

Abstract
Background
The NLR family, pyrin domain containing 3 (NLRP3) inflammasome is an interleukin (IL)‐1β and IL‐18 cytokine processing complex that is activated in inflammatory conditions. The role of the NLRP3 inflammasome in the pathogenesis of atherosclerosis and myocardial infarction is not fully understood.
Methods and Results
Atherosclerotic plaques were analyzed for transcripts of the NLRP3 inflammasome, and for IL‐1β release. The Swedish First‐ever myocardial Infarction study in Ac‐county (FIA) cohort consisting of DNA from 555 myocardial infarction patients and 1016 healthy individuals was used to determine the frequency of 4 single nucleotide polymorphisms (SNPs) from the downstream regulatory region of NLRP3. Expression of NLRP3, Apoptosis‐associated speck‐like protein containing a CARD (ASC), caspase‐1 (CASP1), IL1B, and IL18 mRNA was significantly increased in atherosclerotic plaques compared to normal arteries. The expression of NLRP3 mRNA was significantly higher in plaques of symptomatic patients when compared to asymptomatic ones. CD68‐positive macrophages were observed in the same areas of atherosclerotic lesions as NLRP3 and ASC expression. Occasionally, expression of NLRP3 and ASC was also present in smooth muscle cells. Cholesterol crystals and ATP induced IL‐1β release from lipopolysaccharide‐primed human atherosclerotic lesion plaques. The minor alleles of the variants rs4266924, rs6672995, and rs10733113 were associated with NLRP3 mRNA levels in peripheral blood mononuclear cells but not with the risk of myocardial infarction.
Conclusions
Our results indicate a possible role of the NLRP3 inflammasome and its genetic variants in the pathogenesis of atherosclerosis.
Keywords: inflammasome, interleukin‐1β, myocardial infarction, NLRP3, polymorphism
Subject Categories: Atherosclerosis; Vascular Disease; Genetic, Association Studies
Introduction
Atherosclerosis is a multifactorial, chronic inflammatory disease that causes myocardial infarction (MI) and acute ischemic stroke.1 Enhanced activation of inflammatory pathways and their bidirectional interaction with lipid pathology and accumulation are hallmarks of atherosclerosis. Several inflammatory cytokines are activated during this process, including interleukin (IL)‐1β and IL‐18 that are overexpressed in atherosclerotic lesions.2, 3, 4, 5 The importance of IL‐1β and IL‐18 in atherosclerosis is further supported by studies in mice showing that IL‐1β and IL‐18 deficiency has been shown to attenuate plaque development.6, 7
Pro‐IL‐1β and pro‐IL‐18 are processed by caspase‐1 to their active form upon activation of inflammasomes, of which the NLR family, pyrin domain containing (NLRP) 3 inflammasome is of particular importance.8 Caspase‐1 is a key constituent of this inflammasome together with NLRP3 and an adaptor protein termed Apoptosis‐associated speck‐like protein containing a CARD (ASC).9 The release of IL‐1β and IL‐18 requires 2 signals; signal 1 (eg, Toll‐like receptor [TLR]4) agonists and certain inflammatory cytokines such as tumor necrosis factor that primes the inflammasome by enhancing mRNA levels of NLRP3, IL‐1β, and IL‐18; and signal 2 that results in the assembly of the NLRP3 inflammasome and caspase‐1 activation.10 Several chemically different molecules have been shown to activate NLRP3 (ie, signal 2), including extracellular ATP, monosodium urate crystals, and recently, cholesterol crystals.11, 12, 13
Recent studies have suggested a role for NLRP3 inflammasome in atherosclerosis, although the results are somewhat conflicting. Duewell and colleagues showed that NLRP3‐deficient bone marrow cells transplanted into atherosclerosis‐prone low‐density lipoprotein receptor–deficient mice had reduced atherosclerosis, and that cholesterol crystals promoted IL‐1β release from macrophages via activation of the NLRP3 inflammasome.14 Furthermore, NLRP3 inflammasome activation in cardiac fibroblasts was found to enhance the severity of myocardial ischemia injury,15 and isolated hearts from NLRP3‐deficient mice show attenuated ischemia–reperfusion injury.16 There is also substantial evidence that activation of the NLRP3 inflammasome enhances lipid deposition and migration of macrophages, accelerating foam cell formation.17 However, others suggest that progression of atherosclerosis is independent of inflammasome activation. Menu and coworkers created double knock‐out mice for apolipoprotein E (Apoe) and the NLRP3 inflammasome components NLRP3, Asc, and caspase‐1, but could not identify any difference between Apoe mice and the different double knock‐out genotypes regarding progression, cell infiltration, phenotype, or stability of the atherosclerotic plaque.18 Of note, the literature is virtually devoid of data on NLRP3 expression in human atherosclerosis.
Mutations in the NLRP3 gene, resulting in hyperproduction of IL‐1β, have been associated with rare, hereditary, multisystem inflammatory diseases, such as cryopyrin‐associated periodic syndromes.19 Polymorphisms in the NLRP3 gene were also found to be associated with several common, polygenic, inflammatory disorders including celiac disease, abdominal aortic aneurysms, rheumatoid arthritis, and Crohn's disease.20, 21, 22, 23 Villani and coworkers identified several additional polymorphisms downstream of the NLRP3 gene that were associated with risk of Crohn's disease.24 Except for an association with abdominal aortic aneurysms,21 there is little data on associations between NLRP3 downstream regulatory region polymorphisms and atherosclerotic disorders.
To elucidate the role of the NLRP3 inflammasome in human atherosclerotic disease, we (1) examined the expression and activation of the NLRP3 inflammasome in human atherosclerotic carotid plaques, and (2) assessed 4 SNPs, rs4353135, rs4266924, rs6672995, and rs10733113, located within the downstream regulatory region of the NLRP3 gene for association with the occurrence of MI and expression of NLRP3 and inflammatory markers.
Methods
Study Subjects
(1) Gene expression patterns of NLRP3‐related genes were analyzed in the Biobank of Karolinska Endarterectomies (BiKE) study at Centre for Molecular Medicine (CMM), Karolinska Institute, Stockholm, Sweden.25 Carotid artery plaque tissues, obtained during endarterectomy surgery from 106 patients with ischemic cerebrovascular disease, were used to isolate mRNA. Nonatherosclerotic vessels were obtained from iliac arteries (n=9) and from intima of aorta (n=1) from transplant donors. (2) Peripheral blood mononuclear cells (PBMCs) were collected (n=98) as previously described.26 (3) Cytokine levels were measured in the sera available from the Stockholm Coronary Atherosclerosis Risk Factor (SCARF) cohort, which includes 387 MI patients and 387 controls. (4) The First‐ever myocardial Infarction study in AC‐county (FIA) cohort from northern Sweden, including 1016 controls and 555 MI patients, was genotyped. Information regarding the MI definition, sampling, and baseline characteristics of BiKE, SCARF, and FIA cohorts has been previously reported.26, 27, 28 Ethical approval was granted for each cohort from ethical review boards, the study was ethically performed according to the guidelines of the Helsinki Declaration, and consent was given by the patients.
Immunohistochemistry
Sections of paraffin‐embedded carotid plaque (n=3), collected during endarterectomy surgery, were obtained from the BiKE. Reagents for immunohistochemistry were from Biocare Medical (Concord, CA). The 5‐μm paraffin sections were treated with Tissue Clear for deparaffinization and rehydrated in graded ethanol (99.9–70%). The slides were subjected to antigen retrieval by boiling in DIVA buffer and TE buffer. After blocking with background Sniper, the primary antibodies against NLRP3 (HPA012878; Sigma‐Aldrich, St. Louis, MO; 1:50), ASC (ADI905173100; Enzo Life Sciences, Farmingdale, NY; 1:200), CD68 (NCL‐L‐CD68, Novacastra, Newcastle, UK; 1:50), and smooth muscle actin (SMA; M0851; Dako, Glostrup, Denmark; 1:500) were diluted in Da Vinci Green solution and the slides were incubated with the primary antibody for 1 hour at room temperature. Rabbit and mouse IgG were used as negative control. A double‐stain polymer detection kit with alkaline phosphatase and horseradish peroxidase was used to detect NLRP3 and ASC using Warp Red and Vina green to detect CD68 and SMA. The sections were counterstained with hematoxylin QS (Vector Laboratories, Burlingame, CA), dehydrated, and mounted in Pertex (Histolab, Göteborg, Sweden). Images were taken using a Nikon OPTIPHOT‐2 microscope and NIS‐Element software.
Culturing of Atherosclerotic Carotid Plaques
Biopsies from atherosclerotic carotid plaques (n=9), obtained from patients within 1 month of symptoms (Department of Neurology, Oslo University Hospital Rikshospitalet, Oslo, Norway) were rinsed in PBS and placed in DMEM/F12 (Gibco, Grand Island, NY) enriched with 30 mg/mL endotoxin‐free and fatty acid–free BSA (Sigma, St. Louis, MO) within 4 hours of surgery, and pre‐incubated with or without lipopolysaccharide (LPS) from Escherichia coli 026:B6 (Sigma, 100 ng/mL) as described.5, 29 Before collection, the atherosclerotic area was carefully evaluated. The large section of the samples was segmented into equal pieces macroscopically and was used in the experiments to further minimize the problem with sampling error. After 18 hours, 3 mmol/L ATP (Sigma), 500 μg/mL cholesterol crystals,30 or vehicle was added, and the tissue slices were further incubated for 6 hours before tissue and conditioned media were chilled on ice and harvested before being stored at −80°C. Protein levels of IL‐1β in ex vivo stimulated carotid plaque supernatants were measured by enzyme immunoassays from R&D Systems Inc (Minneapolis, MN).
Expression of NLRP3‐Related Components in Human Atherosclerotic Lesions and Normal Arteries; Role of NLRP3 Variants
Affymetrix HG‐U133 plus 2.0 Gene chip® arrays were used to assess the expression of NLRP3, ASC, CASP1, IL1B, and IL18 mRNA in the samples from the BiKE study. In brief, RNA purified using a Qiagen RNeasy kit was hybridized to arrays at Karolinska Institute Affymetrix core facility and the RMA procedure was used for normalization of the raw data.26 For expression of quantitative trait loci (eQTL) analysis, the SNPs were genotyped and association analysis was carried out as previously described.25
Genotyping
Genotyping was carried out using a Taqman SNP genotype assay and the 7900 HT Fast Realtime polymerase chain reaction system (Applied Biosystems, Foster City, CA). The polymerase chain reaction contained 10 ng of genomic DNA in 10 μL of master mix containing 2X Taqman genotyping Master mix (Applied Biosystems) and 40X Taqman SNP Genotyping Assay with predesigned primer and probes C__26052013, C__28967719, C__28967716, C__30713847 for the SNPs rs4353135, rs4266924, rs6672995, and rs10733113, respectively. Genotyping accuracy was verified by repeating the polymerase chain reaction in 10% randomly selected samples.
Inflammatory Markers in Plasma
Inflammatory cytokines were measured in plasma from SCARF. IL‐18 levels were measured using an ELISA kit from R&D Systems Inc. (Minneapolis, MN) as previously described.31 Concentrations of IL‐1β, tumor necrosis factor‐α, and monocyte chemoattractant peptide (MCP‐1/CCL2) were analyzed using an Evidence® automated biochip array system (Randox Laboratories Ltd., Co Antrim, UK).32 The levels of high‐sensitivity C‐reactive protein were measured in SCARF and FIA as reported by Samnegård et al28 and Wennberg et al,33 respectively.
Statistical Analysis
A 2‐tailed Student t test was used for analyzing mRNA levels of NLRP3, ASC, CASP1, IL1B, and IL18 between carotid lesions and transplant donor vessels, respectively. The carotid lesions consisted of the 106 patients described from the BiKE cohort. The transplant donor vessels consisted of nonatherosclerotic vessels as similarly described. Both tested groups were normally distributed according to a Shapiro–Wilks test (P>0.1). The Mann–Whitney U‐test was used for analyzing the ex vivo experiments of carotid plaques. STATISTICA 7.1 software (StatSoft) was used for the statistical analysis of cytokine profiling. Assessment of association between NLRP3 mRNA expression and genotypes in different tissues was performed using an additive linear regression model as previously mentioned.25 Investigation of possible associations between cytokines and genotyped SNPs was performed using linear regression with an additive genetic model and adjusted for age, sex, and presence of MI in PLINK.34 Association between SNPs and MI was analyzed with the χ2 test using the SPSS software package (SPSS, Inc, Chicago, IL) and EpiInfo software 2008 (Centre for Disease Control and Prevention, Atlanta, GA). Since the analysis was solely directed at the NLRP3 gene‐testing hypotheses, the significance level was here considered to be P<0.05.
Results
Increased NLRP3, ASC, caspase‐1, IL‐1β, and IL‐18 mRNA Expression in Human Carotid Artery Plaques
The mRNA levels of NLRP3 inflammasome components were compared between atherosclerotic (n=106) and nonatherosclerotic (n=10) arteries. The expression of NLRP3, ASC, CASP1, IL1B, and IL18 mRNA was found to be markedly increased in atherosclerotic as compared to nonatherosclerotic vessels (P<0.0001 for all comparisons; Figure 1A through 1E), indicating upregulation in the atherosclerotic lesion of transcripts for NLRP3 inflammasome components. Within carotid plaques, mRNA levels of NLRP3, but not of ASC, CASP1, IL1B, and IL18 (Figure S1), were significantly increased in patients with symptomatic (n=85) compared to patients with asymptomatic lesions (n=40, P<0.05) (Figure 1F). Levels of NLRP3 mRNA also showed a correlation with mRNA levels of the macrophage marker CD68 (r=0.66, P<0.001, data not shown).
Figure 1.
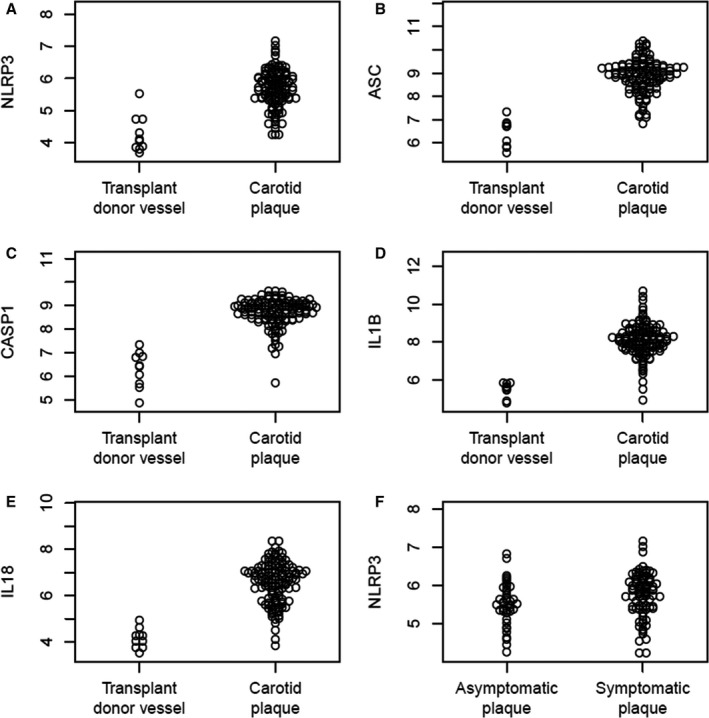
Expression of (A) NLR family, pyrin domain containing 3 (NLRP3) (P<0.00001); (B) Apoptosis‐associated speck‐like protein containing a CARD (ASC) (P<0.00000001); (C) Caspase‐1 (P<0.000001); (D) Interleukin (IL)‐1β (P<0.0000000000001), and (E) IL‐18 (P<0.00000000001) mRNA in human atherosclerotic carotid lesions compared to transplant donor vessels from the Biobank of Karolinska Endarterectomies (BiKE) cohort. F, NLRP3 mRNA expression in asymptomatic plaques compared to symptomatic plaques from the BiKE cohort (P<0.035).
NLRP3 Immunoreactivity in Carotid Plaque
Immunostaining for NLRP3 or ASC was performed in carotid plaques (n=3). In line with the correlation between NLRP3 and the macrophage marker CD68, CD68‐positive macrophages were expressed in the same areas as NLRP3. In addition, ASC was found to be expressed by CD68‐positive macrophages in plaque. Also, NLRP3 and ASC staining was occasionally expressed in smooth muscle cells (SMC) of atherosclerotic lesions (Figure 2).
Figure 2.
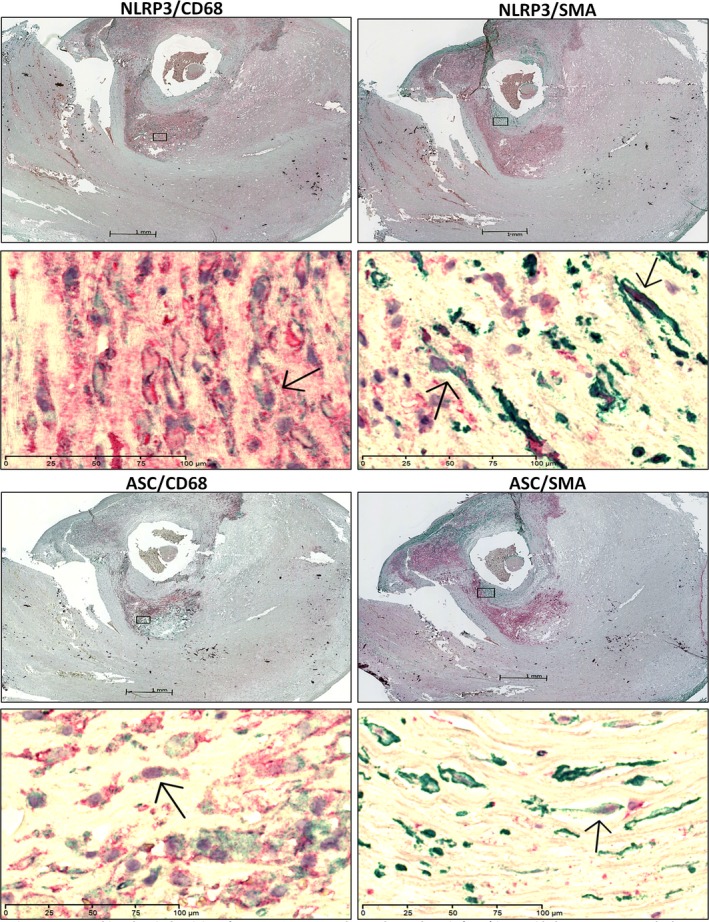
Immunohistochemical staining of NLR family, pyrin domain containing 3 (NLRP3), Apoptosis‐associated speck‐like protein containing a CARD (ASC), CD68 and smooth muscle actin (SMA) in carotid plaque. Lower panels show the magnified image. Left panels show the immunostaining of NLRP3/ASC (red) and CD68 (green). Right panels show immunostaining of NLRP3/ASC (red) and SMA (green). Arrows indicate co‐localization of NLRP3 or ASC in CD68‐ (left) or SMA‐positive cells (right). The scale bars for ×1.25 and ×40 objectives are 1 mm and 100 μm, respectively.
LPS and Cholesterol Crystals Induce IL‐1β Release in Human Carotid Plaque
To further assess the relevance of NLRP3 inflammasome in the atherosclerotic arteries, we examined the induction of IL‐1β protein release from freshly isolated human carotid plaques by using TLR4 activation (ie, LPS) as signal 1 and ATP or cholesterol crystals as signal 2. As shown in Figure 3A, LPS, ATP, and particularly the combination of these 2 stimuli markedly enhanced the release of IL‐1β. Notably, a similar pattern was also seen when cholesterol crystals were used in combination with LPS, with significantly increased release of IL‐1β. (Figure 3A and 3B). However, ATP was found to be more potent than cholesterol crystal stimulation. These findings demonstrate that atherosclerotic carotid plaques contain NLRP3 inflammasomes that could be activated when exposed to TLR4 activation and ATP or cholesterol crystals. Importantly, even without addition of LPS, there was significant release of IL‐1β upon stimulation with ATP or cholesterol crystals, suggesting that plaques were already primed for inflammasome activation.
Figure 3.
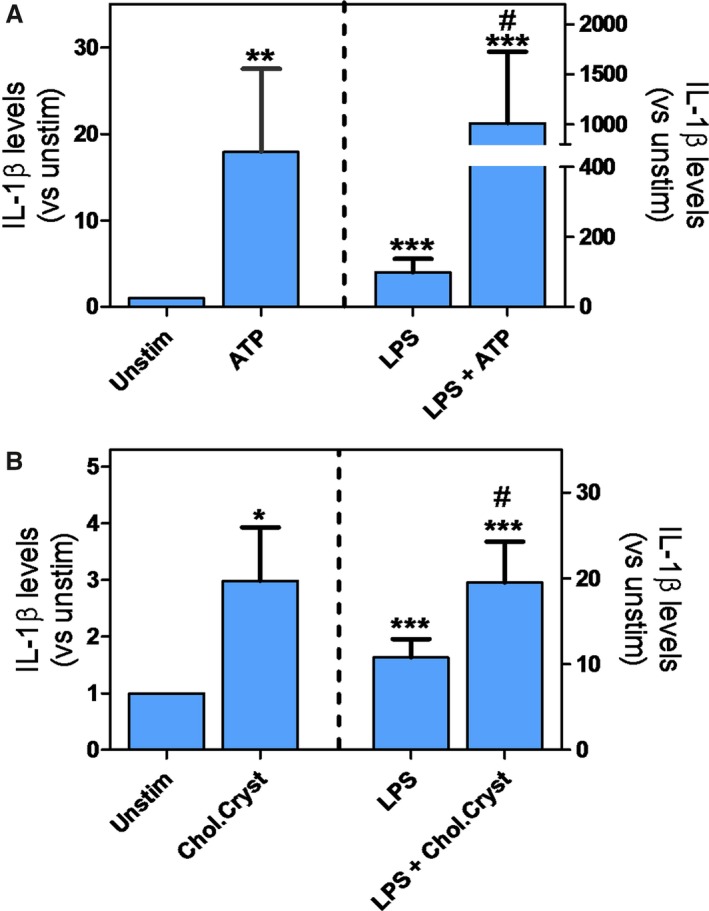
A, IL‐1β release from human carotid plaque (n=9) subjected to lipopolysaccharide (LPS) (113.8±138.6 pg/mL), ATP (78±129.0 pg/mL), or LPS+ATP (413.8±412.7 pg/mL) treatment and in control (9±12.7 pg/mL). B, IL‐1β release from human carotid plaque (n=7) subjected to LPS (145.8±112.7 pg/mL), cholesterol crystal (38.3±8.3 pg/mL), or LPS+ cholesterol crystal (351.1±206.4 pg/mL) treatment and in control (15.2 pg/mL). # P<0.05 vs LPS. **P<0.005 vs unstim. ***P<0.001 vs unstim.
Association Between Genetic Variants and NLRP3 mRNA Level in Plaque and PBMC
To examine cis‐acting regulatory effects of the variants on the NLRP3 gene, we investigated the associations between genotype and gene expression in atherosclerotic plaque (BiKE population, n=106) and PBMC (healthy controls, n=98). The polymorphisms rs6672995 (P=0.002) and rs10733113 (P=0.008) exhibited significant association with NLRP3 expression in PBMCs, but no significant association in PBMCs were evident for rs4353135 or rs4266924 (P=0.30 and 0.15, respectively; Figure 4). In plaque, a trend of moderate association was found between NLRP3 expression and rs4266924 (P=0.06), but no significant association was evident between NLRP3 expression and rs6672995, rs10733113, or rs4353135 in plaque (P=0.08, 0.13 and 0.19, respectively, Figure 4).
Figure 4.
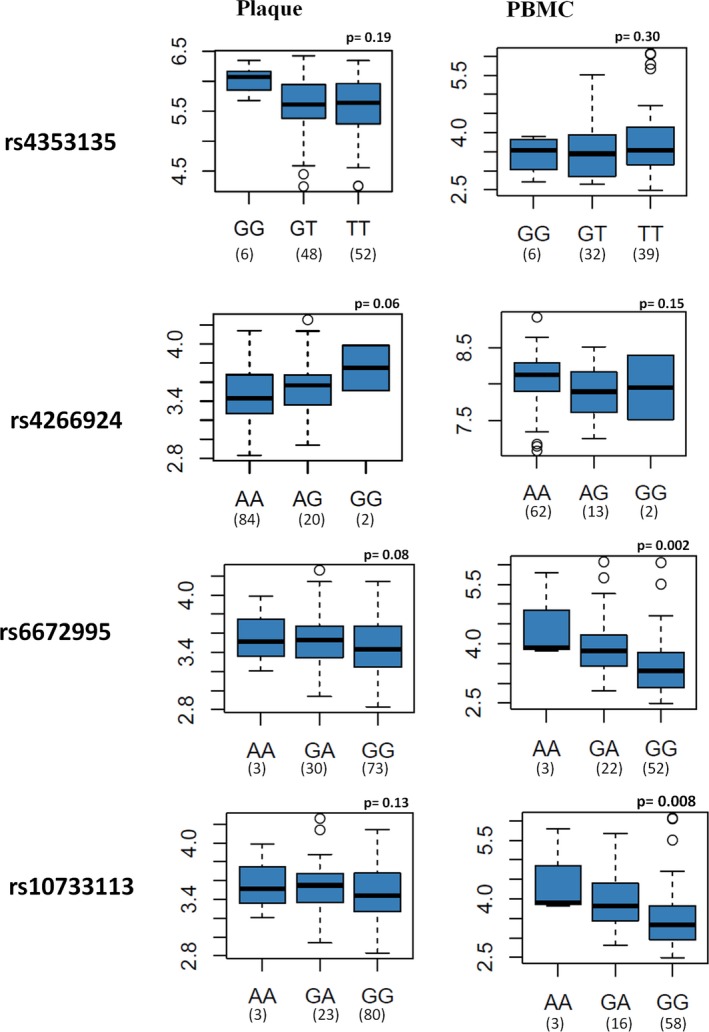
Association between genotype of variants and expression level of the NLR family, pyrin domain containing 3 (NLRP3) gene in plaque and peripheral blood mononuclear cells (PBMCs). The Y axis represents −log10 (P), calculated using additive model for the association between genotype and expression level. The X axis represents different genotypes of the variants. The minimum and maximum values are represented by whiskers, the first and third quartiles (box), the median values (medlines), outliers are displayed as circles. The Y axis represents the mRNA expression level of NLRP3.
Association Between NLRP3 Variants and Cytokines Levels in Plasma
As NLRP3 is involved in IL‐1β processing, we next examined the influence of these NLRP3 variants on IL‐1β levels in the SCARF cohort. We found that the minor allele of rs4266924 and rs10733113 was significantly associated with increased IL‐1β levels in controls (P=0.03 and 0.02, respectively), but not in MI patients (P=0.29, P=0.15; Table 1). No association was found between these variants and IL‐18, tumor necrosis factor‐α, MCP‐1, or C‐reactive protein levels in either patients or controls (data not shown).
Table 1.
Association of SNPs With IL‐1β Level in Plasma From Controls and Patients With MI of the SCARF Cohort Using ELISA
| SNP | Samples | Beta | SEM | P Value |
|---|---|---|---|---|
| Controls | ||||
| rs4353135 | 157 | 2.7 | 3.01 | 0.37 |
| rs4266924 | 160 | 2.29 | 4.26 | 0.03 |
| rs6672995 | 154 | 5.74 | 3.84 | 0.13 |
| rs10733113 | 153 | 1.27 | 0.56 | 0.02 |
| MI patients | ||||
| rs4353135 | 167 | 1.80 | 1.28 | 0.16 |
| rs4266924 | 177 | −1.63 | 1.56 | 0.29 |
| rs6672995 | 171 | −2.12 | 1.28 | 0.09 |
| rs10733113 | 166 | −2.06 | 1.44 | 0.15 |
Beta indicates the magnitude of effect per allele; MI, Myocardial Infarction; SCARF, Stockholm Coronary Atherosclerosis Risk Factor; SNPs, single nucleotide polymorphisms.
Association Between the NLRP3 Polymorphisms and MI
Next, we genotyped 555 MI patients and 1016 healthy controls in the FIA cohort for the SNPs rs4353135, rs4266924, rs6672995, and rs10733113 in the downstream regulatory region of NLRP3 gene.24 Approximately 1.2% of the samples were excluded due to poor genotype calling. The SNPs showed no significant deviation from the Hardy–Weinberg equilibrium, in the case or control group. No overall significant association was observed between the variants and MI; however, the heterozygous genotype of rs4353135 showed a trend toward the higher risk of MI (P=0.06; Table 2).
Table 2.
Overview of the Genotype and Allele Frequencies of NLRP3 Downstream Single Nucleotide Polymorphisms (SNPs) in Patients With Myocardial Infarction and Healthy Controls in the FIA Cohort
| SNP | Genotype | Patients | Controls | Odds Ratio (95% CI) | P Value |
|---|---|---|---|---|---|
| Genotype frequencies | |||||
| rs4353135 | n=459 (%) | n=860 (%) | |||
| TT | 237 (52) | 483 (56) | 1 | ||
| GG | 29 (6) | 63 (7) | 0.93 (0.58–1.49) | 0.78 | |
| GT | 193 (42) | 31 (37) | 1.25 (0.98–1.58) | 0.06 | |
| rs4266924 | n=490 (%) | n=902 (%) | |||
| AA | 380 (78) | 708 (79) | 1 | ||
| GG | 4 (0.8) | 10 (0.1) | 1.3 (0.42–4.36) | 0.61 | |
| AG | 106 (21) | 184 (21) | 1.4 (0.44–4.70) | 0.54 | |
| rs6672995 | n=491 (%) | n=901 (%) | |||
| GG | 355 (72) | 640 (71) | 1 | ||
| AA | 13 (3) | 23 (3) | 1.0 (0.51–2.03) | 0.95 | |
| AG | 123 (25) | 238 (26) | 0.93 (0.72–1.20) | 0.58 | |
| rs10733113 | n=432 (%) | n=802 (%) | |||
| GG | 320 (74) | 592 (74) | 1 | ||
| AA | 6 (1) | 12 (1) | 0.92 (0.34–2.48) | 0.87 | |
| AG | 106 (25) | 198 (25) | 0.99 (0.75–1.30) | 0.94 | |
| Allele frequencies | |||||
| rs4353135 | n=918 (%) | n=1720 (%) | |||
| T | 667 (73) | 1280 (74) | 1 | ||
| G | 251 (27) | 440 (26) | 1.0 (0.91–1.3) | 0.32 | |
| rs4266924 | n=980 (%) | n=1804 (%) | |||
| A | 866 (88) | 1600 (89) | 1 | ||
| G | 114 (12) | 204 (11) | 0.9 (0.7–1.23) | 0.79 | |
| rs6672995 | n=982 (%) | n=1802 (%) | |||
| G | 833 (85) | 1518 (84) | 1 | ||
| A | 149 (15) | 284 (16) | 0.95 (0.7–1.18) | 0.68 | |
| rs10733113 | n=864 (%) | n=1604 (%) | |||
| G | 746 (86) | 1382 (86) | 1 | ||
| A | 118 (14) | 222 (14) | 0.9 (0.7–1.25) | 0.89 | |
FIA indicates First‐ever myocardial Infarction study in Ac‐county; NLRP3, NLR Family, Pyrin Domain Containing 3; SNPs, Single Nucleotide Polymorphisms.
Discussion
Despite evidence of NLRP3 inflammasome activation in experimental atherosclerosis, neither the functional profile of activated NLRP3 inflammasome in the human plaque nor the association between NLRP3 genotype and plaque expression are known. In this study, we show that the mRNA level of NLRP3 inflammasome‐related genes is significantly increased in human atherosclerotic plaques compared to nonatherosclerotic vessels, and for NLRP3, particularly high expression was seen in those with symptomatic lesions. Previously, Zheng et al have shown enhanced expression of NLRP3 in atherosclerotic aorta as compared with control samples.35 In the present study, we extend these previous findings in several ways. First, while Zheng et al examined samples from 36 patients, the present study include samples from 106 patients. Second, in addition to immunohistochemistry, we also performed mRNA analysis, which is a quantitatively more accurate method. Third, the transcripts of the other NLRP3 inflammasome components (ie, ASC, caspase‐1, IL‐1β, and IL‐18) were also analyzed and the pathophysiological relevance of NLRP3 inflammasome in human atherosclerotic lesions was assessed in the cultured biopsies. However, the validation of our results based on microarray without real‐time polymerase chain reaction verification might be a limitation to our study, although microarray analysis is considered a more appropriate method for complex tissue and therefore is preferred.36
In the lesions, NLRP3 and ASC co‐localized with macrophages and SMC. Moreover, our ex vivo experiments suggest that NLRP3 inflammasome generates significant amounts of mature IL‐1β in the plaque. This was not only observed upon LPS/ATP stimulation. LPS/cholesterol crystal activation also produced IL‐1β, and even the addition of ATP or cholesterol crystals alone resulted in increased IL‐1β secretion, which suggests that the cells in the plaque were already primed for inflammasome activation. Moreover, SNP analyses revealed associations between certain gene variants and increased NLRP3 in PBMCs and IL‐1β levels in plasma. Our findings support a role for NLRP3 inflammasome in human atherosclerosis, linking cholesterol accumulation and inflammation within the atherosclerotic lesion.
The upregulated levels of NLRP3, ASC, caspase‐1, IL‐1β, and IL‐18 mRNA in the atherosclerotic plaque can be implicated as a checkpoint for NLRP3 inflammasome activation,37 suggesting the likelihood of activation of this inflammasome in the human atherosclerotic lesion. We also investigated the expression of NLRP3 in relation to plaque vulnerability and found the NLRP3 expression to be significantly increased in patients with symptomatic plaques when compared to patients with asymptomatic lesions, which is in agreement with the results from Zheng et al, who reported a significant correlation between aortic NLRP3 expression and Gensini coronary severity scores.35 Enhanced expression of NLRP3 in the symptomatic plaques indicates high‐grade inflammation that might be due to cholesterol crystallization in the lipid‐rich necrotic core and the presence of inflammatory cell infiltrates often observed in high‐grade carotid stenosis.14, 38 The mRNA levels of NLRP3 were significantly correlated with mRNA levels of CD68, and CD68‐positive macrophages were expressed in the same areas of atherosclerotic lesions as NLRP3 and ASC. In agreement with our results, Peng et al showed the expression of NLRP3 in the human atherosclerotic areas with CD68‐positive macrophage accumulation.39 Moreover, the expression of NLRP3 in SMC in the intima of atherosclerotic lesions may reflect the role of SMC in IL‐1β production, which is in line with our previous studies showing that SMC express NLRP3 and produce IL‐1β.40 Infiltrating macrophages have been linked to plaque progression and destabilization of high‐grade carotid stenosis,41 and our findings in the present study suggest that this may involve activation of NLRP3 inflammasome within these cells.
As NLRP3 is involved in IL‐1β processing, we also analyzed IL‐1β release after stimulating the atherosclerotic plaque with diverse agents such as LPS, extracellular ATP, and cholesterol crystals. Cholesterol crystals, as well as ATP, have previously been shown to activate NLRP3 inflammasome, thereby inducing the release of IL‐1β in LPS‐primed PBMCs.14, 42 In our previous studies we have shown that LPS‐stimulated carotid lesions release IL‐1β.5 In the present study, we extend these findings by showing that human carotid plaques not only contain NLRP3 inflammasome, but also release IL‐1β upon TLR4 activation and exposure to ATP or cholesterol crystals. The finding that cholesterol crystals can activate IL‐1β release from plaques in the absence of a TLR4 priming agent suggests that the cells have already received a priming signal for NLRP3 activation. This may reflect in situ priming of the inflammasome, which is consistent with our mRNA data. The effect of ATP was found to be more pronounced in comparison with cholesterol crystals, especially when these stimuli were combined with LPS. These findings may possibly depend on different mechanisms of signaling/uptake or, more simply, increased accessibility of ATP compared to cholesterol crystals due to different molecular size in our ex vivo experiments. However, whereas release of extracellular ATP may be relevant during plaque destabilization with increased cell death within the lesion, the exposure of cholesterol crystals known to be present in atherosclerotic lesions may be more relevant during the chronic atherosclerotic process. Our data along with other studies indicate that human atherosclerotic lesions contain NLRP3 inflammasomes that are activated and release IL‐1β when exposed to TLR4 activation and ATP or cholesterol crystals. The release of IL‐1β performs a key role in the recruitment of neutrophils, thereby promoting an inflammatory milieu in the atherosclerotic plaque.
We also investigated the genetic association between the genotypes of SNPs in the NLRP3 downstream region and NLRP3 gene expression. The minor alleles of rs4353135, rs4266924, rs6672995, and rs10733113 within the NLRP3 downstream regulatory region were previously found to be associated with risk of the development of Crohn's disease.24 In the present study, we found that the polymorphisms rs6672995 and rs10733113 were significantly associated with elevated NLRP3 expression in PBMCs of healthy controls and a borderline significant association of rs4266924 was evident in plaque. We also found that the expression of IL‐1β was significantly associated with the polymorphisms rs4266924 and rs10733113 in healthy controls. However, no significant association was evident for the polymorphisms investigated and IL‐1β in plasma from MI patients. This is partly in agreement with Villani and coworkers, who showed a significant association between these variants, NLRP3 expression, and IL‐1β production.24 The association of SNPs with increased plasma IL‐1β cytokine in the control group may suggest a role of the SNPs in the regulation of the expression of NLRP3 and IL‐1β in healthy individuals. The lack of association in the plaque of MI patients may suggest that several other factors expressed during the disease may overdominate the effect of these SNPs.
In the present study, we also examined the association of SNPs rs4353135, rs4266924, rs6672995, and rs10733113 with the risk of MI in the FIA cohort. Although no significant association was found between the variants and MI, the heterozygous genotype of rs4353135 showed a trend toward higher risk. Our results are in agreement with previous studies on the absence of genetic association between the above SNPs in Crohn's disease and ankylosing spondylitis.43, 44 The fact that the present study contains a relatively low number of individuals might be a limitation of the study, but we were able to reach statistical power of 78% for rs4353135 (assuming an effect of 1.21) and >95% of power for the additional 3 SNPs to detect an effect (assuming effects of 1.69, 1.53, and 1.78 for rs4266924, rs6672995, and rs10733113, respectively) and minor allele frequency based on publicly available information at dbSNP by NCBI for a European population.24 In particular, the absence of association between the NLRP3 variants and MI in a northern Swedish population should be interpreted cautiously due to the small sample size and the interaction of these loci with environmental exposures. Furthermore, the FIA cohort origins from a region where genetic drift has been proposed, which may explain a possible divergence from other cohorts.45 On the other hand, a strength of the present study is the use of 2 demographically different Swedish cohorts (FIA and SCARF) for the association studies with the combination of a third cohort of plaque (BiKE), since atherosclerosis is a frequent underlying disease for MI. However, ideally it would have been better to perform all analyses on all study subjects, but due to different study design of the different cohorts, this was not an option and is therefore a limitation to the study.
In conclusion, the present study shows that NLRP3 inflammasome‐related genes are highly expressed in atherosclerotic plaque and sensitive to activation when exposed to TLR4 activation and ATP or cholesterol crystals. Although we lack more quantitative protein data (eg, Western blot) and co‐localization of NLRP3 to cholesterol crystals and foam cells, our results suggest the possible role of NLRP3 and its variants in accelerating atherosclerosis.
Sources of Funding
This work was supported by grants from the Swedish Research Council (projects 6816 and K2012‐64X‐12233‐13‐3), the Center of Excellence for Research on Inflammation and Cardiovascular Disease (CERIC) Linnaeus Center (no 8703), the Swedish Heart‐Lung Foundation (20110359), the Foundation for Strategic Research, Uppdrag Besegra Stroke (P581/2011‐123), the Strategic Cardiovascular Programs of Karolinska Institutet, Stockholm County Council (ALF 20110279), Örebro University, Sigurd and Elsa Goljes Foundation, and Magnus Bergvall Foundation.
Disclosures
None.
Supporting information
Figure S1. A, Caspase‐1 (CASP1; P=0.0981); (B) Interleukin (IL)‐1B (P=0.834); (C) IL‐18 (P=0.259), and (D) apoptosis‐associated speck like protein containing a CARD (PYCARD/ASC; P=0.0842) mRNA expression in asymptomatic plaques compared to symptomatic plaques.
Acknowledgments
We thank the individuals in the Biobank of Karolinska Endarterectomies, Stockholm Coronary Atherosclerosis Risk Factor, and First‐ever myocardial Infarction study in Ac‐county cohorts for their participation.
(J Am Heart Assoc. 2016;5:e003031 doi: 10.1161/JAHA.115.003031)
References
- 1. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, Majmudar MD, Lasitschka F, Etzrodt M, Waterman P, Waring MT, Chicoine AT, van der Laan AM, Niessen HW, Piek JJ, Rubin BB, Butany J, Stone JR, Katus HA, Murphy SA, Morrow DA, Sabatine MS, Vinegoni C, Moskowitz MA, Pittet MJ, Libby P, Lin CP, Swirski FK, Weissleder R, Nahrendorf M. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin‐1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16:1000–1006. [DOI] [PubMed] [Google Scholar]
- 3. Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schonbeck U. Expression of interleukin (IL)‐18 and functional IL‐18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med. 2002;195:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mallat Z, Corbaz A, Scoazec A, Graber P, Alouani S, Esposito B, Humbert Y, Chvatchko Y, Tedgui A. Interleukin‐18/interleukin‐18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res. 2001;89:E41–E45. [DOI] [PubMed] [Google Scholar]
- 5. Olofsson PS, Sheikine Y, Jatta K, Ghaderi M, Samnegård A, Eriksson P, Sirsjö A. A functional interleukin‐1 receptor antagonist polymorphism influences atherosclerosis development. The interleukin‐1beta:interleukin‐1 receptor antagonist balance in atherosclerosis. Circ J. 2009;73:1531–1536. [DOI] [PubMed] [Google Scholar]
- 6. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin‐1beta decreases the severity of atherosclerosis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–660. [DOI] [PubMed] [Google Scholar]
- 7. Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, Bayard F, Hansson GK. Reduced atherosclerosis in interleukin‐18 deficient apolipoprotein E‐knockout mice. Cardiovasc Res. 2003;59:234–240. [DOI] [PubMed] [Google Scholar]
- 8. Barker BR, Taxman DJ, Ting JPY. Cross‐regulation between the IL‐1β/IL‐18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol. 2011;23:591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Mol Cell. 2002;10:417–426. [DOI] [PubMed] [Google Scholar]
- 10. Franchi L, Munoz‐Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. [DOI] [PubMed] [Google Scholar]
- 12. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. [DOI] [PubMed] [Google Scholar]
- 13. Rajamaki K, Lappalainen J, Oorni K, Välimäki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. [DOI] [PubMed] [Google Scholar]
- 16. Sandanger O, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G, Christensen G, Fitzgerald KA, Lien E, Valen G, Espevik T, Aukrust P, Yndestad A. The NLRP3 inflammasome is up‐regulated in cardiac fibroblasts and mediates myocardial ischaemia‐reperfusion injury. Cardiovasc Res. 2013;99:164–174. [DOI] [PubMed] [Google Scholar]
- 17. Li X, Zhang Y, Xia M, Gulbins E, Boini KM, Li PL. Activation of Nlrp3 inflammasomes enhances macrophage lipid‐deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS One. 2014;9:e87552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J. Atherosclerosis in ApoE‐deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011;2:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu JR, Leslie KS. Cryopyrin‐associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pontillo A, Vendramin A, Catamo E, Fabris A, Crovella S. The missense variation Q705K in CIAS1/NALP3/NLRP3 gene and an NLRP1 haplotype are associated with celiac disease. Am J Gastroenterol. 2011;106:539–544. [DOI] [PubMed] [Google Scholar]
- 21. Roberts RL, Van Rij AM, Phillips LV, Young S, McCormick SP, Merriman TR, Jones GT. Interaction of the inflammasome genes CARD8 and NLRP3 in abdominal aortic aneurysms. Atherosclerosis. 2011;218:123–126. [DOI] [PubMed] [Google Scholar]
- 22. Kastbom A, Verma D, Eriksson P, Skogh T, Wingren G, Soderkvist P. Genetic variation in proteins of the cryopyrin inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project). Rheumatology. 2008;47:415–417. [DOI] [PubMed] [Google Scholar]
- 23. Schoultz I, Verma D, Halfvarsson J, Törkvist L, Fredrikson M, Sjöqvist U, Lördal M, Tysk C, Lerm M, Söderkvist P, Söderholm JD. Combined polymorphisms in genes encoding the inflammasome components NALP3 and CARD8 confer susceptibility to Crohn's disease in Swedish men. Am J Gastroenterol. 2009;104:1180–1188. [DOI] [PubMed] [Google Scholar]
- 24. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, Baba N, Libioulle C, Belaiche J, Bitton A, Gaudet D, Cohen A, Langelier D, Fortin PR, Wither JE, Sarfati M, Rutgeerts P, Rioux JD, Vermeire S, Hudson TJ, Franchimont D. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat Genet. 2009;41:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Folkersen L, van't Hooft F, Chernogubova E, Agardh HE, Hansson GK, Hedin U, Liska J, Syvänen AC, Paulsson‐Berne G, Franco‐Cereceda A, Hamsten A, Gabrielsen A, Eriksson P; BiKE and ASAP study groups . Association of genetic risk variants with expression of proximal genes identifies novel susceptibility genes for cardiovascular disease. Circ Cardiovasc Genet. 2010;3:365–373. [DOI] [PubMed] [Google Scholar]
- 26. Folkersen L, Persson J, Ekstrand J, Agardh HE, Hansson GK, Gabrielsen A, Hedin U, Paulsson‐Berne G. Prediction of ischemic events on the basis of transcriptomic and genomic profiling in patients undergoing carotid endarterectomy. Mol Med. 2012;18:669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Guelpen B, Hultdin J, Johansson I, Witthöft C, Weinehall L, Eliasson M, Hallmans G, Palmqvist R, Jansson JH, Winkvist A. Plasma folate and total homocysteine levels are associated with the risk of myocardial infarction, independently of each other and of renal function. J Intern Med. 2009;266:182–195. [DOI] [PubMed] [Google Scholar]
- 28. Samnegard A, Silveira A, Lundman P, Boquist S, Odeberg J, Hulthe J, McPheat W, Tornvall P, Bergstrand L, Ericsson CG, Hamsten A, Eriksson P. Serum matrix metalloproteinase‐3 concentration is influenced by MMP‐3‐1612 5A/6A promoter genotype and associated with myocardial infarction. J Intern Med. 2005;258:411–419. [DOI] [PubMed] [Google Scholar]
- 29. Olofsson PS, Jatta K, Wagsater D, Gredmark S, Hedin U, Paulsson‐Berne G, Söderberg‐Nauclér C, Hansson GK, Sirsjö A. The antiviral cytomegalovirus inducible gene 5/viperin is expressed in atherosclerosis and regulated by proinflammatory agents. Arterioscler Thromb Vasc Biol. 2005;25:e113–e116. [DOI] [PubMed] [Google Scholar]
- 30. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, Lappegård KT, Brekke OL, Lambris JD, Damås JK, Latz E, Mollnes TE, Espevik T. Cholesterol crystals induce complement‐dependent inflammasome activation and cytokine release. J Immunol. 2014;192:2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hulthe J, McPheat W, Samnegard A, Tornvall P, Hamsten A, Eriksson P. Plasma interleukin (IL)‐18 concentrations is elevated in patients with previous myocardial infarction and related‐to severity of coronary atherosclerosis independently of C‐reactive protein and IL‐6. Atherosclerosis. 2006;188:450–454. [DOI] [PubMed] [Google Scholar]
- 32. Samnegård A, Hulthe J, Silveira A, Ericsson CG, Hamsten A, Eriksson P. Gender specific associations between matrix metalloproteinases and inflammatory markers in post myocardial infarction patients. Atherosclerosis. 2009;202:550–556. [DOI] [PubMed] [Google Scholar]
- 33. Wennberg P, Wensley F, Di Angelantonio E, Johansson L, Boman K, Rumley A, Lowe G, Hallmans G, Danesh J, Jansson JH. Haemostatic and inflammatory markers are independently associated with myocardial infarction in men and women. Thromb Res. 2012;129:68–73. [DOI] [PubMed] [Google Scholar]
- 34. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng F, Xing SS, Gong ZS, Xing QC. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 2013;22:746–750. [DOI] [PubMed] [Google Scholar]
- 36. Folkersen L, Kurtovic S, Razuvaev A, Agardh HE, Gabrielsen A, Paulsson‐Berne G. Endogenous control genes in complex vascular tissue samples. BMC Genomics. 2009;10:516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes‐Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carr SC, Farb A, Pearce WH, Virmani R, Yao JST. Activated inflammatory cells are associated with plaque rupture in carotid artery stenosis. Surgery. 1997;122:757–763. [DOI] [PubMed] [Google Scholar]
- 39. Peng K, Liu L, Wei D, Lv Y, Wang G, Xiong W, Wang X, Altaf A, Wang L, He D, Wang H, Qu P. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int J Mol Med. 2015;35:1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tangi TN, Elmabsout AA, Bengtsson T, Sirsjo A, Fransén K. Role of NLRP3 and CARD8 in the regulation of TNF‐alpha induced IL‐1beta release in vascular smooth muscle cells. Int J Mol Med. 2012;30:697–702. [DOI] [PubMed] [Google Scholar]
- 41. Jander S, Sitzer M, Schumann R, Schroeter M, Siebler M, Steinmetz H, Stoll G. Inflammation in high‐grade carotid stenosis: a possible role for macrophages and T cells in plaque destabilization. Stroke. 1998;29:1625–1630. [DOI] [PubMed] [Google Scholar]
- 42. Griffiths RJ, Stam EJ, Downs JT, Otterness IG. ATP induces the release of IL‐1 from LPS‐primed cells in vivo. J Immunol. 1995;154:2821–2828. [PubMed] [Google Scholar]
- 43. Lewis GJ, Massey DCO, Zhang H, Bredin F, Tremelling M, Lee JC, Berzuini C, Parkes M. Genetic association between NLRP3 variants and Crohn's disease does not replicate in a large UK panel. Inflamm Bowel Dis. 2011;17:1387–1391. [DOI] [PubMed] [Google Scholar]
- 44. Kastbom A, Klingberg E, Verma D, Carlsten H, Forsblad‐d'Elia H, Wesamaa J, Cedergren J, Eriksson P, Söderkvist P.Genetic variants in CARD8 but not in NLRP3 are associated with ankylosing spondylitis. Scand J Rheumatol. 2013;42:465–468. [DOI] [PubMed] [Google Scholar]
- 45. Lappalainen T, Hannelius U, Salmela E, von Döbeln U, Lindgren CM, Huoponen K, Savontaus ML, Kere J, Lahermo P. Population structure in contemporary Sweden—a Y‐chromosomal and mitochondrial DNA analysis. Ann Hum Genet. 2009;73:61–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A, Caspase‐1 (CASP1; P=0.0981); (B) Interleukin (IL)‐1B (P=0.834); (C) IL‐18 (P=0.259), and (D) apoptosis‐associated speck like protein containing a CARD (PYCARD/ASC; P=0.0842) mRNA expression in asymptomatic plaques compared to symptomatic plaques.