ECG in a man aged 47 years showing atrioventricular block type 1 (PR interval 220 ms) associated with incomplete left bundle branch block (RS pattern in right precordial leads, RS pattern in left precordial leads, QRS complex 100 ms, QRS axis −35°). E, ECG in a girl aged 7 years showing bradycardia caused by complete atrioventricular block with narrow QRS complex (heart rate 40 beats/min, QRS complex 80 ms). F, Holter ECG in a boy aged 6 months showing severe bradycardia caused by complete atrioventricular block with narrow QRS complexes (heart rate 55 beats/min, QRS complex 65 ms). G, Holter ECG in a boy aged 2 years showing severe bradycardia caused by 2/1 atrioventricular block with narrow QRS complexes (heart rate 40 beats/min, QRS 80 ms).
TRPM4 Variants Show Different Protein Expression Levels
We assessed whether the TRPM4 variants have any effect on protein expression when expressed transiently in HEK293 cells. We performed cell surface biotinylation experiments to determine whether they affect TRPM4 expression at the cell membrane. As reported previously,19 we observed that WT and TRPM4 variants were expressed in fully and core‐glycosylated forms (Figure 4A). We expressed the E7K variant to serve as a control. The E7K variant was reported previously in a family with a cardiac conduction defect8 and was shown to lead to increased expression levels in HEK293 cells.8 As illustrated in Figure 4A and 4B, we also observed increased expression at the cell membrane for the E7K variant (184±26% of WT; quantification done on 4 different Western blots), and the 2 variants A432T and A432T/G582S showed decreased expression (28.8±4.5% and 16.5±6.9% of WT, respectively; quantification done on 4 different Western blots). Interestingly, the variant G582S alone showed an increase in expression (176.5±15% of WT; quantification done on 4 different Western blots), whereas the variants D198G, T677I, and V921I showed no significant changes in expression (Figure 4A and 4B) (quantification done on 4 different Western blots). We performed real time‐PCR experiments to determine whether these differences may have been caused by variable mRNA levels; however, no significant differences were observed (data not shown).
Figure 4.
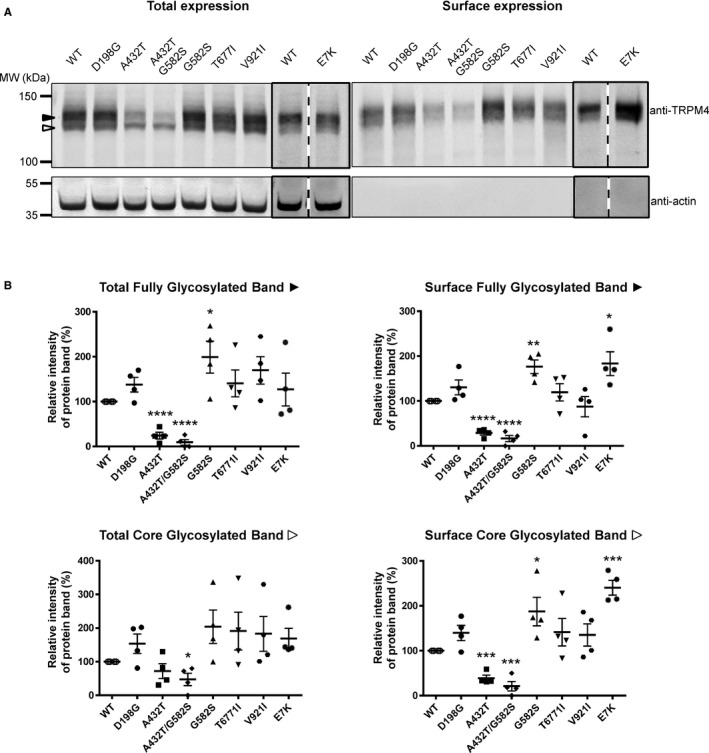
Expression of TRPM4 WT and atrioventricular block variants. A, Western blots showing the expression of TRPM4 at the total (left panel) and surface levels (right panel), with black and white arrowheads representing fully glycosylated and core‐glycosylated forms of TRPM4, respectively. B, Quantification of the Western blots is shown as relative intensity of protein bands for both fully and core‐glycosylated forms of TRPM4 in each fraction. Quantification done on 4 different Western blots. *P<0.05, **P<0.01, ***P<0.001. WT indicates wild type.
Electrophysiological Properties of TRPM4 Variants Involved in AVB
As recently reported using the patch‐clamp technique in whole‐cell configuration with HEK293 cells,20 TRPM4 currents recorded over time showed 2 distinct phases (Figure 5A). There is a transient phase of approximately <1 minute that appears quickly after the rupture of the membrane patch, and a steady plateau phase that appears 4 to 8 minutes later (Figure 5A). To investigate the functional consequences of the TRPM4 variants, we measured the transient and plateau current densities using a ramp protocol (Figure 5A). The transient phase showed widely variable current amplitudes, possibly because of a known phenomenon of channel desensitization.21, 22 We considered the more stable plateau phase for further functional analysis. We observed decreased current density with the variant A432T (42±4% of WT; WT, n=20, and A432T, n=6; P<0.05) and increased current density with the variants G582S (171±20% of WT; WT, n=20, and G582S, n=6; P<0.05) and E7K (238±46% of WT; WT, n=20, and E7K, n=6; P<0.05). The rest of the variants did not show any significant changes in current densities in comparison to WT (WT, n=20; D198G, n=6; A432T/G582S, n=5; T677I, n=5; and V921I, n=5) (Figure 5B and 5C).
Figure 5.
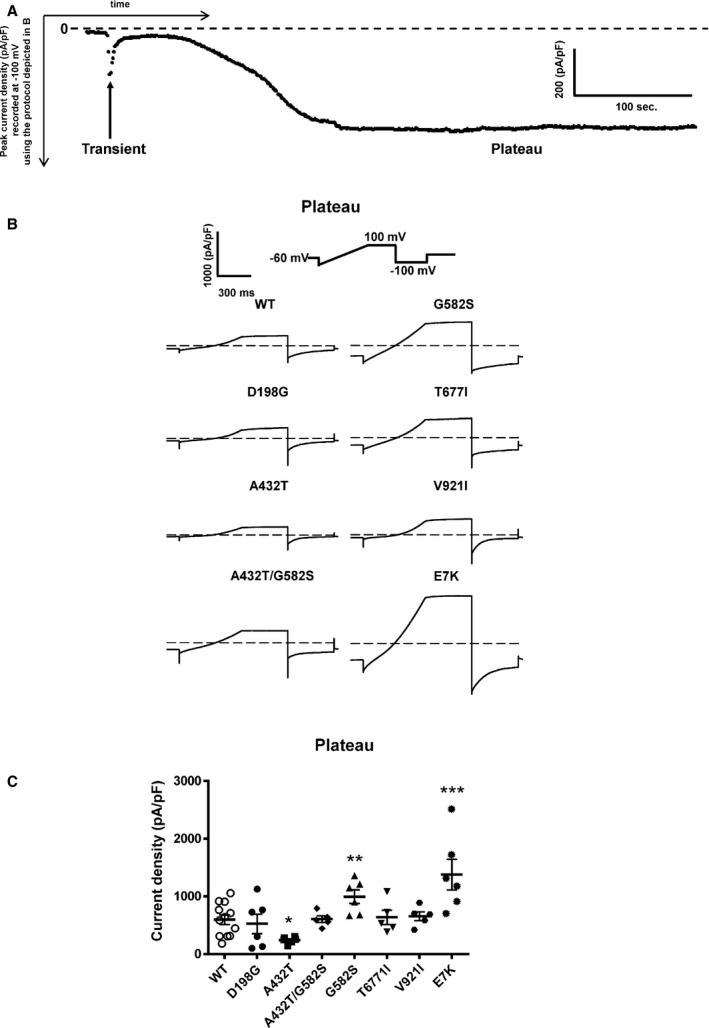
Whole‐cell patch‐clamp recording of WT and TRPM4 variants. A, Time course recording of TRPM4 current. B, Individual current traces of each WT and TRPM4 variants recorded as plateau phases. C, Quantification of current density of WT and atrioventricular block variants. *P<0.05, **P<0.01, ***P<0.001 (WT, n=20; D198G, n=6; A432T, n=6; A432T/G582S, n=5; G582S, n=6; T677I, n=5; V921I, n=5; and E7K, n=6). WT indicates wild type.
No Evidence of Direct SUMOylation of TRPM4
In the initial report linking TRPM4 to cardiac conduction defects,8 it was proposed that the gain of function demonstrated by the E7K variant, observed as increased protein expression and current density, was caused by augmentation of channel SUMOylation. A variant in the present study, G582S, also showed increased expression at the plasma membrane (Figure 4B) and current density (Figure 5C). We performed immunoprecipitation experiments and subsequent Western blots using anti‐SUMO1 and anti‐SUMO2/3 antibodies to investigate its SUMOylation status. The Ran GTPase‐activating protein 1 (RanGAP1) was used as a positive control because it is a well‐known protein that is SUMOylated with SUMO1, SUMO2, and SUMO3.23 As illustrated in Figure 6A, RanGAP1 (left panel) and both TRPM4 (WT and G582S; right panel) were immunoprecipitated. RanGAP1 showed a doublet, corresponding to its SUMOylated (≈90 kDa) and non‐SUMOylated (≈70 kDa) forms.24 The immunoprecipitated RanGAP1 and TRPM4 were then blotted with anti‐SUMO1 and anti‐SUMO2/3. As shown in Figure 6B, RanGAP1 showed clear signals for anti‐SUMO1 (upper panel) and anti‐SUMO2/3 (lower panel) at ≈90 kDa, whereas TRPM4 WT and the mutant G582S did not (Figure 6B). Based on these results, we came to the conclusion that TRPM4 is not directly SUMOylated with SUMO1, SUMO2, or SUMO3.
Figure 6.
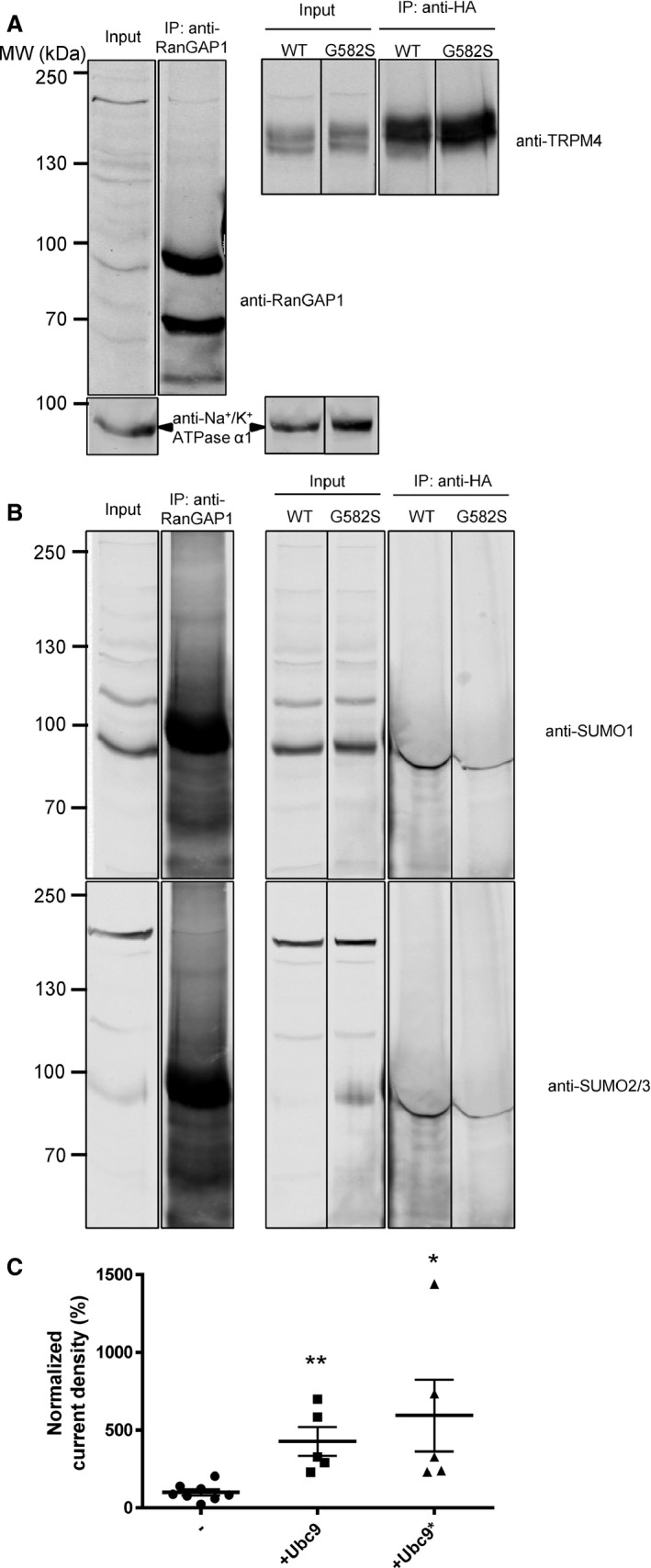
Role of SUMOylation. Immunoprecipitation with anti‐RanGAP1 and anti‐HA. A, Western blots on the upper panel show immunoprecipitation of RanGAP1 and TRPM4, (B) whereas the lower panels show their SUMOylation status. All samples were run on the same gel and were blotted multiple times on the same membrane with different antibodies. The lanes were rearranged for clarity, and each experiment was performed 3 times. C, Normalized current density of WT TRPM4 coexpressed with either Ubc9 or the catalytically inactive mutant Ubc9*. *P<0.05 (WT, n=8; Ubc9, n=5; and Ubc9*, n=5). WT indicates wild type.
Because Ubc9 is the E3 ligase responsible for SUMOylation,8, 9 we tested whether this enzyme was able to increase the current density of WT TRPM4, as previously shown by Kruse et al.8 As seen in Figure 6C, the coexpression of Ubc9 increased the TRPM4 current density by 3‐fold (WT, n=8, and Ubc9, n=5; P<0.05). Nevertheless, because a similar increase was observed with the catalytic inactive mutant (Ubc9*) of the E3 ligase, we concluded that this effect was not dependent on the SUMOylation activity of the protein (WT, n=8, and Ubc9*, n=5; P<0.05). These experiments suggest that direct SUMOylation of TRPM4 is unlikely to be linked to the increased expression and gain of function of the TRPM4 G582S variant.
TRPM4 Loss‐of‐Function Variants Are Rescued by Incubation at Low Temperature
TRPM4 variants A432T and A432T/G582S showed reduced protein expression at the cell membrane, a phenomenon that has not been reported previously (Figure 4B). Because decreased expression may be a result of protein misfolding, we tested whether lowering the incubation temperature could possibly rescue the misfolded membrane proteins and restore their expression at the cell surface, as already shown by the others.25 As illustrated in Figure 7A, lowering the incubation temperature of cells expressing the variants to 28°C for 24 hours increased their expression both at the total level (A432T 78±3% and A432T/G582S 69±5% of WT; quantification done on 3 different Western blots) and at the cell surface level (A432T 72±8% and A432T/G582S 65±7% of WT; quantification done on 3 different Western blots). This partial rescue was also observed at the functional level with a significant increase in the current density of A432T (599±104 pA/pF [n=6] at 28°C versus 230±27 pA/pF [n=6] at 37°C; P<0.05), whereas WT showed no significant change (639±101 pA/pF [n=4] at 28°C versus 553±74 pA/pF [n=6] at 37°C; P>0.05) (Figure 7B).
Figure 7.
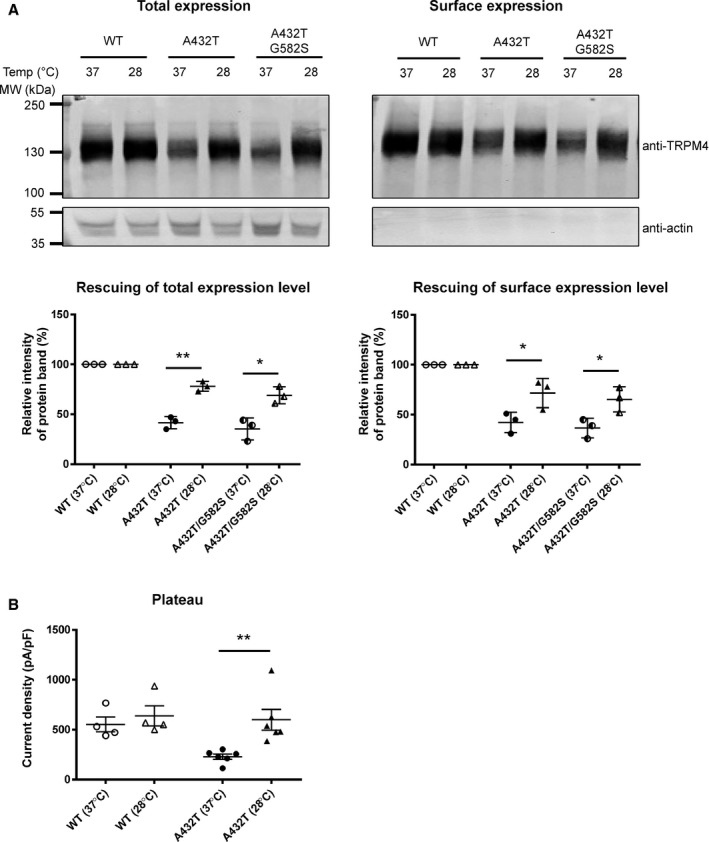
Rescue experiments of WT and TRPM4 variants. A, Western blot and quantification showing expression of WT and TRPM4 variants at the total and surface levels at 37°C and 28°C. Quantification was done on 3 different Western blots. B, Current density of WT and A432T at 37°C and 28°C recorded as the plateau phase. *P<0.05, **P<0.01 (WT at 28°C, n=4; WT at 37°C, n=6; A432T at 28°C, n=6; and A432T at 37°C, n=6). Temp indicates temperature; WT, wild type.
A Fraction of TRPM4 Protein Is Ubiquitylated
As presented in Figure 7, protein misfolding seems to be involved in decreased protein levels of selected TRPM4 variants. It may be postulated that the ubiquitylation–proteasome system might also play a role in this phenomenon.26 To see whether TRPM4 can be ubiquitylated, we performed a GST pull‐down experiment using GST‐S5a, a GST‐tagged proteasome subunit that recognizes ubiquitylated proteins.27 With this GST‐S5a, we were able to pull down bulk ubiquitylated proteins that were otherwise absent when pulled down with GST only (Figure 8A). As presented in Figure 8A, the pull‐down fractions showed bands that correspond to TRPM4 when revealed with antibody against TRPM4; however, quantification of input and pull‐down fractions did not show any significant difference between the 2 fractions for the WT and mutant TRPM4 channels (Figure 8B) (quantification done on 3 different Western blots). Our results suggest a fraction of TRPM4 to be ubiquitylated; however, we did not observe any further increase in the ubiquitylation to explain the loss‐of‐function variants.
Figure 8.
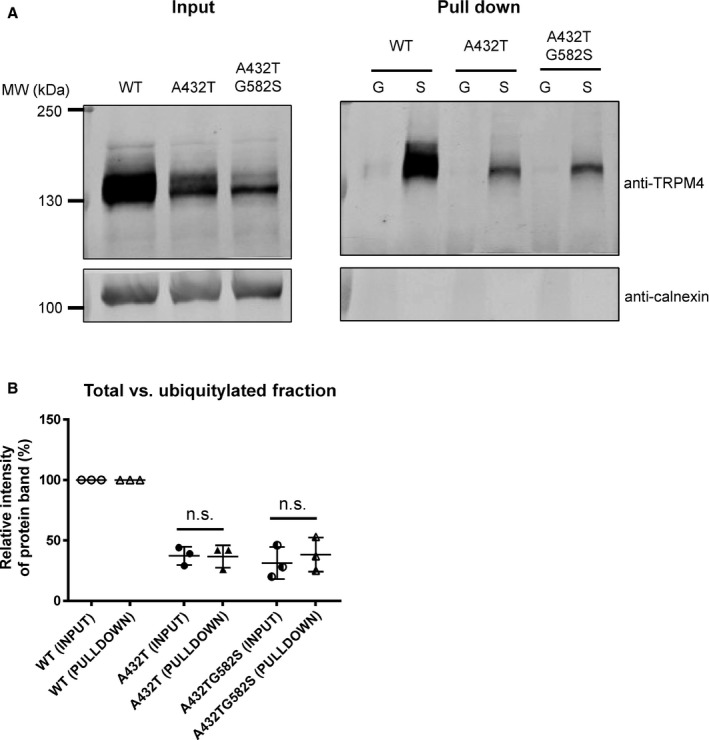
Ubiquitylation of TRPM4. A, GST pull‐down experiment using GST‐tagged S5a (S), a subunit proteasome that recognizes ubiquitylated proteins, and GST alone (G) to pull down TRPM4 from HEK293 cells transiently transfected with WT TRPM4, A432T, and A432T/G582S individually. The input fraction is shown at left, and the pull‐down fraction is shown at right. B, Quantification of each fraction and comparison of WT to variant ratios between fractions. Quantification was done on 3 different Western blots. GST indicates glutathione‐S‐transferase; n.s., not significant; WT, wild type.
Discussion
The study had several main findings. First, the study identified 5 rare variants in the gene encoding TRPM4 channel in patients with congenital or childhood AVB. Second, functional and expression studies in a heterologous expression system identified 2 of these variants, A432T and A432T/G582S, with loss of function and variant G582S with gain of function. Third, unlike previous reports, we did not find any evidence to confirm a direct or indirect role of SUMOylation in the gain‐of‐function TRPM4 variants. Fourth, the loss‐of‐function TRPM4 variants could likely be caused by protein misfolding and retention in the endoplasmic reticulum because a reduced incubation temperature was able to partially rescue their expression and function; however, the loss of function could not be explained by an increase of ubiquitylation of TRPM4 protein.
Roles of TRPM4 in Cardiac Function
The localization and role of the TRPM4 channel in the heart is still not completely understood. The fact that the pathogenic TRPM4 variant E7K, among others, leads to conduction disorders8 and that immunoreactivity was observed in Purkinje fibers of the bovine heart9 strongly suggest that the TRPM4 channel is expressed and plays an important role in the cardiac conduction pathway. Furthermore, several studies9, 28 have provided evidence of the expression and function of TRPM4 in the atrial myocardium and mouse sinoatrial node cells.29 Mathar et al14 recently recorded TRPM4 currents from isolated ventricular myocytes using Trpm4 knockout mice. These Ca2+‐activated currents were found to prolong the late phase of the mouse action potential.14 Interestingly, the silencing of TRPM4 increased the β‐adrenergic–dependent inotropic response of the ventricles. The ECG characteristics of Trpm4 knockout mice are controversial. Mathar et al14 did not observe any difference in the standard ECG parameters, whereas Demion et al30 reported prolonged PR and QRS durations, reduced conduction times above and below the His‐bundle, and episodes of intermittent AVB. This discrepancy may be related to the different genetic backgrounds of the 2 different knockout mouse lines or other methodological differences. Further studies using cardiac‐specific knockout combined with potent pharmacological tools will help decipher the physiological role of TRPM4 in heart.
To date, 4 studies8, 9, 10, 11 have reported rare TRPM4 variants in patients and families with various types of cardiac conduction disorders and BrS. The present study further supports the role of this channel in cardiac conduction, particularly in atrioventricular conduction; therefore, we propose adding TRPM4 to the list of candidate genes to be screened in patients with conduction disease and BrS.
Altered Expression With TRPM4 AVB Variants
Several disease‐causing variants in ion channels have helped unravel their role in physiology. The present work, with identification of 5 new variants observed in patients with either congenital or childhood AVB, further strengthens the significance of TRPM4 in the cardiac conduction system. In analyzing the functional consequences of TRPM4 AVB variants (A432T and A432T/G582S), we determined a reduction in cell membrane expression and function. A possible explanation for such a loss of function could be protein misfolding. To test our hypothesis about misfolding of TRPM4 AVB variants, we performed temperature rescue experiments. A similar temperature‐dependent rescue of misfolded protein was demonstrated previously in CFTR31 and hERG.32 Indeed, we observed a partial rescue of the expression and function of the TRPM4 variants (A432T and A432T/G582S) at lower temperature. To the best of our knowledge, this report is the first showing possible rescue of TRPM4 variants with incubation at low temperature. This finding also provides a clinical implication of using TRPM4 as a target for potent pharmacological chaperones in rescuing loss‐of‐function variants.33 These misfolded proteins could be tagged by ubiquitylation to be further processed by the endoplasmic reticulum–associated protein degradation pathway.34 Using a GST‐tagged proteasome subunit, we pulled down ubiquitylated fractions of TRPM4 WT and its variants; however, our data did not suggest increased ubiquitylation as the cause of loss of function. When the variant G582S was expressed individually, increased expression and current density was observed. Previous studies reported impairment of channel endocytosis and an increase in SUMOylation for TRPM4 gain‐of‐function mutants.8, 9 We investigated the hypothesis that TRPM4 could be SUMOylated and the gain‐of‐function variant (G582S) could be explained by increase in its SUMOylation status, as shown by Kruse et al.8 On the contrary, despite using reliable positive controls in our study, we did not observe any evidence of direct SUMOylation of either the TRPM4 WT or G582S variant. We observed a stimulatory effect on WT TRPM4 with the coexpression of the SUMO‐conjugating enzyme Ubc9, similar to Kruse et al,8 but this increase was also observed when a catalytic inactive mutant of Ubc9 was used. The existing disparity between our studies and those previously reported by Kruse et al illustrates the complexity of the regulation of TRPM4 and its expression and trafficking to the cell membrane; however, the existence of indirect SUMOylation of TRPM4 via an ancillary protein may be possible. Our findings suggest that the role of SUMOylation in the mechanism of gain of expression and function of TRPM4 variants should be reconsidered.
Both Gain and Loss of Function Were Observed
The observation that, as in BrS,11 these variants led to either gain or loss of function is puzzling. Furthermore, the mechanism by which these rare variants may reduce impulse propagation in the conduction pathway is more intriguing. A possibility is a mechanism analogous to the supernormal excitability and conduction phenomenon35 that has been described in atrioventricular conduction.36 Because TRPM4 generates a net inward depolarizing current, assuming a tonic activity under resting conditions, its increase would depolarize the resting membrane potential, whereas its reduction would hyperpolarize it. Consequently, the availability of the excitable Na+ and Ca2+ channels would depend on TRPM4 activity. Both TRPM4‐dependent hyperpolarization and depolarization of the resting membrane potential could reduce excitability and conduction; however, no changes in the resting membrane potential of ventricular cardiomyocytes were reported in the study of Mathar et al,14 suggesting that this model should be further investigated using cardiac tissues from WT and Trpm4 knockout mice, with particular attention to its roles in the conduction pathway.
Limitations of the Study
A main limitation of this study is that no direct causality between the TRPM4 variants and congenital AVB could be demonstrated. Whether or not a loss or gain of function is related to the phenotype is still under debate. The small pedigree size along with the sporadic nature of the presence of the variants with the phenotype precludes any statistical associations. In addition, the 2 TRPM4 variants that reveal a functional impairment of the TRPM4 channel have been identified in patients that also carry a mutation in other genes known to be involved in AVB (SCN5A, NKX2.5), thus it cannot be excluded that the AVB phenotype might not just be related to mutations in TRPM4. Finally, 3 variants identified did not lead to any defects in our conditions (D198G, T677I, and V921I). Future genetic studies with larger cohorts of patients with different types of conduction disorders will help clarify the precise role of TRPM4 in these phenotypes. In addition, caution must be taken when extrapolating the results of heterologous expression studies, as performed in this study, to the in vivo situation. Further studies using knockout and knock‐in mouse models expressing similar variants are required to better ascertain the proposed molecular and cellular pathological mechanisms.
Conclusions
In conclusion, this study further supports the role of TRPM4 genetic variants in genetically determined cardiac conduction disorders such as congenital and childhood AVB. Future studies addressing the location and physiological and pathophysiological roles of this channel in cardiac function are necessary.
Author Contributions
Conception and design of the experiments: Syam, Chatel, Sottas, Rougier, Baruteau, Baron, Amarouch, Daumy, Probst, Schott, and Abriel. Collection, analysis and interpretation of data: Syam, Chatel, Sottas, Rougier, Baruteau, Baron, Amarouch, Daumy, Probst, Schott, and Abriel. Drafting the article or revising it critically for important intellectual content: Syam, Chatel, Ozhathil, Sottas, Rougier, Baruteau, Baron, Amarouch, Daumy, Probst, Schott, and Abriel. All authors approved the final version of the manuscript and all authorships are listed.
Sources of Funding
This work was supported by the Association Française contre les Myopathies grant 14305, and ANR‐08‐GENO‐006‐01, project R08147DS, the Swiss National Science Foundation to Abriel (310030B_14706035693), and the TransCure NCCR Network, the Berne University Research Foundation, PHRC PROG/11/33 – RNI and the Coeur et Recherche Foundation to Probst and the Leducq Foundation (CVD‐05; Alliance Against Sudden Cardiac Death) to Schott.
Disclosures
None.
Acknowledgments
We thank Dr A. Felley and the members of the Hugues Abriel's group for their help and useful comments on this manuscript. The authors would like to thank the contribution of pediatric cardiologists (listed below), as well as the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL‐102923), the WHI Sequencing Project (HL‐102924), the Broad GO Sequencing Project (HL‐102925), the Seattle GO Sequencing Project (HL‐102926) and the Heart GO Sequencing Project (HL‐103010). The following pediatric cardiologists that provided patients are acknowledged: Albin Behaghel, Philippe Mabo, Elisabeth Villain, Jean‐Benoit Thambo, Francois Marcon,Veronique Gournay, Francis Rouault, Alain Chantepie, Sophie Guillaumont, Francois Godart, Caroline Bonnet, Alain Fraisse, Jean‐Marc Schleich, Jean‐Rene Lusson, Yves Dulac, Christophe Leclercq, Jean‐Claude Daubert.
(J Am Heart Assoc. 2016;5:e001625 doi: 10.1161/JAHA.114.001625)
References
- 1. Baruteau AE, Behaghel A, Fouchard S, Mabo P, Schott JJ, Dina C, Chatel S, Villain E, Thambo JB, Marcon F, Gournay V, Rouault F, Chantepie A, Guillaumont S, Godart F, Martins RP, Delasalle B, Bonnet C, Fraisse A, Schleich JM, Lusson JR, Dulac Y, Daubert JC, Le Marec H, Probst V. Parental electrocardiographic screening identifies a high degree of inheritance for congenital and childhood nonimmune isolated atrioventricular block. Circulation. 2012;126:1469–1477. [DOI] [PubMed] [Google Scholar]
- 2. Xiao GQ, Hu K, Boutjdir M. Direct inhibition of expressed cardiac L‐ and T‐type calcium channels by IgG from mothers whose children have congenital heart block. Circulation. 2001;103:1599–1604. [DOI] [PubMed] [Google Scholar]
- 3. Xiao GQ, Qu Y, Hu K, Boutjdir M. Down‐regulation of L‐type calcium channel in pups born to 52 kDa SSA/Ro immunized rabbits. FASEB J. 2001;15:1539–1545. [DOI] [PubMed] [Google Scholar]
- 4. Korb D, Tng PY, Milenkovic VM, Reichhart N, Strauss O, Ritter O, Fischer T, Benz PM, Schuh K. Identification of PDZ domain containing proteins interacting with 1.2 and PMCA4b. ISRN Cell Biol. 2013;2013:16. [Google Scholar]
- 5. Prall OW, Elliott DA, Harvey RP. Developmental paradigms in heart disease: insights from tinman. Ann Med. 2002;34:148–156. [PubMed] [Google Scholar]
- 6. Lupoglazoff JM, Cheav T, Baroudi G, Berthet M, Denjoy I, Cauchemez B, Extramiana F, Chahine M, Guicheney P. Homozygous SCN5A mutation in long‐QT syndrome with functional two‐to‐ one atrioventricular block. Circ Res. 2001;89:E16–E21. [DOI] [PubMed] [Google Scholar]
- 7. Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F, Escande D, Wiesfeld AC, Pfeufer A, Kaab S, Wichmann HE, Hasdemir C, Aizawa Y, Wilde AA, Roden DM, Bezzina CR. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kruse M, Schulze‐Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, Ohmert I, Brink P, Pongs O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest. 2009;119:2737–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu H, El Zein L, Kruse M, Guinamard R, Beckmann A, Bozio A, Kurtbay G, Megarbane A, Ohmert I, Blaysat G, Villain E, Pongs O, Bouvagnet P. Gain‐of‐function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ Cardiovasc Genet. 2010;3:374–385. [DOI] [PubMed] [Google Scholar]
- 10. Stallmeyer B, Zumhagen S, Denjoy I, Duthoit G, Hebert JL, Ferrer X, Maugenre S, Schmitz W, Kirchhefer U, Schulze‐Bahr E, Guicheney P. Mutational spectrum in the Ca(2+) ‐activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum Mutat. 2012;33:109–117. [DOI] [PubMed] [Google Scholar]
- 11. Liu H, Chatel S, Simard C, Syam N, Salle L, Probst V, Morel J, Millat G, Lopez M, Abriel H, Schott JJ, Guinamard R, Bouvagnet P. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS One. 2013;8:e54131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gonzales AL, Garcia ZI, Amberg GC, Earley S. Pharmacological inhibition of TRPM4 hyperpolarizes vascular smooth muscle. Am J Physiol Cell Physiol. 2010;299:C1195–C1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simard JM, Kahle KT, Gerzanich V. Molecular mechanisms of microvascular failure in central nervous system injury—synergistic roles of NKCC1 and SUR1/TRPM4. J Neurosurg. 2010;113:622–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mathar I, Kecskes M, Van der Mieren G, Jacobs G, Camacho Londono JE, Uhl S, Flockerzi V, Voets T, Freichel M, Nilius B, Herijgers P, Vennekens R. Increased beta‐adrenergic inotropy in ventricular myocardium from Trpm4−/− mice. Circ Res. 2014;114:283–294. [DOI] [PubMed] [Google Scholar]
- 15. Abriel H, Syam N, Sottas V, Amarouch MY, Rougier JS. TRPM4 channels in the cardiovascular system: physiology, pathophysiology, and pharmacology. Biochem Pharmacol. 2012;84:873–881. [DOI] [PubMed] [Google Scholar]
- 16. Baruteau AE, Fouchard S, Behaghel A, Mabo P, Villain E, Thambo JB, Marcon F, Gournay V, Rouault F, Chantepie A, Guillaumont S, Godart F, Bonnet C, Fraisse A, Schleich JM, Lusson JR, Dulac Y, Leclercq C, Daubert JC, Schott JJ, Le Marec H, Probst V. Characteristics and long‐term outcome of non‐immune isolated atrioventricular block diagnosed in utero or early childhood: a multicentre study. Eur Heart J. 2012;33:622–629. [DOI] [PubMed] [Google Scholar]
- 17. Bazett H. An analysis of the time‐relations of electrocardiograms. Heart. 1920;7:353–370. [Google Scholar]
- 18. Dentice M, Cordeddu V, Rosica A, Ferrara AM, Santarpia L, Salvatore D, Chiovato L, Perri A, Moschini L, Fazzini C, Olivieri A, Costa P, Stoppioni V, Baserga M, De Felice M, Sorcini M, Fenzi G, Di Lauro R, Tartaglia M, Macchia PE. Missense mutation in the transcription factor NKX2‐5: a novel molecular event in the pathogenesis of thyroid dysgenesis. J Clin Endocrinol Metab. 2006;91:1428–1433. [DOI] [PubMed] [Google Scholar]
- 19. Syam N, Rougier JS, Abriel H. Glycosylation of TRPM4 and TRPM5 channels: molecular determinants and functional aspects. Front Cell Neurosci. 2014;8:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amarouch MY, Syam N, Abriel H. Biochemical, single‐channel, whole‐cell patch clamp, and pharmacological analyses of endogenous TRPM4 channels in HEK293 cells. Neurosci Lett. 2013;541:105–110. [DOI] [PubMed] [Google Scholar]
- 21. Nilius B, Mahieu F, Prenen J, Janssens A, Owsianik G, Vennekens R, Voets T. The Ca2+‐activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5‐biphosphate. EMBO J. 2006;25:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z, Okawa H, Wang Y, Liman ER. Phosphatidylinositol 4,5‐bisphosphate rescues TRPM4 channels from desensitization. J Biol Chem. 2005;280:39185–39192. [DOI] [PubMed] [Google Scholar]
- 23. Flotho A, Werner A. The RanBP2/RanGAP1*SUMO1/Ubc9 complex: a multisubunit E3 ligase at the intersection of sumoylation and the RanGTPase cycle. Nucleus. 2012;3:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mahajan R, Gerace L, Melchior F. Molecular characterization of the SUMO‐1 modification of RanGAP1 and its role in nuclear envelope association. J Cell Biol. 1998;140:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keller DI, Rougier JS, Kucera JP, Benammar N, Fressart V, Guicheney P, Madle A, Fromer M, Schlapfer J, Abriel H. Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res. 2005;67:510–519. [DOI] [PubMed] [Google Scholar]
- 26. Diaz‐Villanueva JF, Diaz‐Molina R, Garcia‐Gonzalez V. Protein folding and mechanisms of proteostasis. Int J Mol Sci. 2015;16:17193–17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994;269:7059–7061. [PubMed] [Google Scholar]
- 28. Guinamard R, Chatelier A, Demion M, Potreau D, Patri S, Rahmati M, Bois P. Functional characterization of a Ca(2+)‐activated non‐selective cation channel in human atrial cardiomyocytes. J Physiol. 2004;558:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Demion M, Bois P, Launay P, Guinamard R. TRPM4, a Ca2+‐activated nonselective cation channel in mouse sino‐atrial node cells. Cardiovasc Res. 2007;73:531–538. [DOI] [PubMed] [Google Scholar]
- 30. Demion M, Thireau J, Gueffier M, Finan A, Khoueiry Z, Cassan C, Serafini N, Aimond F, Granier M, Pasquie JL, Launay P, Richard S. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS One. 2014;9:e115256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature‐sensitive. Nature. 1992;358:761–764. [DOI] [PubMed] [Google Scholar]
- 32. Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem. 1999;274:31123–31126. [DOI] [PubMed] [Google Scholar]
- 33. Naik S, Zhang N, Gao P, Fisher MT. On the design of broad based screening assays to identify potential pharmacological chaperones of protein misfolding diseases. Curr Top Med Chem. 2012;12:2504–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rougier JS, Albesa M, Abriel H, Viard P. Neuronal precursor cell‐expressed developmentally down‐regulated 4‐1 (NEDD4‐1) controls the sorting of newly synthesized Ca(V)1.2 calcium channels. J Biol Chem. 2011;286:8829–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spear JF, Moore EN. Supernormal excitability and conduction in the His‐Purkinje system of the dog. Circ Res. 1974;35:782–792. [DOI] [PubMed] [Google Scholar]
- 36. Damato AN, Wit AL, Lau SH. Observations on the mechanism of one type of so‐called supernormal A‐V conduction. Am Heart J. 1971;82:725–730. [DOI] [PubMed] [Google Scholar]