

Abstract
Background
Proprotein convertase subtilisin kexin 9 (PCSK9) inhibitors reduce low‐density lipoprotein cholesterol (LDL‐C) and improve outcomes in the general population. HIV‐infected individuals are at increased risk for cardiovascular events and have high rates of dyslipidemia and hepatitis C virus (HCV) coinfection, making PCSK9 inhibition a potentially attractive therapy.
Methods and Results
We studied 567 participants from a clinic‐based cohort to compare PCSK9 levels in patients with HIV/HCV coinfection (n=110) with those with HIV infection alone (n=385) and with uninfected controls (n=72). The mean age was 49 years, and the median LDL‐C level was 100 mg/dL (IQR 77–124 mg/dL); 21% were taking statins. The 3 groups had similar rates of traditional risk factors. Total cholesterol, LDL‐C, and high‐density lipoprotein cholesterol levels were lower in coinfected patients compared with controls (P<0.001). PCSK9 was 21% higher in HIV/HCV‐coinfected patients versus controls (95% CI 9–34%, P<0.001) and 11% higher in coinfected individuals versus those with HIV infection alone (95% CI 3–20%, P=0.008). After adjustment for cardiovascular risk factors, HIV/HCV coinfection remained significantly associated with 20% higher PCSK9 levels versus controls (95% CI 8–33%, P=0.001). Interleukin‐6 levels increased in a stepwise fashion from controls (lowest) to HIV‐infected to HIV/HCV‐coinfected individuals (highest) and correlated with PCSK9 (r=0.11, P=0.018).
Conclusions
Despite having lower LDL‐C, circulating PCSK9 levels were increased in patients coinfected with HIV and HCV in parallel with elevations in the inflammatory, proatherogenic cytokine interleukin‐6. Clinical trials should be conducted to determine the efficacy of targeted PCSK9 inhibition in the setting of HIV/HCV coinfection.
Keywords: hepatitis C virus, HIV, low‐density lipoprotein cholesterol, proprotein convertase subtilisin kexin 9
Subject Categories: Lipids and Cholesterol, Biomarkers, Inflammation, Cardiovascular Disease, Clinical Studies
Introduction
Dyslipidemia is common in individuals infected with HIV and likely contributes to the higher rates of cardiovascular complications that have been described in this patient population, including acute myocardial infarction (MI) and sudden cardiac death.1, 2, 3, 4 Approximately 25% of all HIV‐infected patients are also infected with hepatitis C virus (HCV).5 Several studies have reported that HIV/HCV coinfection is associated with an even higher risk of adverse cardiovascular outcomes compared with individuals with HIV infection alone.6, 7 Further, both HIV and HCV infections can alter lipid metabolism. HIV infection and certain antiretroviral medications have been associated with higher low‐density lipoprotein cholesterol (LDL‐C), lower high‐density lipoprotein cholesterol (HDL‐C), and higher triglyceride (TG) levels.3, 8, 9 In contrast, HCV infection alone has been associated with lower total cholesterol (TC) and TG levels compared with uninfected controls3 and HIV/HCV coinfection attenuates some of the atherogenic lipid changes observed with HIV infection.10 One potential mechanism for these apparently lower atherogenic lipids is the compromised synthetic capacity of the liver in the setting of chronic viral infection that affects protein synthesis. For example, our group has previously reported that HIV/HCV coinfection is associated with a lower concentration of liver‐derived acute phase reactants, including C‐reactive protein (CRP), compared with HIV‐infected individuals.11 Another mechanism may relate to chronic systemic inflammation.12 Given the high prevalence of HIV/HCV coinfection, increased rates of cardiovascular complications in this setting, and the challenges of using statin therapy in the setting of chronic hepatic disease, the discovery of novel and safe therapies for treating dyslipidemia in such patients is an important therapeutic goal.
Proprotein convertase subtilisin kexin 9 (PCSK9) modulates cell‐surface expression of LDL receptor (LDL‐R), which binds and removes LDL‐C. Upregulation of PCSK9 leads to increased degradation of LDL‐R within the hepatocyte and, therefore, higher circulating concentrations of LDL‐C. Pharmacologic inhibition of PCSK9 with the use of a monoclonal antibody has resulted in substantial lowering of LDL‐C by ≈50% to 80% without any serious adverse effects in multiple short‐term placebo‐controlled trials among individuals without HIV or HCV infection.13, 14, 15, 16, 17, 18, 19 It has also been shown to improve cardiovascular outcomes in uninfected individuals in preliminary studies.20, 21 Because of their favorable safety profile, lack of drug–drug interactions with antiretroviral or HCV medications, and absence of hepatic toxicity, PCSK9 antibody inhibitors may be an advantageous lipid‐lowering therapy for individuals coinfected with HIV and HCV.
We chose to study PCSK9 in the setting of HIV/HCV coinfection for 2 reasons. First, PCSK9 is increased in experimental models of inflammation.22 Second, PCSK9 is synthesized in the liver and levels may be altered in the setting of HCV infection because of hepatic injury or chronic systemic inflammation, and, thus, PCSK9 levels may be dysregulated when HCV infection is present along with HIV infection. Therefore, the purpose of this study was to compare PCSK9 levels among HIV/HCV‐coinfected individuals, HIV‐monoinfected individuals, and uninfected controls. In addition, as PCSK9 may be increased in systemic inflammatory states, we sought to determine if PCSK9 levels in HIV/HCV‐coinfected individuals correlate with measures of systemic inflammation (eg, interleukin [IL]‐6) or with measures of hepatic injury (eg, transaminase elevations).
Methods
Study Population
Four hundred ninety‐five HIV‐infected participants (385 with HIV monoinfection, 110 with HIV/HCV coinfection) from the SCOPE cohort (Studies of the Consequences of the Protease Inhibitor Era) and 72 uninfected controls were included in this study. The SCOPE cohort is a clinic‐based cohort consisting of well‐characterized HIV‐infected patients enrolled at different sites within San Francisco. Our study included SCOPE subjects who agreed to participate in a carotid ultrasound substudy; these individuals were not preselected for cardiovascular disease or symptoms in any way. SCOPE recruitment included the following groups: (1) untreated: no previous antiretroviral therapy [ART] or off ART for 6 months; (2) treated/virologically suppressed: undetectable HIV viral load for previous 6 months while on ART; (3) treated/virologic noncontrolled: patients on ART, with HIV RNA level ≥500 copies/mL; (4) elite controllers: untreated individuals with ≥3 documented HIV RNA <2000 copies/mL during a 12‐month period; and (5) HIV‐uninfected subjects were recruited separately to provide age‐ and demographic‐matched controls. Control subjects were friends and acquaintances of HIV‐infected individuals and have been well characterized by our research group previously.23 All controls were tested and documented to be HIV negative before study enrollment. The protocol, including the blood collection, was approved by the University of California, San Francisco Committee on Human Research, and written consent was obtained from all patients.
PCSK9, Lipid, and Biomarker Measurement
Fasting blood samples were obtained at enrollment, and plasma was isolated within 60 minutes of sample acquisition. Plasma samples were stored in plastic cryovials at a centralized storage facility (San Francisco, CA) where they were maintained at −80°C during storage and shipping. Soluble PCSK9 was measured by using a high‐affinity ELISA (Amgen Inc) at an Amgen testing facility in Richmond, VA, by personnel blinded to clinical characteristics of the patients. The CV of the assay was ≤4%, with a range of quantification of 15 to 1200 ng/mL and a K d of 16 pmol/L.
Fasting blood samples were also used to measure TC, LDL‐C, HDL‐C, and TG levels. The San Francisco General Hospital clinical laboratory was used to measure CD4+ T‐cell count and HIV RNA levels, and the nadir CD4+ T cell count was the lowest laboratory‐confirmed value before study enrollment. IL‐6 and high‐sensitivity CRP (hsCRP) levels were measured using by standard ELISAs.
Participants were followed until August 2015 or until time of death as determined by using the National Death Index.
Statistical Analysis
All analyses were conducted with use of SAS version 9.4 (SAS Institute Inc). The baseline characteristics of control, HIV‐monoinfected, and HIV/HCV‐coinfected subjects were compared by using χ2 tests for categorical predictors, ANOVA for normally distributed age, and Wilcoxon rank sum test for all other continuous predictors, which were not normally distributed even after log transformation. PCSK9 was found to be right‐skewed and showed evidence of heteroskedasticity, even after log transformation. Use of log linear ordinary least squares in such circumstances has been shown to lead to biased estimates.24, 25 We therefore used generalized linear regression with a log link function and Poisson pseudo‐maximum likelihood estimator with a robust variance estimator24, 25, 26 to determine associations of HIV‐ and HCV‐infection status with levels of continuous PCSK9. Based on this analysis, percentage differences and the corresponding 95% CIs were estimated. Models were adjusted for demographic variables (age, sex, race), statin use, and smoking status as these variables are known to be associated with PCSK9 levels.27, 28, 29 LDL‐C was not adjusted for as it is directly related to PCSK9 levels via regulation of the LDL‐R. Sensitivity analyses were conducted to determine the effect of HIV and HCV status among individuals with treated and virologically suppressed HIV disease only. We also directly compared PCSK9 levels between uninfected controls and HIV‐infected subjects, uninfected controls and HIV/HCV‐coinfected subjects, and HIV‐monoinfected with HIV/HCV‐coinfected individuals.
Results
Baseline Characteristics
The median age of the overall cohort was 50 years (IQR 43–55 years), with 87% men, 35% subjects who were smokers, and 26% subjects with hypertension. The median LDL‐C level was 100 mg/dL (IQR 77–124), and 21% were taking statins. HIV‐monoinfected, HIV/HCV‐coinfected, and uninfected controls were generally similar in age, body mass index, blood pressure, and rates of diabetes, with some exceptions such as sex and race. Coinfected persons were more likely to be female, black, and smokers compared with HIV‐infected individuals and controls. HIV/HCV‐coinfected persons had lower rates of statin use relative to uninfected and HIV‐monoinfected persons (Table 1).
Table 1.
Baseline Characteristics of Uninfected Controls, HIV‐Monoinfected Patients, and HIV/HCV‐Coinfected Patients From the SCOPE Cohort
| Uninfected (n=72) | HIV Infected (n=385) | HIV/HCV Coinfected (n=110) | P Value | |
|---|---|---|---|---|
| Age, y | 52 (41–56) | 50 (43–56) | 50 (43–54) | 0.84 |
| Sex | <0.001 | |||
| Male | 72 (100) | 347 (90.1) | 85 (77.3) | |
| Female | 0 | 30 (7.8) | 21 (19.1) | |
| Transgender | 0 | 8 (2.1) | 4 (3.6) | |
| Race | 0.006 | |||
| White | 37 (51.4) | 235 (61) | 58 (52.7) | |
| African American | 23 (31.9) | 86 (22.3) | 43 (39.1) | |
| Latino | 6 (8.3) | 37 (9.6) | 8 (7.3) | |
| Other | 6 (8.3) | 27 (7) | 1 (0.9) | |
| BMI, kg/m2 | 25.8 (26.5–28.8) | 25.1 (22.4–29.6) | 25.4 (23.1–28.4) | 0.67 |
| Systolic blood pressure, mm Hg | 121 (113–134) | 120 (111–131) | 122 (111–132) | 0.65 |
| Diastolic blood pressure, mm Hg | 78 (71–86) | 76 (68–82) | 73 (68–81) | 0.16 |
| Diabetes mellitus | 3 (4.2) | 23 (6) | 7 (6.4) | 0.81 |
| Current smoker | 24 (33.3) | 112 (29.1) | 61 (55.5) | <0.001 |
| History of smoking | 40 (55.6) | 207 (53.8) | 86 (78.2) | <0.001 |
| CAD or CVA | 0 | 25 (6.5) | 8 (7.3) | 0.08 |
| Hypertension meds | 12 (16.7) | 98 (25.5) | 30 (27.3) | 0.22 |
| Cholesterol meds | 11 (15.3) | 124 (32.2) | 13 (11.8) | <0.001 |
| Statins | 9 (12.5) | 101 (26.2) | 8 (7.3) | <0.001 |
| Alanine transaminase, IU/L | 27 (21–36) | 32 (23–43) | 39 (26–61) | 0.001 |
| Aspartate transaminase, IU/L | 28 (24–35) | 31 (25–40) | 41 (29–55) | <0.001 |
All values shown are median (IQR) or N (%) number (percentage). BMI indicates body mass index; CAD indicates coronary artery disease; CVA, cerebrovascular accident; HCV, hepatitis C virus infection; HIV, HIV infection; Meds, medications.
HIV‐Related Features
HIV‐monoinfected and HIV/HCV‐coinfected individuals had similar HIV disease characteristics (Table 2). Overall, the median duration of HIV infection was 15 years (IQR 8–21) with a median CD4+ count of 527 cells/mm3 (IQR 346–732) and a median HIV RNA level of 75 copies/mL. More than two‐thirds of the individuals (67.5%) were currently taking ART, and 56% of these treated individuals had an undetectable HIV RNA level of <75 copies/mL.
Table 2.
HIV Characteristics in Monoinfected and HCV‐Coinfected Individuals
| HIV Infected (n=385) | HIV/HCV Coinfected (n=110) | P Value | |
|---|---|---|---|
| HIV duration, y | 15 (7–21) | 15 (9–20) | 0.87 |
| HIV viral load, copies/mL | 75 (40–860) | 75 (40–1450) | 0.07 |
| Current CD4 count, cells/mm3 | 533 (357–739) | 516 (303–701) | 0.32 |
| Nadir CD4 count, cells/mm3 | 239 (100–399) | 227 (93–365) | 0.67 |
| Off current ART | 111 (28.8) | 40 (45.5) | <0.001 |
| Opportunistic infection | 199 (51.7) | 63 (57.3) | <0.001 |
| Off ART, viral load <75 copies | 19 (4.9) | 13 (11.8) | 0.002 |
| Off ART, viral load >75 copies | 92 (23.9) | 37 (33.6) | |
| On ART, viral load >75 copies | 41 (10.7) | 14 (12.7) | |
| On ART, viral load <75 copies | 233 (60.5) | 46 (41.8) |
All values expressed as median (IQR) or N (%): number (percentage). ART indicates antiretroviral therapy; HCV, hepatitis C virus.
Lipid Profiles
As shown in Figure 1, there were notable differences in the lipid profiles of the 3 patient groups. Specifically, HIV/HCV‐coinfected patients had significantly lower levels of TC, LDL‐C, and HDL‐C compared with HIV‐monoinfected patients and uninfected controls. TG levels were highest in HIV‐infected individuals, with a median concentration of 310 mg/dL (IQR 213–496), and lowest in uninfected controls, with a median concentration of 239 mg/dL (IQR 186–328).
Figure 1.
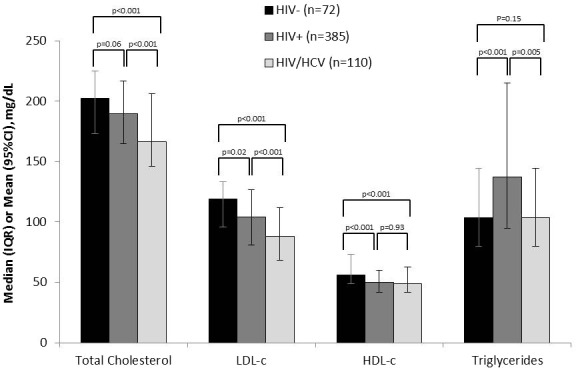
Lipid profiles in uninfected (black), HIV‐infected (dark gray), and HIV/HCV‐coinfected (light gray) individuals. HDL‐C indicates high‐density lipoprotein cholesterol; LDL, low‐density lipoprotein cholesterol; HCV, hepatitis C virus. Bars on each column reflect the 95% CIs. HIV/HCV‐coinfected individuals had lower levels of total cholesterol, LDL‐C, and HDL‐C compared with both HIV‐infected individuals and controls. Triglycerides were highest among individuals with HIV infection only and lowest in controls.
PCSK9 Levels
Compared with controls, HIV/HCV‐coinfected patients had 21% higher PCSK9 (95% CI 9–34%, P<0.001), while HIV‐monoinfected individuals had 9% higher PCSK9 (95% CI −1% to +19%, P=0.07) (Figure 2). Compared with HIV‐monoinfected subjects, HIV/HCV‐coinfected individuals had 11% higher PCSK9 (95% CI 3–20%, P=0.008). In a fully adjusted model that accounted for age, sex, race, smoking status, and statin use, HIV/HCV coinfection remained associated with 21% higher PCSK9 (95% CI 9–35%, P<0.001) compared with controls and 14% higher in coinfected patients compared with individuals with HIV monoinfection (95% CI 5–24%, P=0.001). Because of colinearity with HCV, we did not adjust for intravenous drug use.
Figure 2.
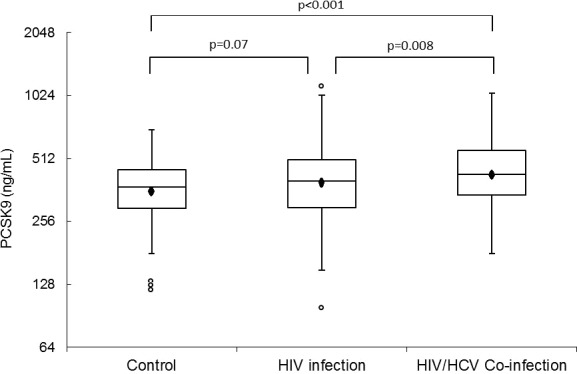
PCSK9 levels in uninfected controls, HIV‐infected, and HIV/HCV‐coinfected individuals. The boxes represent IQR, the outer bars represent the 1.5 IQR below 25th percentile and 1.5 IQR above 75th percentile. The circles beyond the outer bars represent extreme values. The marker inside the box indicates the mean value. The line inside the box indicates the median value. Compared with controls, HIV/HCV‐coinfected individuals had 21% higher PCSK9 levels while HIV‐infected individuals had 9% higher PCSK9 levels (P=0.07). When compared with HIV‐infected subjects, HIV/HCV‐coinfected individuals had 11% higher PCSK9 levels (P=0.008). PCSK9 indicates proprotein convertase subtilisin kexin 9; HCV, hepatitis C virus.
These findings persisted when the analysis was restricted to treated and suppressed HIV‐infected individuals only; namely, HIV‐monoinfected patients had 10% (95% CI 0.4–21%, P=0.04) and coinfected patients had 20% (95% CI 5.8–36.7%, P=0.005) higher PCSK9 levels compared with controls in a univariate model. In a fully adjusted model that adjusted for age, sex, race, smoking status, and statin use, HIV/HCV coinfection remained independently associated with 19% higher PCSK9 levels relative to controls (95% CI 5–36%, P=0.006). However, in the restricted analysis of treated and suppressed HIV patients, the association between higher PCSK9 with HIV monoinfection alone was no longer significant (percent different 6.7%, 95% CI −2.6% to 17.0%, P=0.16).
Association Between Inflammatory Markers (IL‐6, hsCRP) and LDL‐C and PCSK9
Median concentrations of the proinflammatory cytokine IL‐6 increased in a stepwise fashion, from 1.5 to 2.2 to 2.9 pg/mL, from uninfected controls to HIV‐monoinfected individuals to HIV/HCV‐coinfected individuals, respectively. This increase paralleled the stepwise increase in PCSK9 levels (Figure 3). Among all individuals, IL‐6 had a modest positive correlation with PCSK9 (r=0.11, P=0.018) (Figure S1A) and negative correlation with LDL‐C (r=−0.12, P=0.01) (Figure S1B); IL‐6 remained modestly correlated with PCSK9 levels when restricted to HIV‐monoinfected patients (r=0.14, P=0.009). In a model that was adjusted for demographics, statin use, smoking, and IL‐6, PCSK9 levels remained significantly higher among HIV/HCV‐coinfected individuals compared with controls (percent different 21%, 95% CI 8.4–35.6%, P=0.001) and compared with HIV‐infected individuals (percent different 14%, 95% CI 4.5–25.0%, P=0.003).
Figure 3.
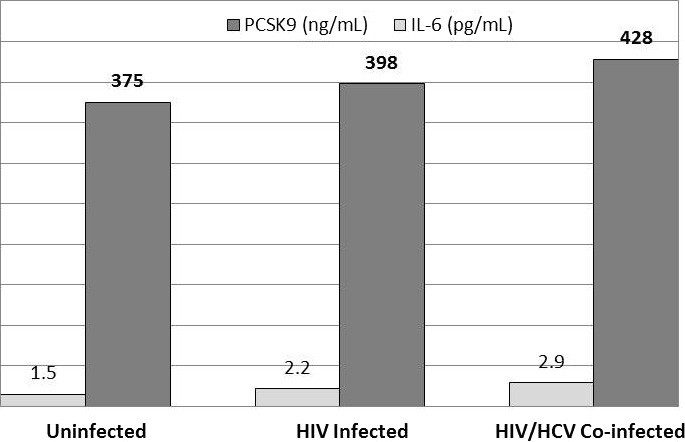
IL‐6 and PCSK9 are elevated in a stepwise and parallel fashion in uninfected controls, HIV infection, and HIV/HCV coinfection. IL‐6 levels are shown in light gray; PCSK9 levels are shown in dark grey. As shown, levels of IL‐6 and PCSK9 increase in stepwise from uninfected individuals, HIV‐infected, and HIV/HCV‐coinfected individuals. PCSK9 indicates proprotein convertase subtilisin kexin 9; HCV, hepatitis C virus; IL, interleukin.
In contrast to IL‐6, there was no significant difference in baseline levels of hs‐CRP among controls, HIV‐infected individuals, and HIV/HCV‐coinfected individuals (median 1.3 mg/L [IQR 0.65–3.1] versus 1.5 mg/L [IQR 0.8–3.5] versus 1.3 mg/L [IQR 0.6–3.7], respectively, P=0.17); correlations of hs‐CRP with PCSK9 were weak overall in the cohort (r=−0.03, P=0.42) and did not reach statistical significance in any of the 3 groups.
There was no correlation between LDL‐C and PCSK9 levels in any of the 3 groups or overall (data not shown).
Hepatic Injury Markers: Alanine Transaminase and Aspartate Transaminase
In all patients, the correlation coefficient r between alanine transaminase (ALT) and PCSK9 was 0.03 (P=0.49) and that between aspartate transaminase (AST) and PCSK9 was 0.09 (P=0.05). When restricted to only those patients with HIV/HCV coinfection, these values attenuated to r=0.02 (P=0.82) and r=0.03 (P=0.74) for ALT and AST, respectively. Both ALT and AST, when added to the fully adjusted models separately, reduced the association with PCSK9 from 21% to 16% for HIV/HCV‐coinfected patients compared with the controls and from 14% to 13% for HIV/HCV‐coinfected patients compared with the HIV‐monoinfected. Neither ALT nor AST reached statistical significance, while the association between HIV/HCV coinfection and PCSK9 levels remained highly significant in the multivariable models.
PCSK9 Levels and Mortality
A total of 55 (9.7%) individuals died since the beginning of the study: 33 HIV‐monoinfected, 21 coinfected, and 1 control subject followed for median of 3 years. Individuals who died had 10% higher PCSK9 levels compared with those who lived (95% CI −1% to 22%, P=0.066), which was reduced to 7% after adjustment for HIV/HCV status (95% CI −4% to 18%, P=0.23). Similar findings were present for PCSK9 levels and baseline CAD/CVA (findings not shown).
Discussion
In this study, we demonstrate that HIV/HCV coinfection is independently associated with high levels of PCSK9 compared with HIV monoinfection and compared with uninfected controls. We also describe a finding that we have termed the apparent “PCSK9–lipid paradox,” which is also observed when individuals are treated with statin therapy. That is, despite the elevation in circulating PCSK9 levels that our study demonstrates, lipids are lower in patients with HIV/HCV coinfection. The mechanism of these findings may be related at least in part to heightened inflammation, as evidenced by inflammatory markers such as IL‐6, which are nearly doubled in the setting of HIV/HCV coinfection compared with uninfected controls. This chronic inflammation may affect the ability of the liver to synthesize lipoproteins and explain the lower lipid levels we observed. The lipid paradox may also be secondary to lower intracellular cholesterol, which can drive a compensatory increase in PCSK9, similar to the physiologic response observed with statin therapy. To our knowledge, this is the first study to demonstrate a perturbation of PCSK9 and LDL‐C homeostasis in the setting of an inflammatory state (HIV/HCV coinfection) that is physiologically similar to what occurs with statin therapy. Our findings have important implications for the use of PCSK9 inhibitors in HIV/HCV‐coinfected patients as well as in those with other inflammatory diseases, which may also be subject to such alterations in PCSK9–LDL‐C regulation. Finally, individuals who died had higher PCSK9 levels than those who lived, suggesting that PCSK9 inhibition may be beneficial in the setting of HIV/HCV coinfection, a finding that will need to be demonstrated in prospective randomized controlled studies.
Because of the high rates of dyslipidemia, cardiovascular disease, and mortality associated with HIV and HCV coinfection, identifying therapies to lower risk in this unique patient population is critical. Targeting dyslipidemia is one obvious way to modulate this risk. The use of statins, which are typically the first‐line therapy in the general population to treat dyslipidemia, may be somewhat limited among individuals with HIV/HCV coinfection for many reasons. First, preexisting liver fibrosis, cirrhosis, or abnormalities in liver function tests may preclude the safe use of this therapy in some patients.5 Second, significant drug–drug interactions of particular statins with both HIV‐ and HCV‐specific protease inhibitors limit the type and dose of statin that can be used in some patients with no other apparent contraindications.30, 31, 32 Most protease inhibitors inhibit the cytochrome P450 system, resulting in elevated concentration–time curve for statins,33 which has resulted in death from rhabdomyolysis reported in rare cases.34 For this reason, the use of lovastatin or simvastatin in combination with protease inhibitor medication is contraindicated and the maximum dose of atorvastatin is reduced among individuals taking a protease inhibitor. Drug–drug interactions have also been reported for statins such as rosuvastatin, whose metabolism is independent of the cytochrome P450 system.35 HIV/HCV‐coinfected individuals are at increased risk for cardiovascular disease, and among coinfected individuals, hyperlipidemia is independently associated with cardiovascular disease events,36 suggesting that lipid lowering may reduce cardiovascular risk in this patient population. While statin therapy may have an impact on HCV replication, fibrosis, and hepatocellular carcinoma in the setting of older HCV regimens,37 high‐dose atorvastatin therapy, which is commonly used among individuals with acute coronary syndromes or coronary artery disease, has been associated with severe hepatic injury in the setting of chronic liver disease.38
Therefore, there is an unmet need to for identification of novel agents that target dyslipidemia in this patient population. Therapies involving PCSK9 inhibition are promising, but the physiology of PCSK9 needs to be better understood in HIV/HCV‐coinfected individuals before their use in clinical trials, because PCSK9–LDL‐C homeostasis may be altered in the setting of inflammation.
PCSK9–Lipid Paradox in HIV/HCV Coinfection
No correlation between LDL‐C and PCSK9 has been previously described in the context of HCV infection, except in a small study of individuals with HCV genotypes 1 and 3.39 In our study, we are able to validate the findings of this small study and further demonstrate that HIV/HCV‐associated inflammation (as mediated by IL‐6) is most likely responsible for this apparent paradox. We also show that hepatic injury and necrosis, with “release” of PCSK9 from hepatocytes into the bloodstream (akin to the release of transaminases during liver injury), are unlikely to explain our findings.
IL‐6 as a Mediator of the Apparent PCSK9–Lipid Paradox
In the setting of a chronic inflammatory disease of the liver, such as HCV, systemic inflammatory cytokines, such as IL‐6, are released from multiple inflammatory cell types, including monocytes. Although the exact mechanism of regulation has not been characterized, PCSK9 and IL‐6 are known to be closely interrelated. In healthy individuals with a PCSK9 loss of function allele, less IL‐6 is produced in response to lipopolysaccharide‐induced inflammation.40 In our study, we report that PCSK9 and IL‐6 are modestly correlated and increase in a parallel, stepwise fashion with increasing inflammation. In aggregate, these findings suggest that IL‐6 may be associated with increased PCSK9 expression and/or secretion.
Lower Lipids in HIV/HCV Coinfection Despite a Higher PCSK9
More of the HIV‐monoinfected individuals were taking statin therapy compared with controls, which is the likely reason why their LDL‐C levels were lower. The unexpected finding of lower LDL‐C in HIV/HCV‐coinfected patients has several possible underlying mechanisms. One possible explanation is that the increased liver fibrosis that exists in HIV/HCV coinfection5 results in reduced function of the liver and decreased production of lipoproteins. Prior studies from our group have already demonstrated that levels of atherogenic lipids, such as LDL‐C, are impaired in the setting of HIV/HCV coinfection.10 In this study, we also demonstrate that TC, LDL‐C, and HDL‐C levels were all lower in HIV/HCV‐coinfected subjects compared with controls, consistent with this hypothesis and confirming our previous findings.
The lower lipid levels that we observed may also be because of suppression of lipoprotein production from the liver in the setting of chronic inflammation. In other clinical settings, serum lipids are an inverse acute phase reactant and thus are decreased during states of inflammation.41, 42 Higher IL‐6 levels were associated with decreased serum lipids in the setting of HIV infection.12 HIV and HCV infections are both chronic inflammatory diseases that may result in decreased lipid production. The elevation in PCSK9 may reflect “uncoupling” of this regulatory mechanism or, more likely, a compensatory increase in PSCK9 in response to less intracellular cholesterol. Given the modest negative association of IL‐6 with LDL‐C and our findings correlating IL‐6 and PCSK9 levels, IL‐6 may play a key role in the lipid paradox observed in our study. Finally, as oxidized LDL levels are elevated in the setting of HIV43 another mechanism underling the low LDL levels we observed may be accelerated removal of oxidatively modified LDL.
Roles of IL‐6 and hsCRP
Both IL‐6 and hsCRP are inflammatory markers independently associated with cardiovascular risk in HIV infection.44 IL‐6 is higher in HIV/HCV coinfection compared with HIV monoinfection and is an inflammatory marker independently associated with cardiovascular risk in both HIV45 and HCV infection.46 IL‐6 has also been thought to play a role in HCV‐ and HIV‐associated liver fibrosis and inflammation.47 Even among treated and suppressed HIV‐infected individuals, elevated IL‐6 is independently predictive of adverse cardiovascular outcomes45 as well as overall mortality.48
Our findings suggest that heightened inflammation, as mediated by IL‐6, may indeed represent the biochemical link between HIV/HCV coinfection and elevated PCSK9 levels as well as decreased LDL‐C levels. This relationship may be bidirectional (PCSK9 regulating levels of IL‐6) because healthy individuals with PCSK9 loss of function mutations have less IL‐6 in response to lipopolysaccharide‐induced inflammation.40 While randomized clinical trials of PCSK9 inhibition in this unique patient population will be required for ultimate proof‐of‐concept, it is biologically plausible that PCSK9 and IL‐6 are coregulated and inhibition of PCSK9 in HIV infection may lead to decreased levels of IL‐6, which may have an impact on cardiovascular risk and mortality in HIV infection. Conversely, inhibition of IL‐6 might reduce PCSK9 levels and thus potentially affect lipoprotein metabolism.
In contrast, we were unable to demonstrate a significant association between hsCRP and PCSK9 levels in HIV‐infected individuals or among HIV/HCV‐coinfected individuals. Previous studies have reported that hsCRP does not correlate well with degree of HIV control, as assessed based on viral load or CD4 count.49 Further, in HIV/HCV‐coinfected patients, hsCRP levels tend to be lower because of impaired synthetic function of the liver11 and, therefore, are not good markers of systemic inflammation in this particular clinical setting. Our results suggest that PCSK9 and hsCRP are not likely to be linked. This is not a surprising finding as previous clinical trials of PCSK9 inhibition among individuals without HIV infection have not shown a significant impact on hsCRP,50 suggesting that the regulation of these 2 proteins is likely mechanistically unrelated.
Limitations
Our analysis is subject to the usual limitations of any observational analysis: although we have rigorously adjusted our model for multiple factors, including demographic factors, statin use, and smoking, there is always the possibility that residual confounding remains. In addition, although PCSK9 has a dose‐dependent relationship with statin use, we were not able to adjust for the dose and potency of statin used. We did not have HCV viral load or genotype available for the majority of our coinfected patients and were not able to adjust for degree of HCV viral activity. Finally, because this was an observational study, we are only able to report associations, and not causation, from our findings.
Clinical Perspective
PCSK9 inhibitors have been used to lower LDL‐C in a number of patient populations, including statin‐intolerant patients and those with familial hypercholesterolemia, and have a safety profile that is extremely favorable along with no known drug–drug interactions. However, their role in unique populations, such as those with chronic inflammatory diseases, remains uncertain. HIV/HCV coinfection provides a unique clinical scenario that may mimic what occurs during statin therapy, where PCSK9 is increased while LDL‐C is decreased. This may, in part, be mediated by IL‐6, which may have important implications for anti‐PCSK9 therapies in other inflammatory diseases in which IL‐6 is upregulated (ie, rheumatoid arthritis).
Translational implications
Additional clinical studies are warranted to confirm our findings and to test if PCSK9 inhibition will be safely tolerated, lower lipid levels, and potentially reduce IL‐6 levels among individuals with HIV and HCV coinfection.
Author Contributions
Drs Kohli, Ganz, and Hsue had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Drs Kohli, Ganz, and Hsue were responsible for study concept and design. All authors were responsible for acquisition, analysis, or interpretation of data. Dr Kohli was responsible for drafting of the manuscript. Drs Kohli, Ganz, Scherzer, Wasserman, Scott, and Hsue and Mr Ma were responsible for critical revision of the manuscript for important intellectual content. Mr Ma and Dr Scherzer were responsible for statistical analysis. Drs Kohli, Wasserman, and Scott obtained funding. Mr Weigel and Ms Hur provided administrative, technical, and material support. Drs Ganz, Hsue, Wasserman, and Scott were responsible for study supervision.
Sources of Funding
The study was sponsored by the San Francisco General Hospital and the National Institutes of Health (K24AI112393 to Dr Hsue) and funded by Amgen, Inc.
Disclosures
Drs Kohli, Ganz, and Hsue have served on an advisory board for Amgen. Dr. Kohli has received consulting fees from Pfizer and honoraria for continuing medical education from Consultant Live. No other disclosures were reported. Drs Wasserman and Scott were employees of Amgen when this research was conducted.
Supporting information
Figure S1. Correlation plot showing correlation of PCSK9 with interleukin‐6 (IL‐6) (A) and LDL‐C (B) in all patients from the study cohort.
(J Am Heart Assoc. 2016;5:e002683 doi: 10.1161/JAHA.115.002683)
Results were presented in an abstract and the authors were awarded second place in the American College of Cardiology Young Investigators Award Competition at the American College of Cardiology Scientific Sessions, March 14–16, 2015, in San Diego, CA.
References
- 1. Tseng ZH, Secemsky EA, Dowdy D, Vittinghoff E, Moyers B, Wong JK, Havlir DV, Hsue PY. Sudden cardiac death in patients with human immunodeficiency virus infection. J Am Coll Cardiol. 2012;59:1891–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, Butt AA, Bidwell GM, Leaf D, Oursler KA, Rimland D, Rodriguez BM, Brown S, Gibert C, Crothers K, Sico J, Crane H, Warner A, Gottlieb S, Gottdiener J, Tracy RP, Budoff M, Watson C, Armah KA, Doebler D, Bryant K, Justice AC. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med. 2013;173:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Butt AA, Xiaoqiang W, Budoff M, Leaf D, Kuller LH, Justice AC. Hepatitis C virus infection and the risk of coronary disease. Clin Infect Dis. 2009;49:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roed T, Lebech AM, Kjaer A, Weis N. Hepatitis C virus infection and risk of coronary artery disease: a systematic review of the literature. Clin Physiol Funct Imaging. 2012;32:421–430. [DOI] [PubMed] [Google Scholar]
- 5. Thomas DL. The challenge of hepatitis C in the HIV‐infected person. Annu Rev Med. 2008;59:473–485. [DOI] [PubMed] [Google Scholar]
- 6. Bedimo R, Westfall AO, Mugavero M, Drechsler H, Khanna N, Saag M. Hepatitis C virus coinfection and the risk of cardiovascular disease among HIV‐infected patients. HIV Med. 2010;11:462–468. [DOI] [PubMed] [Google Scholar]
- 7. Freiberg MS, Chang CC, Skanderson M, McGinnis K, Kuller LH, Kraemer KL, Rimland D, Goetz MB, Butt AA, Rodriguez BM, Gibert C, Lead D, Brown ST, Samet J, Kazis L, Bryant K, Justice AC. The risk of incident coronary heart disease among veterans with and without HIV and hepatitis C. Circ Cardiovasc Qual Outcomes. 2011;4:425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feingold KR, Krauss RM, Pang M, Doerrler W, Jensen P, Grunfeld C. The hypertriglyceridemia of acquired immunodeficiency syndrome is associated with an increased prevalence of low density lipoprotein subclass pattern B. J Clin Endocrinol Metab. 1993;76:1423–1427. [DOI] [PubMed] [Google Scholar]
- 9. Sosner P, Wangermez M, Chagneau‐Derrode C, Le Moal G, Silvain C. Atherosclerosis risk in HIV‐infected patients: the influence of hepatitis C virus co‐infection. Atherosclerosis. 2012;222:274–277. [DOI] [PubMed] [Google Scholar]
- 10. Wheeler AL, Scherzer R, Lee D, Delaney JA, Bacchetti P, Shlipak MG, Sidney S, Grunfeld C, Tien PC. HIV/hepatitis C virus coinfection ameliorates the atherogenic lipoprotein abnormalities of HIV infection. AIDS. 2014;28:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reingold J, Wanke C, Kotler D, Lewis C, Tracy R, Heymsfield S, Tien P, Bacchetti P, Scherzer R, Grunfeld C, Shlipak M. Association of HIV infection and HIV/HCV coinfection with C‐reactive protein levels: the fat redistribution and metabolic change in HIV infection (FRAM) study. J Acquir Immune Defic Syndr. 2008;48:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Borges AH, O'Connor JL, Phillips AN, Ronsholt FF, Pett S, Vjecha MJ, French MA, Lundgren JD. Factors associated with plasma IL‐6 levels during HIV infection. J Infect Dis. 2015;212:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giugliano RP, Desai NR, Kohli P, Rogers WJ, Somaratne R, Huang F, Liu T, Mohanavelu S, Hoffman EB, McDonald ST, Abrahamsen TE, Wasserman SM, Scott R, Sabatine MS. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE‐TIMI 57): a randomised, placebo‐controlled, dose‐ranging, phase 2 study. Lancet. 2012;380:2007–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koren MJ, Scott R, Kim JB, Knusel B, Liu T, Lei L, Bolognese M, Wasserman SM. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double‐blind, placebo‐controlled, phase 2 study. Lancet. 2012;380:1995–2006. [DOI] [PubMed] [Google Scholar]
- 15. McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–2353. [DOI] [PubMed] [Google Scholar]
- 16. Raal F, Scott R, Somaratne R, Bridges I, Li G, Wasserman SM, Stein EA. Low‐density lipoprotein cholesterol‐lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL‐C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–2417. [DOI] [PubMed] [Google Scholar]
- 17. Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–1900. [DOI] [PubMed] [Google Scholar]
- 18. Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, Wu R, Pordy R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low‐density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. [DOI] [PubMed] [Google Scholar]
- 19. Sullivan D, Olsson AG, Scott R, Kim JB, Xue A, Gebski V, Wasserman SM, Stein EA. Effect of a monoclonal antibody to PCSK9 on low‐density lipoprotein cholesterol levels in statin‐intolerant patients: the GAUSS randomized trial. JAMA. 2012;308:2497–2506. [DOI] [PubMed] [Google Scholar]
- 20. Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–1509. [DOI] [PubMed] [Google Scholar]
- 21. Robinson JG, Farnier M, Krempf M, Bergerson J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJ. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–1499. [DOI] [PubMed] [Google Scholar]
- 22. Feingold KR, Moser AH, Shigenaga JK, Patzek SM, Grunfeld C. Inflammation stimulates the expression of PCSK9. Biochem Biophys Res Commun. 2008;374:341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsue PY, Scherzer R, Hunt PW, Schnell A, Bolger AF, Kalapus SC, Maka K, Martin JN, Ganz P, Deeks SG. Carotid intima‐media thickness progression in HIV‐infected adults occurs preferentially at the carotid bifurcation and is predicted by inflammation. J Am Heart Assoc. 2012;1:e000422 doi: 10.1161/JAHA.111.000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Manning WG, Mullahy J. Estimating log models: to transform or not to transform? J Health Econ. 2001;20:461–494. [DOI] [PubMed] [Google Scholar]
- 25. Silva JS, Tenreyro S. The log of gravity. Rev Econ Stat. 2006;88:641–658. [Google Scholar]
- 26. Silva JS, Tenreyro S. On the existence of the maximum likelihood estimates in Poisson regression. Econ Lett. 2010;107:310–312. [Google Scholar]
- 27. Yin RX, Wu DF, Wu JZ, Cao XL, Aung LH, Miao L, Long XJ, Liu WY, Zhang L, Li M. Interactions of several lipid‐related gene polymorphisms and cigarette smoking on blood pressure levels. Int J Biol Sci. 2012;8:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mayne J, Dewpura T, Raymond A, Cousins M, Chaplin A, Lahey KA, LaHaye SA, Mbikay M, Ooi TC, Chretien M. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 2008;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. [DOI] [PubMed] [Google Scholar]
- 30. Hare CB, Vu MP, Grunfeld C, Lampiris HW. Simvastatin‐nelfinavir interaction implicated in rhabdomyolysis and death. Clin Infect Dis. 2002;35:e111–e112. [DOI] [PubMed] [Google Scholar]
- 31. Lees RS, Lees AM. Rhabdomyolysis from the coadministration of lovastatin and the antifungal agent itraconazole. N Engl J Med. 1995;333:664–665. [DOI] [PubMed] [Google Scholar]
- 32. Chauvin B, Drouot S, Barrail‐Tran A, Taburet A. Drug‐drug interactions between HMG‐CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin Pharmacokinet. 2013;52:815–831. [DOI] [PubMed] [Google Scholar]
- 33. Fichtenbaum CJ, Gerber JG, Rosenkranz SL, Segal Y, Aberg JA, Blaschke T, Alston B, Fang F, Kosel B, Aweeka F. Pharmacokinetic interactions between protease inhibitors and statins in HIV seronegative volunteers: ACTG Study A5047. AIDS. 2002;16:569–577. [DOI] [PubMed] [Google Scholar]
- 34. Bastida C, Also MA, Pericas JM, Letang E, Tuset M, Miro JM. [Rhabdomyolysis and severe hepatotoxicity due to a drug‐drug interaction between ritonavir and simvastatin. Could we use the most cost‐effective statin in all human immunodeficiency virus‐infected patients?] Enferm Infecc Microbiol Clin. 2014;32:579–582. [DOI] [PubMed] [Google Scholar]
- 35. Bellosta S, Paoletti R, Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109:III50–III57. [DOI] [PubMed] [Google Scholar]
- 36. Freiberg MS, Cheng DM, Kraemer KL, Saitz R, Kuller LH, Samet JH. The association between hepatitis C infection and prevalent cardiovascular disease among HIV‐infected individuals. AIDS. 2007;21:193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simon TG, Butt AA. Lipid dysregulation in hepatitis C virus, and impact of statin therapy upon clinical outcomes. World J Gastroenterol. 2015;21:8293–8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang CH, Chang YC, Lee YC, Liu YC, Chuang LM, Lin JW. Severe hepatic injury associated with different statins in patients with chronic liver disease: a nationwide population‐based cohort study. J Gastroenterol Hepatol. 2015;30:155–162. [DOI] [PubMed] [Google Scholar]
- 39. Bridge SH, Sheridan DA, Felmlee DJ, Crossey MM, Fenwick FI, Lanyon CV, Dubuc G, Seidah NG, Davignon J, Thomas HC, Taylor‐Robinson SD, Toms GL, Neely RD, Bassendine MF. PCSK9, apolipoprotein E and lipoviral particles in chronic hepatitis C genotype 3: evidence for genotype‐specific regulation of lipoprotein metabolism. J Hepatol. 2015;62:763–770. [DOI] [PubMed] [Google Scholar]
- 40. Walley KR, Thain KR, Russell JA, Reilly MP, Meyer NJ, Ferguson JF, Christie JD, Nakada TA, Fjell CD, Thair SA, Cirstea MS, Boyd JH. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci Transl Med. 2014; 6: 258ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bismuth J, Kofoed SC, Jensen AS, Sethi A, Sillesen H. Serum lipids act as inverse acute phase reactants and are falsely low in patients with critical limb ischemia. J Vasc Surg. 2002;36:1005–1010. [DOI] [PubMed] [Google Scholar]
- 42. Miller M. Lipid levels in the post‐acute coronary syndrome setting: destabilizing another myth? J Am Coll Cardiol. 2008;51:1446–1447. [DOI] [PubMed] [Google Scholar]
- 43. Zidar DA, Juchnowski S, Ferrari B, Clagett B, Pilch‐Cooper HA, Rose S, Rodriguez B, McComsey GA, Sieg SF, Mehta NN, Lederman MM, Funderburg NT. Oxidized LDL levels are increased in HIV infection and may drive monocyte activation. J Acquir Immune Defic Syndr. 2015;69:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Armah KA, Quinn EK, Cheng DM, Tracy RP, Baker JV, Samet JH, Freiberg MS. Human immunodeficiency virus, hepatitis C, and inflammatory biomarkers in individuals with alcohol problems: a cross‐sectional study. BMC Infect Dis. 2013;13:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duprez DA, Neuhaus J, Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, Ledergerber B, Lundgren J, Nixon D, Paton NI, Prineas RJ, Neaton JD. Inflammation, coagulation and cardiovascular disease in HIV‐infected individuals. PLoS One. 2012;7:e44454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oliveira CP, Kappel CR, Siqueira ER, Lima VM, Stefano JT, Michalczuk MT, Marini SS, Barbeiro HV, Soriano FG, Carrilho FJ, Pereira LM, Alvares‐da‐Silva MR. Effects of hepatitis C virus on cardiovascular risk in infected patients: a comparative study. Int J Cardiol. 2013;164:221–226. [DOI] [PubMed] [Google Scholar]
- 47. Fuster D, Tsui JI, Cheng DM, Quinn EK, Armah KA, Nunes D, Freiberg MS, Samet JH. Interleukin‐6 is associated with noninvasive markers of liver fibrosis in HIV‐infected patients with alcohol problems. AIDS Res Hum Retroviruses. 2013;29:1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, Ledergerber B, Lundgren J, Neuhaus J, Nixon D, Paton NI, Neaton JD. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guimaraes MM, Greco DB, Figueiredo SM, Foscolo RB, Oliveira AR Jr, Machado LJ. High‐sensitivity C‐reactive protein levels in HIV‐infected patients treated or not with antiretroviral drugs and their correlation with factors related to cardiovascular risk and HIV infection. Atherosclerosis. 2008;201:434–439. [DOI] [PubMed] [Google Scholar]
- 50. Stroes E, Colquhoun D, Sullivan D, Civeira F, Rosenson RS, Watts GF, Bruckert E, Cho L, Dent R, Knussel B, Xue A, Scott R, Wasserman SM, Rocco M. Anti‐PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS‐2 randomized, placebo‐controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–2548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Correlation plot showing correlation of PCSK9 with interleukin‐6 (IL‐6) (A) and LDL‐C (B) in all patients from the study cohort.