

Abstract
Background
Ascending thoracic aortic aneurysm (ATAA) is driven by angiotensin II (AngII) and contributes to the development of left ventricular (LV) remodeling through aortoventricular coupling. We previously showed that locally available leptin augments AngII‐induced abdominal aortic aneurysms in apolipoprotein E–deficient mice. We hypothesized that locally synthesized leptin mediates AngII‐induced ATAA.
Methods and Results
Following demonstration of leptin synthesis in samples of human ATAA associated with different etiologies, we modeled in situ leptin expression in apolipoprotein E–deficient mice by applying exogenous leptin on the surface of the ascending aorta. This treatment resulted in local aortic stiffening and dilation, LV hypertrophy, and thickening of aortic/mitral valve leaflets. Similar results were obtained in an AngII‐infusion ATAA mouse model. To test the dependence of AngII‐induced aortic and LV remodeling on leptin activity, a leptin antagonist was applied to the ascending aorta in AngII‐infused mice. Locally applied single low‐dose leptin antagonist moderated AngII‐induced ascending aortic dilation and protected mice from ATAA rupture. Furthermore, LV hypertrophy was attenuated and thickening of aortic valve leaflets was moderated. Last, analysis of human aortic valve stenosis leaflets revealed de novo leptin synthesis, whereas exogenous leptin stimulated proliferation and promoted mineralization of human valve interstitial cells in culture.
Conclusions
AngII‐induced ATAA is mediated by locally synthesized leptin. Aortoventricular hemodynamic coupling drives LV hypertrophy and promotes early aortic valve lesions, possibly mediated by valvular in situ leptin synthesis. Clinical implementation of local leptin antagonist therapy may attenuate AngII‐induced ATAA and moderate related LV hypertrophy and pre–aortic valve stenosis lesions.
Clinical Trial Registration
URL: https://www.clinicaltrials.gov/. Unique identifier: NCT00449306.
Keywords: angiotensin II, aortic aneurysm, aortic valve stenosis, left ventricular hypertrophy, leptin, leptin antagonist, vascular remodeling
Subject Categories: Animal Models of Human Disease, Aneurysm, Valvular Heart Disease, Translational Studies
Ascending thoracic aortic aneurysms (ATAAs) represent 60% of all thoracic aortic aneurysms. Progressively expanding ATAA mandates surgical intervention to prevent vessel dissection or free rupture.1 ATAA is age related and is frequently encountered in patients with hypertension, hypercholesterolemia, or diabetes mellitus.2 It is also diagnosed in young patients with genetic disorders associated with intrinsic structural abnormalities such as Marfan syndrome and bicuspid aortic valve and as a result of aortic inflammatory diseases.3, 4, 5 Much remains unknown about ATAA pathology, largely because of the wide heterogeneity underlying these abnormalities. Nevertheless, most aortic aneurysms are associated with augmented angiotensin II (AngII) activity,6, 7, 8 and AngII‐infusion murine models have been established for both ATAA and abdominal aortic aneurysm (AAA).9, 10, 11
The AngII hormone is a key component of the renin–angiotensin–aldosterone system. AngII promotes hypertension, atherosclerosis, and vascular remodeling, inducing aortic aneurysms in mice and humans by activating mechanisms of aortic wall stiffening and medial degeneration.12, 13, 14 Aortic aneurysms are elicited in part through modulation of transforming growth factor β (TGF‐β) activity. In mice, AAA formation is associated with decreased TGF‐β gene expression, whereas the pathogenesis of ATAA in mice and humans is promoted by increased TGF‐β signaling.15
Vascular remodeling is also elicited by leptin,16 a regulatory hormone of food intake and energy expenditure. Leptin is mostly synthesized by adipocytes, and its systemic levels correlate with body fat mass.17 Leptin synthesis is induced to a lesser extent in macrophages and vascular smooth muscle cells (SMCs) by cytokines and AngII, resulting in local cardiovascular pathological conditions.18 Hyperleptinemia is a risk factor for symptomatic cardiovascular disease, mainly through increased arterial blood pressure, augmented oxidative stress, stiffening of the vascular wall, and vascular cell calcification.19, 20, 21, 22, 23
In the clinical setting, aging‐related arterial stiffening24 promotes left ventricular hypertrophy (LVH) via complex interactions among elevated systemic blood pressure, increased pulse pressure, diminished arterial distensiblity, and augmented left ventricular (LV) afterload. This hemodynamic system, termed aortoventricular coupling (AVC), leads to LVH and diastolic dysfunction.25 Moreover, augmented rigidity and aneurysmal dilation per se in the ascending aorta are also sufficient to alter cardiac morphology and function.26, 27, 28 Hemodynamic perturbations at the LV outlet augment pulse wave velocity that increases LV workload. The resulting elevated intracardiac pressure stretches LV cardiomyocytes, which respond by upregulating AngII and AngII type 1 receptor expression.29, 30, 31, 32 Finally, leptin synthesized by LV cardiomyocytes mediates AngII‐driven LVH.33
AngII activity and hyperleptinemia are associated clinically with aortic valve stenosis (AVS).34, 35 A recent study in apolipoprotein E–deficient (apoE−/−) mice revealed that AngII infusion promotes thickening of aortic valve leaflets and simultaneously promotes LVH.36 These valvular lesions may represent early valve leaflet remodeling preceding AVS. In addition, mice exposed to hyperlipidemia and type 2 diabetes were found to exhibit aortic valve calcification and reduced LV function.37 This latter observation correlates with the association of hyperlipidemia with hypertension through increased levels of endothelin 1, AngII, and hyperleptinemia, thereby contributing to LVH and aortic valve remodeling.22, 38
We recently demonstrated de novo leptin synthesis in human AAAs. Moreover, using apoE−/− mice, we found that local application of leptin to the abdominal aorta potentiates AngII‐induced AAA and promotes aneurysm formation when acting as a sole local stimulant.14 Those results were the basis for our current hypothesis that locally synthesized leptin in the ascending aorta drives AngII‐induced aortic remodeling and local aneurysm formation.
In the present study, we analyzed human ATAA samples and used a novel mouse model that simulates local leptin synthesis in the ascending aorta to examine the impact of leptin on the vessel wall and subsequent LVH and cardiac valve remodeling. To test the pathophysiological relevance of locally synthesized endogenous leptin, we applied a leptin antagonist (LepA)39 to the ascending aorta in AngII‐infused mice. Our results implicated leptin activity in AngII‐driven vascular remodeling. Moreover, local blocking of leptin may present a novel therapeutic approach to attenuate formation of ATAA, control of related LVH, and remodeling of aortic valve leaflets.
Materials and Methods
Mouse Models
Animal experiments were performed according to protocols approved by the institutional animal care and use committee, Harvard Medical School (protocol no. 05004), and compiled with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication no. 85‐23, revised 1996). Male apoE−/− mice (C57BL/6 background; Jackson Laboratory) aged 16 weeks were used in all experiments (n=70; 36 for leptin application, 34 for AngII administration). Mice underwent left minithoracotomy, providing an exposure of the proximal ascending aorta. To simulate local leptin synthesis at the ascending aorta, a slow‐release film made of polylactic co–glycolic acid matrix containing 2 μg mouse leptin or no protein (control) was applied to the outer surface of the proximal ascending aortic. This model was used in 2 separate experiments. In the first experiment, mice were fed postoperatively with a high‐fat diet (HFD) and followed up for 45 days. In the second experiment mice were fed with a normal chow diet and monitored for 30 or 60 days. To examine the effect of leptin activity inhibition at the ascending aorta, a miniature polylactic co–glycolic acid film carrying 5 μg mouse LepA (superactive mouse leptin antagonist; Protein Laboratories Rehovot) 39 was applied to the surface of the proximal ascending aorta. This was followed by subcutaneous implantation of an osmotic minipump (pump model 2004; Alzet) for AngII (human AngII, A9525; Sigma‐Aldrich) infusion of 1000 ng/kg per minute over 28 days. Mice were followed up for 28 days and were assessed at baseline and periodically during the 28‐day follow‐up period for body weight, systemic blood pressure, and echocardiography of the ascending aorta and left ventricle.
Systemic blood pressure was measured via tail cuff using the BP‐2000 Series II blood pressure analysis system (Visitech Systems). Systemic blood pressure was measured at baseline prior to surgery and then weekly for 1 month and biweekly during the second month (in the leptin application model).
High‐resolution echocardiography was performed at baseline, weekly or biweekly in the leptin application model, biweekly in the AngII infusion/LepA application model, and before termination of each experiment, using the Vevo 2100 system (VisualSonics). To measure the inner diameter of the ascending aortic wall and assess local distensibility, high‐resolution echocardiography was performed 2 mm distal to the aortic valve, and measurements were performed in the long‐axis view. Distensibility was defined as percentage of change between aortic cross‐sectional area in systole versus diastole.
In each mouse, long‐ and short‐axis B‐mode views and videos were generated. Peak systolic velocity (PSV) was measured using Doppler mode at the LV outlet (aortic valve level). Accompanying software was used to measure aortic diameter and PSV and velocity–time integral and peak velocity of aortic valve regurgitation jet. LV wall width and fractional area change were extracted from videos generated by Vevo 2100. The thickness of aortic valve and mitral valve leaflets was measured by light microscopy. The thickest segment was assessed, and measurements were presented as mean thickness in at least 2 leaflets per valve. All measurements were performed in a blinded fashion.
Mouse tissue slides underwent histological analysis with elastic Van Gieson staining and immunohistochemistry for α‐smooth muscle actin (α‐SMA), TGF‐β1, TGF‐β2, Mac‐2, Mac‐3, leptin, and Ki‐67.
Human Tissues
The use of human tissues was approved by the institutional review board of Sheba Medical Center, Tel Hashomer, Israel, and tissues were collected with informed consent (ethics committee approval no. 2354; ClinicalTrials.gov identifier NCT00449306).
Surgical samples of ascending aortic aneurysm were collected (n=11) from patients with a variety of associated diseases, including hypertension (n=10), hypercholesterolemia (n=5), diabetes mellitus (n=2), Marfan syndrome (n=4), bicuspid aortic valve (n=2), and ankylosing spondylitis (n=1) (Table). Four patients were operated urgently for type A dissection. Samples of human AVS (n=11) were collected from patients undergoing aortic valve replacement surgery. Normal aortic valves (n=3) were obtained from explanted hearts of patients undergoing heart transplantation.
Table 1.
Ascending Aortic Aneurysm in Patients Undergoing Surgery: Demographics, Comorbidities, and Associated or Underlying Diseases
| Diagnosis | Age, y | Sex | Hypertension | Dyslipidemia | Diabetes Mellitus | Cardiac and Other Comorbidities | Surgery |
|---|---|---|---|---|---|---|---|
| Ankylosing Spondylitis | 46 | Male | Elective resection of ATAA | ||||
| Ascending aortic aneurysm | 80 | Male | + | + | Mild aortic regurgitation; left ventricular ejection fraction 60%; hypothyroidism; smoking | Elective resection of ATAA | |
| Ascending aortic aneurysm | 82 | Female | + | + | Left ventricular ejection fraction 45%; atrial fibrillation; polymialgia rheumatica; hypothyroidism; chronic obstructive pulmonary disease | Elective resection of ATAA | |
| Ascending aortic aneurysm | 68 | Male | + | + | Ischemic heart disease | Type A dissection; resection of ATAA | |
| Ascending aortic aneurysm | 68 | Male | + | + | + | Type A dissection; resection of ATAA | |
| Bicuspid aortic valve | 46 | Male | + | Aortic valve stenosis | Elective resection of ATAA; aortic valve replacement | ||
| Bicuspid aortic valve | 48 | Male | + | Aortic valve stenosis; left ventricular hypertrophy | Elective plication of ATAA; aortic valve replacement | ||
| Marfan syndrome | 44 | Male | Type A dissection; resection of ATAA | ||||
| Marfan syndrome | 22 | Male | + | Mitral valve prolapse | Elective resection of ATAA | ||
| Marfan syndrome | 46 | Female | + | Type A dissection; resection of ATAA | |||
| Marfan syndrome | 45 | Male | + | + | + | Type A dissection; resection of ATAA |
ATAA indicates ascending thoracic aortic aneurysm.
Human tissues were analyzed histologically by routine hematoxylin and eosin staining and modified Movat's pentachrome connective tissue stain. Immunohistochemistry was performed for α‐SMA, CD68, leptin, and leptin receptor, as described previously.17 For in situ mRNA hybridization, leptin RNA sense and antisense probes were transcribed in vitro and labeled with digoxigenin. Reverse transcriptase quantitative polymerase chain reaction was performed for the lep and lepr genes using TaqMan probes.
In Vitro Studies
Valve interstitial cell culture
Human valve interstitial cells (VICs; generously provided by Kristyn S. Masters) were grown in MEMα plus 15% FCS,1.5% Penicillin‐Streptomycin‐Neomycin 1% glutamine, and 2.5 μg/mL amphotericin B (complete medium).
Expression of leptin and leptin receptor
Cells were grown in complete medium and treated with 1, 2.5 and 10 nmol/L AngII (Sigma‐Aldrich) for 4 or 24 hours, as indicated. The mRNA was extracted, and lep and lepr expression was quantified by reverse transcriptase quantitative polymerase chain reaction using TaqMan probes.
Proliferation experiments
Cells were seeded in complete medium and grown in 24‐well plates (15 000–20 000 cells per well) for 3 to 5 days, and then the medium was changed to 1.5% FCS containing media (starvation medium) for 48 hours. Proliferation assay was performed in quadruplicate in fresh starvation medium supplemented with the tested factor (leptin 1 ng/mL, AngII 2.5 nmol/L) for 24 hours. The degree of proliferation was measured by the XTT cell proliferation kit (Biological Industries). Cells grown in starvation media only were used as control, and their absorbance was 0.268±0.030 optical density (considered as 100%).
Mineralization experiments
Human VICs were plated in 35‐mm dishes (75 000 cells per dish) in complete medium supplemented with 10 nmol/L dexamethasone, 10 mmol/L β glycerophosphate, 8 mmol/L CaCl2, 50 mg/mL ascorbate, 1 mmol/L insulin, without leptin or in the presence of leptin at concentrations of 1 or 10 ng/mL for 3 weeks. Medium was charged twice a week. After 21 days, cultures were fixed in formalin and then stained by alizarin red. Quantitation of the colored mineralized area was performed using ImageJ (National Institutes of Health).
Statistics
A 2‐sided Mann–Whitney test was used to assess differences between control and leptin‐treated animals or AngII versus AngII and LepA‐treated animals. To overcome baseline variability between physiological parameters of apoE−/− animals, each echocardiograph measurement at the end of the experiment was compared with a baseline measurement prior to surgery. All data are shown as mean±SE, and the typical number of samples was 10 for mouse samples and 5 for in vitro samples, as detailed in the figure captions. The Fisher exact test was used to assess the effect of local LepA treatment on thoracic aortic aneurysm rupture and death on AngII‐treated mice. The Bonferroni correction was used for correction of proliferation and mineralization results in in vitro studies. The Student t test was used for analysis of quantitative polymerase chain reaction and mineralization data. The radar plot scales physiological parameters by standard deviations in the untreated group. Detailed data on materials and methods are provided in Data S1.
Results
Human ATAA Wall Exhibits Abundant Expression of Leptin mRNA and Protein
Movat's pentachrome staining of ascending aortic aneurysms that were collected from patients presenting with different diseases show extensive medial degeneration including multiple fragmentations, loss of elastic lamellas, depletion of SMCs, and abundance of proteoglycans (Figure 1A, 1D, 1G, S1A, and S1D). Macrophage infiltration was variably demonstrated in the different samples but was more prominent in Marfan syndrome cases (Figure S2H). Leptin antigen was demonstrated in medial SMCs of all samples (Figure 1B, 1E, 1H, and S2E) and in medial and adventitial macrophages (Figures S1E and S2E). The lep mRNA was identified by in situ hybridization in all analyzed ATAA samples, generally correlating with the abundance of leptin antigen (Figure 1C, 1F, and 1I). Control samples of nondilated aortas exhibited few scattered leptin‐positive cells with low level of leptin mRNA (Figure 1L). Human retroperitoneal fat was used as a positive control (Figure 1J and 1K). Leptin receptor antigen was also detected in ATAA specimens and expressed by SMCs and macrophages (Figure S2F). These findings confirmed that leptin is de novo synthesized in multiple ATAA samples, associated with a variety of systemic diseases.
Figure 1.
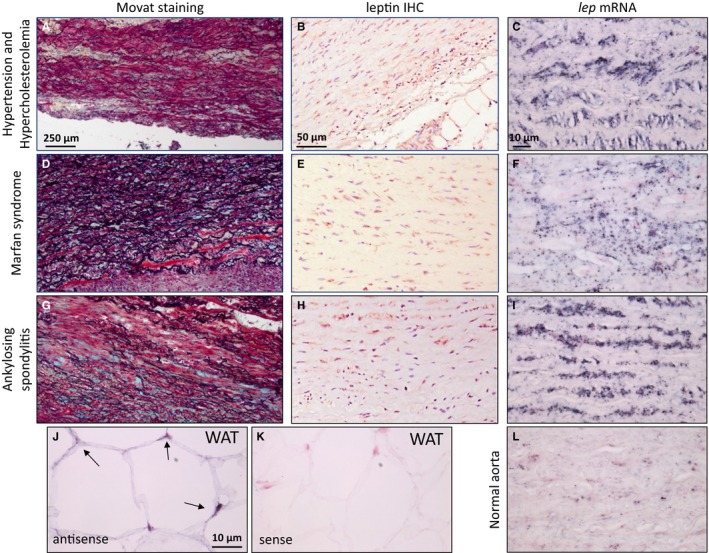
Analysis of surgical ATAA samples collected from patients with a variety of diseases underlying ATAA. A through C, Hypertension and hypercholesterolemia. D through F, Marfan syndrome. G through I, Ankylosing spondylitis. A, D, and G, Diffuse elastic fiber fragmentation with glycosaminoglycan deposition (bluish‐green), matrix clearing, and media degeneration, Movat's pentachrome. Scale bar=250 μm. B, E, and H, Leptin immunostaining in medial smooth muscle cells. Scale bar=50 μm. C, F, I, and L, The lep mRNA (black staining) is present in diseased aortas but is scarcely present in normal aorta. Scale bar=10 μm. J and K, Positive (antisense) (J) and negative (sense) (K) control for lep mRNA. In situ hybridization in human WAT. Scale bar=10 μm. ATAA indicates ascending thoracic aortic aneurysm; IHC, immunohistochemistry; WAT, white adipose tissue.
Perivascular Leptin Applied at the Proximal Ascending Aorta Reduces Aortic Wall Distensibility and Causes Local Aortic Dilation
A polylactic co–glycolic acid film containing leptin or no protein was surgically applied at the proximal ascending aorta of apoE−/− mice (described under Methods) (Figure 2A and S3A). All mice recovered from surgery uneventfully and gained weight equally during the follow‐up period, suggesting that leptin had no systemic effect (Figure S3B). Mean systolic blood pressure in mice fed normal chow diet was 100 mm Hg during the first 4 weeks, with an increase to 120 mm Hg at weeks 6 and 8 in both leptin‐treated and control mice (Figure S3C). This increase in blood pressure may be attributed to aging processes (age 20–22 weeks). In general, feeding mice with an HFD did not affect the results substantially; however, some of the modulated features related to local leptin application were more pronounced in mice fed an HFD compared with a normal chow diet and may be related to augmented AngII synthesis in hypercholesterolemia.40
Figure 2.
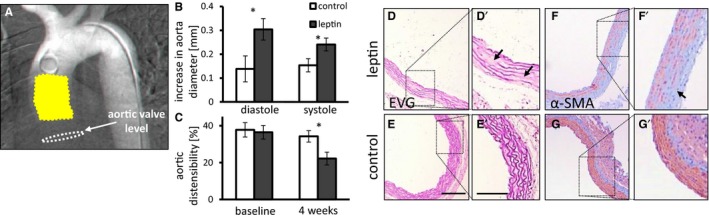
The effects of locally applied leptin on the ascending aorta. A, Human arch arteriogram depicting mouse aortic anatomy. Note the location of leptin slow‐release film (yellow) in relation to the aortic valve level (white dashed lines). B, Increase in maximal aortic diameter from baseline following treatment with leptin in diastole and systole (P=0.02 and P=0.045, respectively). Baseline aortic diameter was 1.3 mm. C, Percentage of aortic wall distensibility assessed at baseline and 4 weeks after surgery (P=0.01; n=10 to 11). Control mice in white bars, leptin treated in gray bars. *P<0.05. D through G, Histologic cross‐sections of the ascending aortic at the location of leptin application, as shown in (A). Elastic Van Gieson (EVG) staining shows fragmentations of elastic lamellae (arrows) in leptin treated (D) and control mice (E). Depletion of α‐smooth muscle actin (α‐SMA) immunostaining (arrow) in leptin treated (F) and control mice (G). Scale bar=100 μm, 200 μm for (D′ and E′).
Echocardiography was performed 2 mm distal to the aortic valve and revealed increased aortic diameter at the site of slow‐release leptin film attachment (P<0.05 for normal chow diet and P<0.05 for HFD) (Figure 2B and S4A). Aortic wall distensibility, defined as percentage of change between aortic cross‐sectional area in systole versus diastole, was reduced in the involved aortic segment in leptin‐treated mice (Figure 2C) (P<0.05). There was no significant difference in aortic diameter farther distally on the ascending segment. Despite the aortic dilation in response to locally applied leptin in the proximal aorta, there was no dilation of the aortic valve annulus. Histological analysis of the ascending aorta at the site of leptin application revealed medial hypertrophy, fragmentation of elastic lamellas (Figure 2D and 2E), and depletion of α‐SMA in mice, regardless of diet (Figure 2F, 2G, and S4B–S4G).
Leptin Application at the Proximal Ascending Aorta Causes LVH and Leaflet Thickening in Aortic and Mitral Valve Leaflets
PSV reflecting hemodynamic perturbation at the LV outlet was increased in leptin‐treated mice receiving HFD (P=0.17 for normal chow diet and P<0.05 for HFD) (Figure 3A and S5A). Leptin did not increase the velocity–time integral or peak velocity of the regurgitation flow across the aortic valve (Figure S3D and S3E). Echocardiography used to assess cardiac function identified LV remodeling with concentric hypertrophy (P<0.05 and P<0.01 for HFD) (Figure 3B and S5B). LV diameter of leptin‐treated mice was significantly increased in systole (Figure 3C) (P<0.05), leading to marginal reduction in LV fractional area change. This parameter was defined as the relative increase in LV cross‐sectional area in systole versus diastole (Figure 3D) (P=0.09) and no change in stroke area, defined as the area difference between systole versus diastole (Figure S3F).
Figure 3.
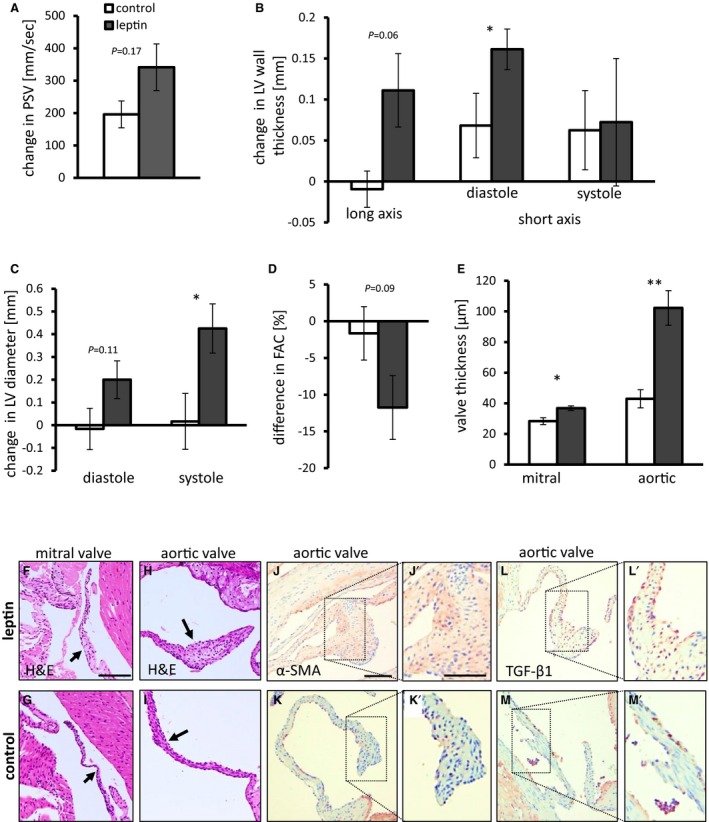
Leptin applied at the ascending aorta induces remodeling of LV wall and valve leaflets. A, Leptin‐induced increase in PSV measured at the aortic valve. P=0.17. Mean baseline PSV was 1150 mm/s. B, Leptin induced an increase in LV wall thickness, as measured in the long‐axis view (P=0.06), and in diastole, as measured in the short‐axis view (P=0.045). Mean LV wall thickness at baseline was 0.83 mm at anterior and posterior walls and 0.55 mm at the septum. C, Increase in LV diameter following treatment with leptin in diastole (P=0.11) and in systole (P=0.04). Mean LV diameter at baseline was 2.2 mm. D, Difference in FAC following treatment with leptin (P=0.09). Mean FAC at baseline was 63%. E, Thickness of mitral and aortic valve leaflets following leptin treatment (P=0.02 and P=0.006, respectively). A through E, n=9 to 11. Control mice shown in white bars; leptin‐treated mice shown in dark gray bars. F and G, H&E staining of mitral valve from leptin‐treated (F) and control (G) mice. Arrows point to the valve. Scale bar=100 μm. H and I, H&E staining of aortic valve from leptin‐treated (H) and control (I) mice. Arrows point to the valve. J and K, α‐SMA staining in aortic valves of leptin‐treated mice (J) and controls (K). L and M, TGF‐β1 expression in aortic valves of leptin‐treated mice (L) and controls (M). Scale bar: Low magnification=50 μm, enlargements=100 μm. *P<0.05, **P<0.01. α‐SMA indicates α‐smooth muscle cell actin; FAC, fractional area change; H&E, hematoxylin and eosin; LV, left ventricular; PSV, peak systolic velocity; TGF‐β1, transforming growth factor β1.
Leptin‐treated mice exhibited thickening of mitral and aortic valve leaflets (P<0.05, P<0.01, respectively) (Figure 3E and S5C for HFD). Interestingly, mitral valve leaflets were diffusely thickened, whereas aortic valve leaflets demonstrated thickening in their free edge. These hyperplastic lesions were mostly composed of extracellular matrix and multiple stromal cells (Figure 3F–3I and S5D–S5G). These cells are assumed to be analogous to human VICs. A few clusters of stromal cells within the hyperplastic lesions were positive for α‐SMA and TGF‐β1 antigen by immunohistochemistry (Figure 3J–3M and S5H–S5K), suggesting activation of VICs. Ki‐67 immunohistochemistry revealed no significant increase in VIC proliferation at the time of euthanasia (Figure S3E). No macrophage infiltration or tumor necrosis factor α antigen were detected in aortic valve leaflets from mice receiving leptin application. Hyperplastic aortic and mitral valve leaflets exhibited no calcifications by alizarin red staining.
Collectively, these experiments suggest that leptin, when available at the proximal ascending aorta, causes local loss of elasticity and induces medial degeneration, thereby promoting aortic dilation without affecting the diameter of the aortic annulus. Ascending aortic stiffening and dilation underlie perturbation of hemodynamics, driving hypertrophy of the LV wall and aortic and mitral valve leaflet thickening, possibly through the AVC axis.
LepA Applied at the Proximal Aorta Attenuates Aortic Dilation and Protects Mice From Rupture of AngII‐Induced Thoracic Aortic Aneurysm
AngII is the key hormone of the renin–angiotensin–aldosterone system, underlying hypertension and cardiovascular remodeling. The phenotype induced by local application of leptin is similar to AngII‐induced aortic remodeling,14, 18 suggesting that leptin may mediate this phenomenon. To test this hypothesis, apoE−/− mice were infused with AngII for 4 weeks after they received a polylactic co–glycolic acid film containing LepA or no protein that was applied on the surface of the proximal ascending aorta (details under Methods).
Weight gain was similar in both groups, indicating lack of systemic effects of LepA (Figure S6A). As expected, systolic blood pressure in all AngII‐treated mice was increased by ≈20%, reaching 125 mm Hg after 1 week, and was sustained throughout the follow‐up period (Figure S6B). Notably, the addition of LepA did not affect the increase in systolic blood pressure in AngII‐receiving mice.
There was marked thinning of the anterior aspect of the ascending aorta and significant dilation of the segment in mice receiving AngII. This dilation was attenuated in mice that were also treated with local LepA application (P<0.05) (Figure 4A and 4B). Echocardiographic analysis performed 4 weeks after surgery revealed an increase in the inner diameter of the ascending aorta 2 mm distal to the aortic valve in AngII‐infused mice. The increase in aortic diameter in diastole and systole was moderated in mice cotreated with LepA (P<0.01, in systole) (Figure 4C). Distensibility was not affected by LepA treatment because aortic diameter in both systole and diastole was similarly affected by LepA application (Figure 4D). Importantly, aortic valve annulus did not dilate in response to AngII infusion.
Figure 4.
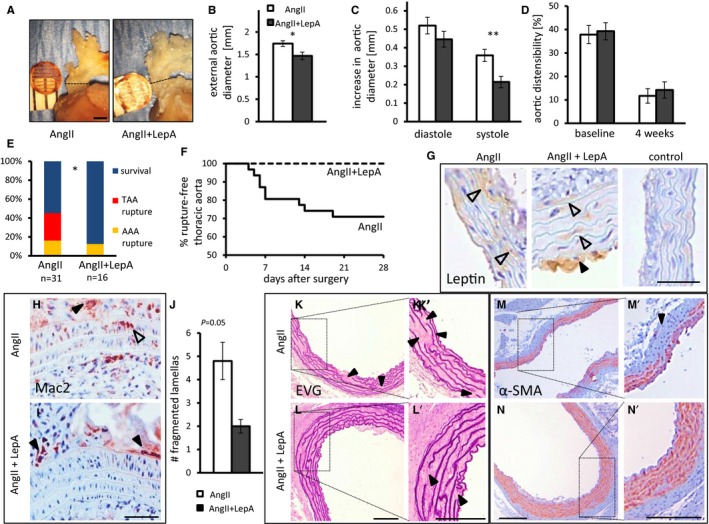
Local application of LepA at the proximal ascending aorta attenuates the effects of AngII infusion on the aortic wall locally. A and B, External aortic diameter 4 weeks after surgery. n=5, P=0.047. C, Increase in aorta diameter in diastole and systole (P<0.01 in systole). Mean internal aortic diameter was 1.3 mm at baseline. D, Aortic distensibility at baseline and 4 weeks after surgery. C and D, n=12 to 13. E, AngII infusion leads to fatal rupture of TAA or AAA. Surviving mice were euthanized 4 weeks after surgery. Application of LepA abolished TAA rupture (P=0.01, 2‐tailed Fisher exact test). F, A Kaplan–Meier survival curve for mice receiving AngII infusion or AngII cotreated with LepA. G, Leptin was detected in smooth muscle cells (open arrowheads) and atherosclerotic plaques (black arrowhead) but was barely detected in control mice treated with an empty polylactic co–glycolic acid film. Scale bar=50 μm. H and I, Macrophage infiltration in AngII (H) and AngII and LepA‐treated mice (I). Open arrowhead points to infiltrating macrophages, black arrowheads point to macrophages in the adventitia. Scale bar=50 μm. We did not detect macrophages infiltrating the media of LepA‐treated mice (I). J, Number of breaks in aortic elastic lamellas. P=0.05, n=5 to 6. K and L′, Ascending aorta from AngII (K) and AngII and LepA‐treated mice (L) stained by EVG; black arrowheads highlight fragmentation of elastic lamellae. Scale bar=100 μm. M and N′, Immunostaining of α‐SMA in similar location as (K and L′); black arrowhead highlights an absence of α‐SMA. Scale bar=100 μm. *P<0.05, **P<0.01. α‐SMA indicates α‐smooth muscle cell actin; AAA, abdominal aortic aneurysm; AngII, angiotensin II; EVG, elastic Van Gieson; LepA, leptin antagonist; TAA, thoracic aortic aneurysm.
We assessed the exclusive impact of AngII infusion on mouse longevity, with or without additional LepA application at the ascending aorta. To this end, we combined data from the current model with data from a previous experiment, which included a similar cohort of apoE−/− mice from the same breeding house (n=13) and receiving the same dose AngII infusion for the same duration.14 We observed 45% overall mortality (referred to as premature death prior to the completion of the experiment) in 31 mice treated with AngII alone or AngII with control film applied on the ascending aortic surface. Death was caused in 9 mice by rupture of thoracic aortic aneurysm (29%) and in 5 by ruptured AAA (16%). There were 5 cases of aortic dissection, 3 diagnosed in mice that succumbed from ruptured thoracic aortic aneurysm. Strikingly, no mouse cotreated with AngII infusion and locally applied LepA died from rupture of thoracic aneurysm (n=16, P<0.05) (Figure 4E and 4F). The death rate in mice receiving AngII and LepA was 12.5%, and was related exclusively to rupture of AAAs. Notably, LepA application at the ascending aorta did not affect the prevalence of AAA rupture in AngII‐receiving mice.
Histological analysis revealed leptin antigen in medial SMCs of all mice receiving AngII or AngII and LepA, and a strong leptin signal was demonstrated in foam cells of aortic luminal atherosclerotic plaques (Figure 4G). Macrophage infiltration was evident in the media of the ascending aorta in AngII‐treated mice, whereas mice that also received LepA demonstrated lack of macrophages in the media (Figure 4H). Furthermore, AngII‐receiving mice expressed TGF‐β2 in medial SMCs and periadventitial macrophages (Figure S7A and S7B). In contrast, ascending aortas of mice that were locally cotreated with LepA were devoid of TGF‐β2 in medial SMCs and displayed reduced expression of TGF‐β2 in perivascular macrophages (Figure S7C and S7D). The ascending aorta in AngII receivers demonstrated medial hypertrophy, multiple fragmentations of elastic lamellas, and extensive medial α‐SMA depletion. LepA application decreased fragmentation of the elastic lamellas and minimized α‐SMA depletion (Figure 4J–4N) (P=0.05).
Our results demonstrate that locally applied LepA at the ascending aorta reduces macrophage infiltration and moderates the extent of AngII‐induced aortic wall degeneration, thereby preventing subsequent rupture of thoracic aortic aneurysms.
LepA Applied at the Proximal Ascending Aorta Attenuates AngII‐Induced LVH and Valve Leaflet Thickening in the Left Ventricle
Increase in PSV as measured at the aortic valve level was significantly moderated by the addition of LepA to AngII‐infused mice (P<0.05) (Figure 5A). Aortic valve regurgitation was recorded in 8 of 12 mice treated with AngII and in 6 of 13 of those cotreated with LepA. There was a trend toward lower velocity–time integral and peak jet velocity of regurgitation flow in LepA‐treated mice; however, it was short of statistical significance (Figure S6C and S6D). Mice cotreated with LepA presented a lesser degree of LV wall hypertrophy by echocardiography (P<0.01) (Figure 5B). LV diameter increased similarly in both groups in diastole, whereas LepA treatment attenuated the increase in LV diameter during systole (P<0.05) (Figure 5C). Accordingly, fractional area change, which represented LV ejection fraction, was reduced in AngII‐treated mice by 15%, whereas cotreatment with LepA preserved fractional area change at near baseline values (P<0.05) (Figure 5D). LV stroke area was more variable, and the effect of LepA treatment was not statistically significant. Histology revealed increased thickening of aortic and mitral valve leaflets in AngII‐treated mice. This effect was moderated in both valves by local LepA application (P<0.05) (Figure 5E–5I). Hyperplastic aortic and mitral valve leaflets collected from AngII‐treated mice did not demonstrate calcification by alizarin red staining.
Figure 5.
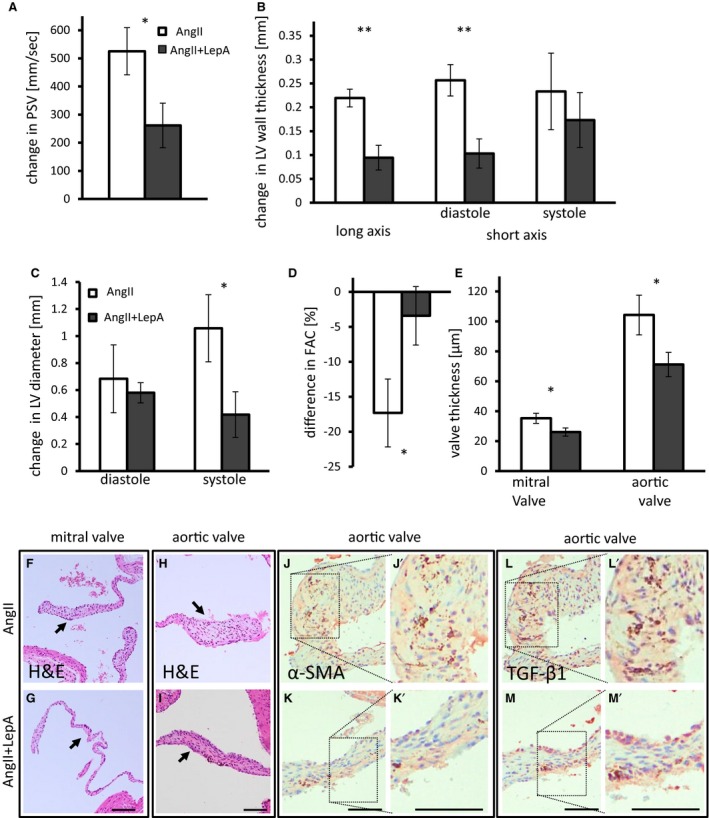
Local application of LepA at the proximal ascending aorta attenuates the effects of AngII infusion on LV remodeling. A, Increase in PSV measured at the aortic valve. P=0.03. AngII‐infused mice shown in white bars; AngII and LepA shown in dark gray bars. Mean PSV at baseline was 1275 mm/s. B, Increase in LV wall thickness as measured by echocardiography in long axis (P<0.01) and in diastole in short axis views (P<0.01). Mean LV wall thickness at baseline was 0.79 mm at anterior and posterior walls and 0.57 mm at the septum. C, Increase in LV diameter in diastole (P=0.047) and systole. Mean LV diameter was 2.3 mm at baseline. D, FAC is reduced in AngII‐infused mice but is not affected in AngII and LepA‐treated mice. P=0.01. Mean FAC was 55% at baseline. E, Mitral and aortic valve leaflets thickness is reduced in AngII and LepA‐treated mice compared with AngII‐infused mice. P=0.03 for both valves. A through E, n=10 to 12. F and G, H&E staining of a mitral valve from AngII (F) and AngII and LepA‐treated mice (G). Arrows point to the valve. H and I, H&E staining of an aortic valve from AngII (H) and AngII and LepA‐treated mice (I). Arrows point to the valve. J and K, α‐SMA is abundant in aortic valves of AngII‐infused mice (J), with relatively weaker expression in AngII and LepA‐treated mice (K). L and M, TGF‐β1 expression is widespread in aortic valves of AngII‐infused mice (L) but is less prevalent in AngII and LepA‐treated mice (M). Scale bar=100 μm (F through M). *P<0.05, **P<0.01. α‐SMA indicates α‐smooth muscle cell actin; AngII, angiotensin II; FAC, fractional area change; H&E, hematoxylin and eosin; LepA, leptin antagonist; LV, left ventricular; PSV, peak systolic velocity; TGF‐β1, transforming growth factor β1.
We observed α‐SMA and TGF‐β1 antigen in stromal cells of aortic valve leaflets (Figure 5J–5M), suggesting activation of those cells. We did not identify statistically significant changes in stromal cell proliferation through Ki‐67 immunohistochemistry (Figure S6D). Mac‐3 and tumor necrosis factor α–positive cells were scarcely found in leaflets from AngII‐treated mice, and fewer cells were positive for those antigens in LepA receivers (Figure S6E and S6F). Collectively, these findings imply that attenuation of remodeling in the ascending aorta by local application of LepA moderates LVH and valvular remodeling.
Leptin Is De Novo Synthesized in Human Aortic Valve Leaflets of AVS Disease
Human samples of advanced AVS disease and normal control valves were analyzed (Figure 6A and 6B). Advanced AVS disease was characterized by the abundance of α‐SMA and CD68‐positive cells, along with strong expression of leptin and leptin receptor (Figure 6C, 6D, and S8A–S8D). Leptin was mostly expressed in valve leaflets by SMC‐like elongated cells and by macrophage‐like round cells (Figure 6E). De novo lep and lepr mRNA synthesis was confirmed by quantitative polymerase chain reaction (Figure 6F) and by in situ hybridization for lep (Figure S8E and S8F). Interestingly, lep and lepr expression was correlated in AVS (Figure 6G).
Figure 6.
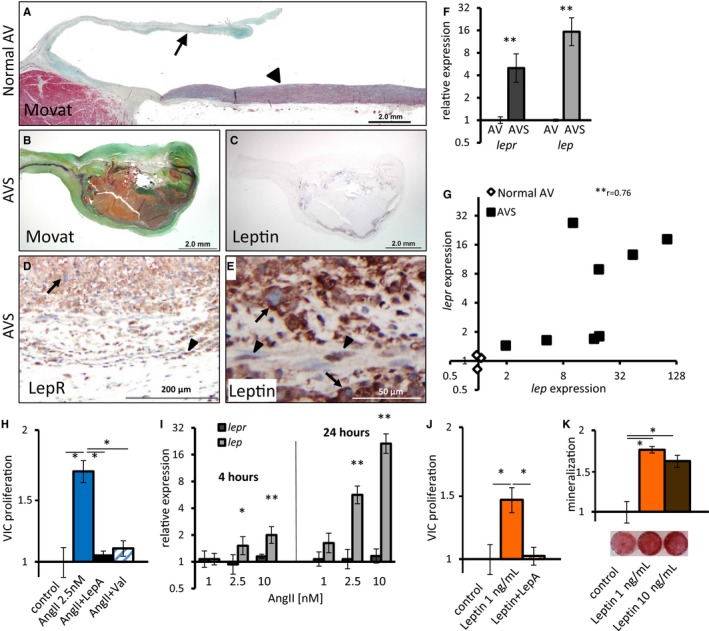
Leptin is expressed in AVs of AVS patients and induces VIC proliferation and calcification in vitro. A, Movat staining of healthy human AV. Arrowhead points to ascending aorta, arrow to AV. B and C, Movat (B) and leptin IHC (C) of AV collected from an AVS patient. D and E, LepR (D) and leptin (E) IHC from the same valve as in (B and C). Arrows point to macrophage‐like cells; arrowheads point to elongated SMC smooth muscle‐like cells. F, qPCR analysis for lep and lepr expression in AVs. n=3 and n=8 for valves of healthy and AVS patients, respectively. G, Positive correlation between lep and lepr expression in AVs. r 2=0.76 (Spearman correlation), P=0.007. H through K, In vitro primary human VICs. H, AngII increases VIC proliferation. This effect is blocked by the AngII type 1 receptor blocker valsartan and LepA. (P=0.01; n=5–6). I, qPCR analysis for lepr and lep expression in VICs treated with AngII for 4 or 24 hours compared with untreated VICs. P=0.046, P=0.007 (4 hours), P<0.001 (24 hours). Error bars represent standard deviation. J, Leptin increases VICs proliferation (P=0.01), and this effect is blocked by LepA (P=0.01; n=6). K, Mineralization of VICs grown in osteogenic medium is increased by leptin (P<0.05; n=3). *P<0.05, **P<0.01. AngII indicates angiotensin II; AV, aortic valve; AVS, aortic valve stenosis; IHC, immunohistochemistry; LepA, leptin antagonist; LepR, leptin receptor; qPCR, quantitative polymerase chain reaction; Val, valsartan; VIC, valve interstitial cell.
Leptin Mediates the Effects of AngII on Human VIC Proliferation
AngII induced VIC proliferation in cell cultures. This effect was blocked by AngII type 1 receptor blocker valsartan and LepA. The latter response indicated that the AngII proliferative effect on VICs is leptin dependent (Figure 6H). Indeed, AngII induced the expression of lep mRNA in VIC cultures in a dose‐ and time‐dependent fashion (Figure 6I). Leptin was sufficient to induce VIC proliferation without AngII (Figure 6J) and enhanced VIC mineralization in osteogenic medium (Figure 6K). Remarkably, a similar leptin dose (1 ng/mL) drove VIC proliferation and mineralization. It should be noted that VICs in culture are susceptible to chemical and mechanical properties of culture media, which may promote mineralization.
Our in vitro data corroborate the human aortic valve analysis, suggesting that AngII‐induced leptin may drive early aortic valve hyperplastic lesions and further stimulate VICs’ osteogenic differentiation.
Discussion
By using a mouse model, we showed that local leptin activity in the proximal ascending aortic wall mediates pathological changes and aneurysm formation. These processes subsequently promote remodeling of the LV wall and valves. Because the effects of leptin are local, we hypothesize that hemodynamic AVC leads to the cardiac effect. Delivery of LepA to the proximal ascending aorta in mice receiving AngII infusion attenuated local aortic aneurysm formation, rescued mice from ATAA rupture, and decreased LV remodeling. This observation suggests that local antileptin treatment is a potential therapeutic avenue for early stage ATAA as well as related LVH and early aortic and mitral valve hyperplastic lesions (Figure 7).
Figure 7.
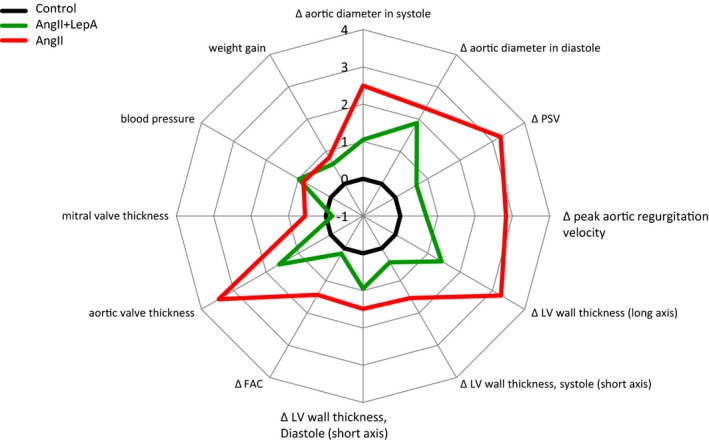
Effects of LepA application at the surface of the ascending aorta on AngII‐induced thoracic aortic aneurysm formation and left ventricular remodeling. A radar plot of quantitative cardiovascular parameters assessed in this study demonstrating the impact of LepA applied on the surface of the ascending aorta in AngII‐infused mice. The distance from center is measured by a Z score calculated from the standard deviation in mice treated with an empty polylactic co–glycolic acid film (black, control). Red line corresponds to the AngII‐infused mice alone, whereas the green line corresponds to the AngII and LepA‐treated mice. Moving clockwise, LepA application attenuates the effects of AngII on ascending aortic diameter, hemodynamics at the LV outlet, LV wall thickness, FAC, and LV valves’ leaflets thickness. There is no effect on blood pressure or weight gain. AngII indicates angiotensin II; FAC, fractional area change; LepA, leptin antagonist; LV, left ventricular; PSV, peak systolic velocity.
Multiple AngII‐related ascending aortic aneurysms that were diagnosed in patients suffering from various diseases displayed de novo leptin synthesis, corroborating the design and results of our mouse model. AngII infusion in mouse models induces leptin synthesis in arterial wall SMCs and macrophages. Leptin was previously shown to modulate multiple gene expression in the aortic wall.14 In addition, leptin elicits macrophage chemotaxis, and macrophages have been shown infiltrating the arterial media and periadventitial tissue in the AngII‐infusion mouse model. In the current study, we also showed that AngII‐infused mice exhibit increased expression of TGF‐β2 in medial SMCs and in periadventitial macrophages at the dilated segment of ascending aorta. These results corroborate other models of ascending aortic aneurysm such as Marfan syndrome. It is conceivable that local LepA application in AngII‐treated mice affects the ascending aorta by reduction of macrophage recruitment and inhibition of local TGF‐β2 synthesis.41
Stiffening and dilation of the proximal ascending aorta in patients with ATAA led to LV remodeling, possibly via AVC activation related to perturbation of hemodynamics at the LV outlet.32 A similar AVC mechanism was observed in mice receiving systemic AngII infusion and in those that received periadventitial low‐dose leptin on the ascending aorta. Increased ascending aortic diameter in mice undergoing local leptin application and in AngII‐treated mice may have generated hemodynamic perturbations that were partially reflected by elevated PSV and promoted LV wall remodeling and valve leaflet thickening. The observed decrease in fractional area change was attributed to concentric LV hypertrophy and represented reduced ejection fraction, correlating with systolic and diastolic dysfunction in the clinical setting.
Stiffening and dilation of the proximal ascending aorta in patients with ATAA led to LV remodeling by AVC via stretch‐related mechanisms.32 Analogously, segmental ascending aortic aneurysm and AVC activation were generated in both of our mouse models. Hemodynamic perturbations were partially reflected by elevated PSV, and both models displayed LV hypertrophy as well as left heart valve leaflet hyperplasia. The leptin application model was designed to evaluate the exclusive impact of locally synthesized leptin on the ascending aortic wall in normotensive mice. The generated ATAA drove LV remodeling, but no aortic valve regurgitation was evident. These results suggest that leptin‐induced ATAA increased impedance to LV outflow that was sufficient to activate the AVC mechanism driving LV remodeling; however, shortcomings of this model, including lack of background hypertension and short follow‐up period, may limit the translation of our findings into the clinical arena. In contrast, our AngII infusion model offers pathophysiological conditions that likely resemble clinical scenarios. Mice that received AngII presented similar changes in the ascending aorta and left ventricle and exhibited a varying degree of aortic regurgitation. These results might be linked, at least in part, to the apoE−/− genetic background42; however, the application of LepA to the ascending aortic wall not only moderated local aneurysmal changes but also caused a trend toward reduced peak jet velocity of regurgitation flow. The short‐duration exposure to AngII infusion likely only partially simulated the clinical circumstances of long‐term exposure encountered in patients that develop ATAA. Consequently, our AngII infusion model results may support the validity of aortic regurgitation in clinical scenarios of ATAA‐related LV remodeling. These findings are consistent with clinical data showing that reduced aortic elasticity and dilation is associated with aortic regurgitation and LV hypertrophy in patients with nonstenotic bicuspid aortic valve.43
Direct impact of the locally applied leptin or leptin antagonist through tissue diffusion on LV remodeling was evidently ruled out because the effect of both substances was not displayed beyond the immediate adjacent ascending aortic wall.
Multiple clinical data implicate AngII in AVS.35 In this light, recent experimental findings of aortic valve leaflet thickening in AngII‐infused mice are consistent with mechanisms of human AVS pathogenesis.36 We observed thickening of aortic and mitral valve leaflets in both AngII‐infused mice and in mice receiving leptin locally on the ascending aorta. This suggests that systemic and hemodynamic conditions within the left ventricle may affect both valves in a similar manner. Increased numbers of stromal cells, which are histologically and functionally analogous to human VICs,44, 45 contributed to thickening of valve leaflets. This is consistent with our in vitro result showing that AngII‐induced human VIC proliferation is leptin dependent. Leptin was found to be both sufficient and necessary for the proliferation of VICs in this context. Moreover, we demonstrated de novo leptin synthesis in advanced human AVS samples.
The clinical, in vitro, and mouse model data are further substantiated by the epidemiological association between chronic hyperleptinemia and advanced AVS disease to support a novel role for leptin in AVS pathogenesis.34 Specifically, thickening of aortic and mitral valve leaflets could be driven by valvular in situ synthesis of leptin. Furthermore, this idea implies that hyperplasia of aortic and mitral valve leaflets may be associated with AVC activation. This process is likely mediated by elevated LV AngII signaling, which in turn promotes valvular in situ leptin synthesis. Leaflet hyperplasia is an early event preceding subsequent macrophage infiltration and cellular mineralization interacting with valve biomechanics,46 constituting a chain of events leading to advanced AVS in humans (Figure 8). Hyperplastic lesions in mouse aortic valves in both models exhibited increased expression of TGF‐β1. Interestingly, TGF‐β signaling promotes VIC proliferation and mineralization in vitro and in a clinical setting.47, 48
Figure 8.
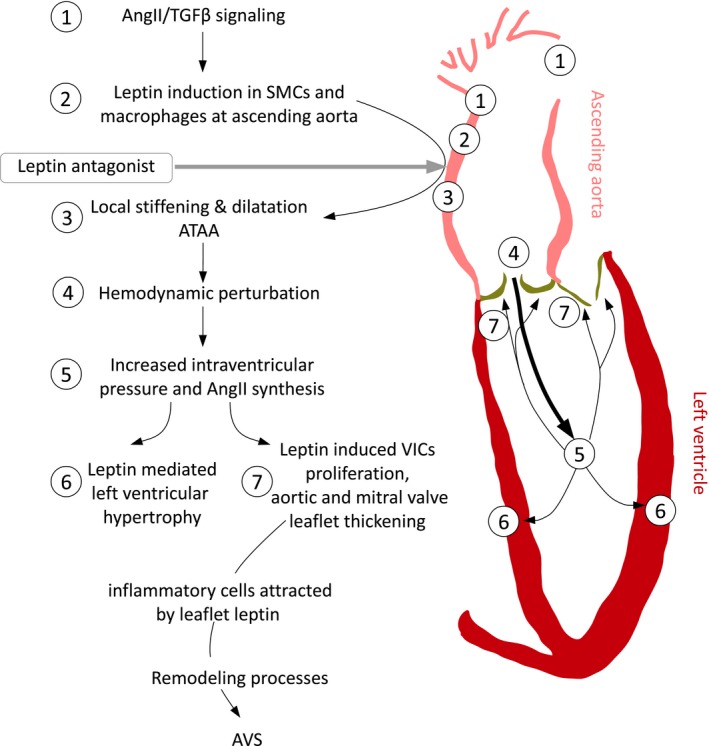
A model of a mechanism for AVS initiation through AngII‐induced ATAA, associated with AVC activation. The numerical sequence of events corresponds to findings discussed and shown in this study: Leptin‐mediated ATAA formation, AVC activation underlying left ventricular hypertrophy and aortic valve thickening through leptin‐induced VIC proliferation. AngII indicates angiotensin II; ATAA, ascending thoracic aortic aneurysm; AVC, aortoventricular coupling; AVS, aortic valve stenosis; SMC, smooth muscle cell; TGF‐β, transforming growth factor β; VIC, valve interstitial cell.
Local leptin application at the ascending aorta in normotensive mice drove LVH and aortic and mitral valve leaflet thickening, whereas concomitant LepA treatment in AngII‐infused mice moderated AVC activation, preventing LV dysfunction and valvular hyperplastic lesions. Remarkably, the inhibitory effects of local LepA on LVH and aortic and mitral valve leaflet thickening occurred despite continuous systemic AngII administration and subsequent hypertension. These results underscore the significance of AVC activation as an underappreciated force driving LV remodeling and aortic valve disease.49, 50
The current study has several limitations. Because we found wide variability between individual mice when assessing their physiological parameters at baseline, we avoided the use of absolute numbers in our analyses and compared each parameter with its baseline value per mouse as an internal control. Inherent limitations of the mouse model did not allow direct measurement of aortic and LV pressure or assessment of pulse wave velocity, and this impaired our ability to directly assess hemodynamics. In addition, confirmation of LVH was based on echocardiography and was not validated by heart weight or histological morphometry of cardiomyocytes. Although we found similarities in mitral and aortic valve leaflet thickening, mitral valve remodeling was not within the scope of the current study. Cytokines and matrix metalloproteinases associated with LepA effects on the vessel wall were not examined in these models, and we did not explore leptin‐independent mechanisms of ascending aortic aneurysm formation that function through macrophage recruitment and signaling.51 These factors warrant a dedicated study. Finally, nonoptimized dose and delivery for both leptin and LepA present a limiting factor whereby fine tuning may result in even more pronounced ATAA‐related benefits. Furthermore, longer term in vivo studies are warranted because our analysis was limited to 4 weeks of AngII infusion and may not represent the prolonged cardiovascular response analogous to the clinical setting.
The efficacy of local LepA therapy in our mouse model and expression of leptin in human ATAA and AVS has several clinical implications. The prominent role of AngII in cardiovascular remodeling has positioned renin angiotensin aldosterone system antagonists as front‐line therapy for ATAA, LVH, and advanced AVS52, 53, 54; however, systemic therapy with AngII receptor blockers or angiotensin‐converting enzyme inhibitors have achieved limited success, especially in attenuating ATAA expansion and AVS progression. In contrast, we have witnessed promising results in AngII‐infused mice receiving LepA at the ascending aorta. These results imply that we may gain control over the course of ascending aortic aneurysm progression and also attenuate AVC‐related cardiac pathologies. Importantly, local LepA benefit would obviate systemic hormonal perturbation. Targeted local LepA application by minimally invasive modes, such as an intravascular approach, may prove effective in slowing down the progression of AngII‐driven aortic aneurysm at any location and control peripheral aneurysms likewise.
In summary, AngII‐induced leptin in the ascending aorta is a prerequisite to driving local aneurysm formation in apoE−/− mice. We propose an analogous AngII‐associated pathway underlying ATAA in patients suffering from hypertension, obesity, diabetes mellitus, or associated genetically linked vascular diseases. This relationship is particularly important considering ATAA‐related hemodynamic perturbations, which are known to promote LVH and hyperplastic lesions in left‐sided heart valves in mice and humans. We further suggest that valvular in situ leptin synthesis could promote early valve leaflet lesions as a precursor to human AVS. Finally, our findings demonstrate that local inhibition of leptin activity at the ascending aorta attenuates ATAA and its associated cardiac pathologies. These results put forward a novel proof of concept that could be implemented clinically to prevent the progression of early aortic aneurysms.
Sources of Funding
This study was funded by a private philanthropic donation to Schneiderman through Brigham and Women's Hospital, Boston, MA; RO1 DK094434‐04 to Virmani at CVpath, Gaithersburg, MD; and a grant from the German Research Foundation (Deutsche Forschungsgemeinschaft; Scha808/7‐1) to Schäfer. D. Ben‐Zvi was supported by Yad Hanadiv post‐doctoral fellowship and by HFSP post‐doctoral fellowship.
Disclosures
A. G. is the CEO of Protein Laboratories Rehovot (PLR) Ltd company that manufactures the mouse mutant leptin antagonist. All other authors declare no conflict of interest.
Supporting information
Data S1. Methods and materials.
Figure S1. Movat's pentachrome and immunohistochemistry for leptin and leptin receptor (LepR) in ascending thoracic aortic aneurysm samples from bicuspid aortic valve (BAV) and Marfan syndrome (MFS) patients: subintimal and medial region. A through C, BAV patient: Movat's pentachrome (A), diffuse elastic fiber fragmentation with glycosaminoglycan deposition (bluish‐green) and media degeneration, leptin (B), and LepR (C) immunohistochemistry, expressed by smooth muscle cells (SMCs). D through F, MFS patient: Movat's pentachrome (D), histology similar to (A), leptin antigen (E) expressed mostly by medial macrophages, LepR antigen (F) prominently expressed in macrophages and SMCs. A and D, Scale bar=500 μm, 200 μm in (B, C, E, and F).
Figure S2. Immunohistochemical analysis of ascending thoracic aortic aneurysm (ATAA) samples from bicuspid aortic valve (BAV) and Marfan syndrome (MFS) patients: subintimal and medial region. A through F, Analyses were performed on BAV‐related ATAA sample from a case shown in Figure S1A through S1C. A and C, α‐Smooth muscle actin (α‐SMA) staining marking medial smooth muscle cells (SMCs) (A) and capillary SMCs (C). B and D, CD68 marking macrophages was not identified in subintimal and medial region cells (B) but was evident in adventitial pericapillary cells (D). E and F, Leptin antigen was expressed by pericapillary macrophages and surrounding adventitial SMCs (E) and leptin receptor expressed mainly by medial SMCs (F). A through F, Scale bar=100 μm. G and H, Analysis performed on MFS‐related ATAA sample (same case, subintimal and medial region like in Figure S1D through S1F). α‐SMA staining marking SMCs (G) and CD68 marking macrophages (H). Scale bar=200 μm.
Figure S3. Effects of local leptin application on weight, systolic blood pressure, and aortic valve proliferation. A, An intraoperative view showing the slow‐release polylactic co–glycolic acid film (yellow outline) placed adjacent to the ascending aorta. This strategy was used to apply the leptin slow‐release film, leptin antagonist, and control films, as described in this study. B, Weight of mice after receiving local leptin or control film. There was no significant difference in the weight between groups. n=10 to 11. C, Systolic blood pressure of mice after receiving local leptin or control film. There was no significant difference in blood pressure between groups. n=10 to 11. D, Increase in aortic valve regurgitation peak velocity following local treatment with leptin or control film. All animals displayed some regurgitation jet. Mean baseline velocity was 1000 mm/s. E, Increase in aortic valve regurgitation velocity–time integral (VTI). One of 12 control animals displayed above baseline regurgitation, defined as VTI at least 10‐fold greater than mean, 0 of 10 treated animals had regurgitation. Mean baseline VTI was 2.8 mm. F, Change in left ventricular stroke area. n=10 to 11. Mean baseline stroke area was 5.9 mm2. G, Percentage of Ki‐67–positive stromal cells in aortic valve leaflets of mice receiving leptin or control film. Although there was a trend of increased Ki‐67–positive cells in leptin receivers vs controls, it did not reach statistical significance, n=4 to 5.
Figure S4. Modulation of the ascending aorta in mice receiving high‐fat diet (HFD) feeding treated with control or leptin film. A, Aortic diameter at peak systole in mice on HFD treated with leptin or control film. After 3 weeks, a significant difference was recorded between the two groups, P<0.01. n=5 to 6. B and C, Elastic Van Gieson staining of the ascending aorta in local leptin‐treated (B) or control (C) mice. Arrows in high magnification show fragmentation of elastic lamellas in leptin‐treated mice. D and E, α‐Smooth muscle actin (α‐SMA) staining of the ascending aorta in local leptin‐treated (D) or control (E) mice. Arrows indicate depletion of α‐SMA. F and G, α‐SMA staining of the descending aorta in local leptin‐treated (F) or control (G) mice. Leptin was applied to the ascending aorta. Note, medial smooth muscle cell layer looks intact. Scale bar=500 μm for all images, 100 μm in high‐magnification images.
Figure S5. Cardiac and valvular modulation in mice on a high‐fat diet (HFD) treated with local leptin or control film. A, Change in peak systolic velocity (PSV) in control and leptin‐treated mice fed with an HFD. P=0.04. Baseline PSV was 1300 mm/s. B, Left ventricular (LV) wall thickness in leptin‐treated vs control HFD‐fed mice, as measured by echocardiography 6 weeks after surgery in the long‐axis view and in short‐axis view during diastole. P<0.01, n=5 to 6. Mean baseline LV wall thickness was 0.85 mm. C, Mitral and aortic valve thickness measured by histology 6 weeks after surgery. P<0.01 (mitral), P=0.047 (aortic). D and E, Hematoxylin and eosin (H&E) staining of mitral valve leaflets in local leptin‐treated (C) and control (E) mice. F and G, H&E staining of aortic valve leaflets in local leptin‐treated (D) and control (F) mice. H and I, Transforming growth factor β2 staining of aortic valve leaflets in local leptin‐treated (G) and control (H) mice. Positive signal indicates activation of stromal cells. J and K, α‐Smooth muscle actin staining of aortic valve leaflets in local leptin‐treated (I) and control (J) mice. Scale bar=500 μm for all images, 100 μm in enlargements.
Figure S6. Effects of local application of leptin antagonist (LepA) to the ascending aorta following angiotensin II (AngII) infusion. A, Weight of mice following treatment. There was no significant difference in weight between groups. n=11 to 12. B, Systolic blood pressure of mice after treatment. There was no significant difference in systolic blood pressure between groups. n=11 to 12. C, Increase in aortic valve regurgitation peak velocity following local treatment with leptin or control film. All animals displayed some regurgitation jet. Mean baseline peak velocity was 1050 mm/s. D, Increase in aortic valve regurgitation velocity–time integral. Eight of 12 AngII animals displayed aortic regurgitation, defined as at least 10‐fold increase over baseline mean, and 6 of 13 animals cotreated with LepA had aortic regurgitation, defined similarly. Mean baseline velocity time integral was 4.2 mm. E, Change in LV stroke area. n=11 to 12. Mean baseline stroke area was 6 mm. F, Percentage of Ki‐67–positive cells in stromal cells in the aortic valve leaflets. n=4 to 5. G and H, Mac3 staining of macrophages in aortic valve leaflets, in mice treated with AngII (G) or AngII and LepA (H). Scale bar=500 μm.
Figure S7. Transforming growth factor β2 (TGF‐β2) localization in the ascending aorta following angiotensin II (AngII) infusion. Immunostaining for TGF‐β2 in ascending aortas of mice treated with AngII (A and B), AngII and leptin antagonist (LepA) (C and D), or an empty film as control (E and F). Note, a weak to moderate TGF‐β2 signal in medial smooth muscle cells (SMCs) and strong expression in periadventitial macrophages in AngII‐receiving mice. When adding concomitant local LepA therapy, TGF‐β2 antigen is absent in SMCs and decreased in perivascular tissue, similar to controls. Scale bar=50 μm.
Figure S8. Immunohistochemical and in situ mRNA hybridization in stenotic aortic valves. A and B, α‐Smooth muscle actin (α‐SMA) (A) and CD68 (B) staining in advanced aortic valve stenosis (AVS) disease. Scale bar=2 mm. C and D, High magnification of α‐SMA (C) and leptin (D) staining in advanced AVS. Scale bar=200 μm. E and F, In situ hybridization for lep mRNA transcript in aortic valve leaflets from advanced AVS. Arrowheads (E) denote round macrophage‐like cells, arrows (F) denote elongated SMC‐like cells. Scale bar=200 μm.
Acknowledgments
We thank C. Keith Ozaki, MD for hosting JS as a visiting research scientist in the Vascular Surgery Research Laboratory, Brigham and Women's Hospital; Kristyn S, Masters, PhD for generous donation of human VICs; Eugenia Shwartz for excellent technical assistance; and Jonathan I. Schneiderman, PhD for critical review of the manuscript.
(J Am Heart Assoc. 2016;5:e003474 doi: 10.1161/JAHA.116.003474)
References
- 1. Davies RR, Goldstein LJ, Coady MA, Tittle SL, Rizzo JA, Kopf GS, Elefteriades JA. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg. 2002;73:17–27; discussion 27‐8. [DOI] [PubMed] [Google Scholar]
- 2. Danyi P, Elefteriades JA, Jovin IS. Medical therapy of thoracic aortic aneurysms: are we there yet? Circulation. 2011;124:1469–1476. [DOI] [PubMed] [Google Scholar]
- 3. Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation. 2005;111:816–828. [DOI] [PubMed] [Google Scholar]
- 4. Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tadros TM, Klein MD, Shapira OM. Ascending aortic dilatation associated with bicuspid aortic valve: pathophysiology, molecular biology, and clinical implications. Circulation. 2009;119:880–890. [DOI] [PubMed] [Google Scholar]
- 6. Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC III. Angiotensin II blockade and aortic‐root dilation in Marfan's syndrome. N Engl J Med. 2008;358:2787–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, van Erp C, Gerber EE, Parker SJ, Sauls K, Judge DP, Cooke SK, Lindsay ME, Rouf R, Myers L, ap Rhys CM, Kent KC, Norris RA, Huso DL, Dietz HC. Angiotensin II‐dependent TGF‐beta signaling contributes to Loeys‐Dietz syndrome vascular pathogenesis. J Clin Invest. 2014;124:448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silverberg D, Younis A, Savion N, Harari G, Yakubovitch D, Sheick Yousif B, Halak M, Grossman E, Schneiderman J. Long‐term renin‐angiotensin blocking therapy in hypertensive patients with normal aorta may attenuate the formation of abdominal aortic aneurysms. J Am Soc Hypertens. 2014;8:571–577. [DOI] [PubMed] [Google Scholar]
- 9. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E‐deficient mice. J Clin Invest. 2000;105:1605–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci. 2010;118:681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Favreau JT, Nguyen BT, Gao I, Yu P, Tao M, Schneiderman J, Gaudette GR, Ozaki CK. Murine ultrasound imaging for circumferential strain analyses in the angiotensin II abdominal aortic aneurysm model. J Vasc Surg. 2012;56:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raaz U, Zollner AM, Schellinger IN, Toh R, Nakagami F, Brandt M, Emrich FC, Kayama Y, Eken S, Adam M, Maegdefessel L, Hertel T, Deng A, Jagger A, Buerke M, Dalman RL, Spin JM, Kuhl E, Tsao PS. Segmental aortic stiffening contributes to experimental abdominal aortic aneurysm development. Circulation. 2015;131:1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tao M, Yu P, Nguyen BT, Mizrahi B, Savion N, Kolodgie FD, Virmani R, Hao S, Ozaki CK, Schneiderman J. Locally applied leptin induces regional aortic wall degeneration preceding aneurysm formation in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2013;33:311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y, Ait‐Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadiere C, Renia L, Johnson JL, Tharaux PL, Tedgui A, Mallat Z. TGF‐beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II‐infused mice. J Clin Invest. 2010;120:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martinez‐Martinez E, Miana M, Jurado‐Lopez R, Bartolome MV, Souza Neto FV, Salaices M, Lopez‐Andres N, Cachofeiro V. The potential role of leptin in the vascular remodeling associated with obesity. Int J Obes (Lond). 2014;38:1565–1572. [DOI] [PubMed] [Google Scholar]
- 17. Rosenbaum M, Nicolson M, Hirsch J, Heymsfield SB, Gallagher D, Chu F, Leibel RL. Effects of gender, body composition, and menopause on plasma concentrations of leptin. J Clin Endocrinol Metab. 1996;81:3424–3427. [DOI] [PubMed] [Google Scholar]
- 18. Schneiderman J, Schaefer K, Kolodgie FD, Savion N, Kotev‐Emeth S, Dardik R, Simon AJ, Halak M, Pariente C, Engelberg I, Konstantinides S, Virmani R. Leptin locally synthesized in carotid atherosclerotic plaques could be associated with lesion instability and cerebral emboli. J Am Heart Assoc. 2012;1:e001727 doi: 10.1161/JAHA.112.001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cecelja M, Chowienczyk P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc Dis. 2012;1:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ejiri J, Inoue N, Tsukube T, Munezane T, Hino Y, Kobayashi S, Hirata K, Kawashima S, Imajoh‐Ohmi S, Hayashi Y, Yokozaki H, Okita Y, Yokoyama M. Oxidative stress in the pathogenesis of thoracic aortic aneurysm: protective role of statin and angiotensin II type 1 receptor blocker. Cardiovasc Res. 2003;59:988–996. [DOI] [PubMed] [Google Scholar]
- 21. Parhami F, Tintut Y, Ballard A, Fogelman AM, Demer LL. Leptin enhances the calcification of vascular cells: artery wall as a target of leptin. Circ Res. 2001;88:954–960. [DOI] [PubMed] [Google Scholar]
- 22. Simonds SE, Pryor JT, Ravussin E, Greenway FL, Dileone R, Allen AM, Bassi J, Elmquist JK, Keogh JM, Henning E, Myers MG Jr, Licinio J, Brown RD, Enriori PJ, O'Rahilly S, Sternson SM, Grove KL, Spanswick DC, Farooqi IS, Cowley MA. Leptin mediates the increase in blood pressure associated with obesity. Cell. 2014;159:1404–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soderberg S, Ahren B, Jansson JH, Johnson O, Hallmans G, Asplund K, Olsson T. Leptin is associated with increased risk of myocardial infarction. J Intern Med. 1999;246:409–418. [DOI] [PubMed] [Google Scholar]
- 24. Sun Z. Aging, arterial stiffness, and hypertension. Hypertension. 2015;65:252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boonyasirinant T, Rajiah P, Setser RM, Lieber ML, Lever HM, Desai MY, Flamm SD. Aortic stiffness is increased in hypertrophic cardiomyopathy with myocardial fibrosis: novel insights in vascular function from magnetic resonance imaging. J Am Coll Cardiol. 2009;54:255–262. [DOI] [PubMed] [Google Scholar]
- 26. Eren M, Gorgulu S, Uslu N, Celik S, Dagdeviren B, Tezel T. Relation between aortic stiffness and left ventricular diastolic function in patients with hypertension, diabetes, or both. Heart. 2004;90:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Redheuil A, Yu WC, Mousseaux E, Harouni AA, Kachenoura N, Wu CO, Bluemke D, Lima JA. Age‐related changes in aortic arch geometry: relationship with proximal aortic function and left ventricular mass and remodeling. J Am Coll Cardiol. 2011;58:1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mottram PM, Haluska BA, Leano R, Carlier S, Case C, Marwick TH. Relation of arterial stiffness to diastolic dysfunction in hypertensive heart disease. Heart. 2005;91:1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cruickshank K, Riste L, Anderson SG, Wright JS, Dunn G, Gosling RG. Aortic pulse‐wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation. 2002;106:2085–2090. [DOI] [PubMed] [Google Scholar]
- 30. Huggins CE, Domenighetti AA, Pedrazzini T, Pepe S, Delbridge LM. Elevated intracardiac angiotensin II leads to cardiac hypertrophy and mechanical dysfunction in normotensive mice. J Renin Angiotensin Aldosterone Syst. 2003;4:186–190. [DOI] [PubMed] [Google Scholar]
- 31. Schunkert H, Dzau VJ, Tang SS, Hirsch AT, Apstein CS, Lorell BH. Increased rat cardiac angiotensin converting enzyme activity and mRNA expression in pressure overload left ventricular hypertrophy. Effects on coronary resistance, contractility, and relaxation. J Clin Invest. 1990;86:1913–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zablocki D, Sadoshima J. Solving the cardiac hypertrophy riddle: the angiotensin II‐mechanical stress connection. Circ Res. 2013;113:1192–1195. [DOI] [PubMed] [Google Scholar]
- 33. Rajapurohitam V, Javadov S, Purdham DM, Kirshenbaum LA, Karmazyn M. An autocrine role for leptin in mediating the cardiomyocyte hypertrophic effects of angiotensin II and endothelin‐1. J Mol Cell Cardiol. 2006;41:265–274. [DOI] [PubMed] [Google Scholar]
- 34. Glader CA, Birgander LS, Soderberg S, Ildgruben HP, Saikku P, Waldenstrom A, Dahlen GH. Lipoprotein(a), Chlamydia pneumoniae, leptin and tissue plasminogen activator as risk markers for valvular aortic stenosis. Eur Heart J. 2003;24:198–208. [DOI] [PubMed] [Google Scholar]
- 35. Helske S, Lindstedt KA, Laine M, Mayranpaa M, Werkkala K, Lommi J, Turto H, Kupari M, Kovanen PT. Induction of local angiotensin II‐producing systems in stenotic aortic valves. J Am Coll Cardiol. 2004;44:1859–1866. [DOI] [PubMed] [Google Scholar]
- 36. Fujisaka T, Hoshiga M, Hotchi J, Takeda Y, Jin D, Takai S, Hanafusa T, Ishizaka N. Angiotensin II promotes aortic valve thickening independent of elevated blood pressure in apolipoprotein‐E deficient mice. Atherosclerosis. 2013;226:82–87. [DOI] [PubMed] [Google Scholar]
- 37. Le Quang K, Bouchareb R, Lachance D, Laplante MA, El Husseini D, Boulanger MC, Fournier D, Fang XP, Avramoglu RK, Pibarot P, Deshaies Y, Sweeney G, Mathieu P, Marette A. Early development of calcific aortic valve disease and left ventricular hypertrophy in a mouse model of combined dyslipidemia and type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2014;34:2283–2291. [DOI] [PubMed] [Google Scholar]
- 38. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shpilman M, Niv‐Spector L, Katz M, Varol C, Solomon G, Ayalon‐Soffer M, Boder E, Halpern Z, Elinav E, Gertler A. Development and characterization of high affinity leptins and leptin antagonists. J Biol Chem. 2011;286:4429–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation. 2004;110:3849–3857. [DOI] [PubMed] [Google Scholar]
- 41. Leung JC, Chan LY, Tang SC, Chu KM, Lai KN. Leptin induces TGF‐beta synthesis through functional leptin receptor expressed by human peritoneal mesothelial cell. Kidney Int. 2006;69:2078–2086. [DOI] [PubMed] [Google Scholar]
- 42. Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386. [DOI] [PubMed] [Google Scholar]
- 43. Grotenhuis HB, Ottenkamp J, Westenberg JJM, Bax JJ, Kroft LJM, de Roos A. Reduced aortic elasticity and dilation are associated with aortic regurgitation and left ventricular hypertrophy in nonstenotic bicuspid aortic valve patients. J Am Coll Cardiol. 2007;49:1660–1665. [DOI] [PubMed] [Google Scholar]
- 44. Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113:1344–1352. [DOI] [PubMed] [Google Scholar]
- 46. Schoen FJ. Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology, and tissue engineering. Circulation. 2008;118:1864–1880. [DOI] [PubMed] [Google Scholar]
- 47. Jian B, Narula N, Li QY, Mohler ER III, Levy RJ. Progression of aortic valve stenosis: TGF‐beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465; discussion 465‐6. [DOI] [PubMed] [Google Scholar]
- 48. Merryman WD, Lukoff HD, Long RA, Engelmayr GC Jr, Hopkins RA, Sacks MS. Synergistic effects of cyclic tension and transforming growth factor‐beta1 on the aortic valve myofibroblast. Cardiovasc Pathol. 2007;16:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic‐valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999;341:142–147. [DOI] [PubMed] [Google Scholar]
- 50. Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–334. [DOI] [PubMed] [Google Scholar]
- 51. Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II‐induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009;29:1458–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bisping E, Wakula P, Poteser M, Heinzel FR. Targeting cardiac hypertrophy: toward a causal heart failure therapy. J Cardiovasc Pharmacol. 2014;64:293–305. [DOI] [PubMed] [Google Scholar]
- 53. Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, Pearson GD, Selamet Tierney ES, Levine JC, Atz AM, Benson DW, Braverman AC, Chen S, De Backer J, Gelb BD, Grossfeld PD, Klein GL, Lai WW, Liou A, Loeys BL, Markham LW, Olson AK, Paridon SM, Pemberton VL, Pierpont ME, Pyeritz RE, Radojewski E, Roman MJ, Sharkey AM, Stylianou MP, Wechsler SB, Young LT, Mahony L; Pediatric Heart Network I . Atenolol versus losartan in children and young adults with Marfan's syndrome. N Engl J Med. 2014;371:2061–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nadir MA, Wei L, Elder DH, Libianto R, Lim TK, Pauriah M, Pringle SD, Doney AD, Choy AM, Struthers AD, Lang CC. Impact of renin‐angiotensin system blockade therapy on outcome in aortic stenosis. J Am Coll Cardiol. 2011;58:570–576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Methods and materials.
Figure S1. Movat's pentachrome and immunohistochemistry for leptin and leptin receptor (LepR) in ascending thoracic aortic aneurysm samples from bicuspid aortic valve (BAV) and Marfan syndrome (MFS) patients: subintimal and medial region. A through C, BAV patient: Movat's pentachrome (A), diffuse elastic fiber fragmentation with glycosaminoglycan deposition (bluish‐green) and media degeneration, leptin (B), and LepR (C) immunohistochemistry, expressed by smooth muscle cells (SMCs). D through F, MFS patient: Movat's pentachrome (D), histology similar to (A), leptin antigen (E) expressed mostly by medial macrophages, LepR antigen (F) prominently expressed in macrophages and SMCs. A and D, Scale bar=500 μm, 200 μm in (B, C, E, and F).
Figure S2. Immunohistochemical analysis of ascending thoracic aortic aneurysm (ATAA) samples from bicuspid aortic valve (BAV) and Marfan syndrome (MFS) patients: subintimal and medial region. A through F, Analyses were performed on BAV‐related ATAA sample from a case shown in Figure S1A through S1C. A and C, α‐Smooth muscle actin (α‐SMA) staining marking medial smooth muscle cells (SMCs) (A) and capillary SMCs (C). B and D, CD68 marking macrophages was not identified in subintimal and medial region cells (B) but was evident in adventitial pericapillary cells (D). E and F, Leptin antigen was expressed by pericapillary macrophages and surrounding adventitial SMCs (E) and leptin receptor expressed mainly by medial SMCs (F). A through F, Scale bar=100 μm. G and H, Analysis performed on MFS‐related ATAA sample (same case, subintimal and medial region like in Figure S1D through S1F). α‐SMA staining marking SMCs (G) and CD68 marking macrophages (H). Scale bar=200 μm.
Figure S3. Effects of local leptin application on weight, systolic blood pressure, and aortic valve proliferation. A, An intraoperative view showing the slow‐release polylactic co–glycolic acid film (yellow outline) placed adjacent to the ascending aorta. This strategy was used to apply the leptin slow‐release film, leptin antagonist, and control films, as described in this study. B, Weight of mice after receiving local leptin or control film. There was no significant difference in the weight between groups. n=10 to 11. C, Systolic blood pressure of mice after receiving local leptin or control film. There was no significant difference in blood pressure between groups. n=10 to 11. D, Increase in aortic valve regurgitation peak velocity following local treatment with leptin or control film. All animals displayed some regurgitation jet. Mean baseline velocity was 1000 mm/s. E, Increase in aortic valve regurgitation velocity–time integral (VTI). One of 12 control animals displayed above baseline regurgitation, defined as VTI at least 10‐fold greater than mean, 0 of 10 treated animals had regurgitation. Mean baseline VTI was 2.8 mm. F, Change in left ventricular stroke area. n=10 to 11. Mean baseline stroke area was 5.9 mm2. G, Percentage of Ki‐67–positive stromal cells in aortic valve leaflets of mice receiving leptin or control film. Although there was a trend of increased Ki‐67–positive cells in leptin receivers vs controls, it did not reach statistical significance, n=4 to 5.
Figure S4. Modulation of the ascending aorta in mice receiving high‐fat diet (HFD) feeding treated with control or leptin film. A, Aortic diameter at peak systole in mice on HFD treated with leptin or control film. After 3 weeks, a significant difference was recorded between the two groups, P<0.01. n=5 to 6. B and C, Elastic Van Gieson staining of the ascending aorta in local leptin‐treated (B) or control (C) mice. Arrows in high magnification show fragmentation of elastic lamellas in leptin‐treated mice. D and E, α‐Smooth muscle actin (α‐SMA) staining of the ascending aorta in local leptin‐treated (D) or control (E) mice. Arrows indicate depletion of α‐SMA. F and G, α‐SMA staining of the descending aorta in local leptin‐treated (F) or control (G) mice. Leptin was applied to the ascending aorta. Note, medial smooth muscle cell layer looks intact. Scale bar=500 μm for all images, 100 μm in high‐magnification images.
Figure S5. Cardiac and valvular modulation in mice on a high‐fat diet (HFD) treated with local leptin or control film. A, Change in peak systolic velocity (PSV) in control and leptin‐treated mice fed with an HFD. P=0.04. Baseline PSV was 1300 mm/s. B, Left ventricular (LV) wall thickness in leptin‐treated vs control HFD‐fed mice, as measured by echocardiography 6 weeks after surgery in the long‐axis view and in short‐axis view during diastole. P<0.01, n=5 to 6. Mean baseline LV wall thickness was 0.85 mm. C, Mitral and aortic valve thickness measured by histology 6 weeks after surgery. P<0.01 (mitral), P=0.047 (aortic). D and E, Hematoxylin and eosin (H&E) staining of mitral valve leaflets in local leptin‐treated (C) and control (E) mice. F and G, H&E staining of aortic valve leaflets in local leptin‐treated (D) and control (F) mice. H and I, Transforming growth factor β2 staining of aortic valve leaflets in local leptin‐treated (G) and control (H) mice. Positive signal indicates activation of stromal cells. J and K, α‐Smooth muscle actin staining of aortic valve leaflets in local leptin‐treated (I) and control (J) mice. Scale bar=500 μm for all images, 100 μm in enlargements.
Figure S6. Effects of local application of leptin antagonist (LepA) to the ascending aorta following angiotensin II (AngII) infusion. A, Weight of mice following treatment. There was no significant difference in weight between groups. n=11 to 12. B, Systolic blood pressure of mice after treatment. There was no significant difference in systolic blood pressure between groups. n=11 to 12. C, Increase in aortic valve regurgitation peak velocity following local treatment with leptin or control film. All animals displayed some regurgitation jet. Mean baseline peak velocity was 1050 mm/s. D, Increase in aortic valve regurgitation velocity–time integral. Eight of 12 AngII animals displayed aortic regurgitation, defined as at least 10‐fold increase over baseline mean, and 6 of 13 animals cotreated with LepA had aortic regurgitation, defined similarly. Mean baseline velocity time integral was 4.2 mm. E, Change in LV stroke area. n=11 to 12. Mean baseline stroke area was 6 mm. F, Percentage of Ki‐67–positive cells in stromal cells in the aortic valve leaflets. n=4 to 5. G and H, Mac3 staining of macrophages in aortic valve leaflets, in mice treated with AngII (G) or AngII and LepA (H). Scale bar=500 μm.
Figure S7. Transforming growth factor β2 (TGF‐β2) localization in the ascending aorta following angiotensin II (AngII) infusion. Immunostaining for TGF‐β2 in ascending aortas of mice treated with AngII (A and B), AngII and leptin antagonist (LepA) (C and D), or an empty film as control (E and F). Note, a weak to moderate TGF‐β2 signal in medial smooth muscle cells (SMCs) and strong expression in periadventitial macrophages in AngII‐receiving mice. When adding concomitant local LepA therapy, TGF‐β2 antigen is absent in SMCs and decreased in perivascular tissue, similar to controls. Scale bar=50 μm.
Figure S8. Immunohistochemical and in situ mRNA hybridization in stenotic aortic valves. A and B, α‐Smooth muscle actin (α‐SMA) (A) and CD68 (B) staining in advanced aortic valve stenosis (AVS) disease. Scale bar=2 mm. C and D, High magnification of α‐SMA (C) and leptin (D) staining in advanced AVS. Scale bar=200 μm. E and F, In situ hybridization for lep mRNA transcript in aortic valve leaflets from advanced AVS. Arrowheads (E) denote round macrophage‐like cells, arrows (F) denote elongated SMC‐like cells. Scale bar=200 μm.