

One of the most important questions in the double-strand break (DSB) repair field is what mechanisms govern the choice between repair pathways. Early studies established that the cell cycle influences the balance between homologous recombination (HR) and non-homologous end joining (NHEJ) via cyclin-dependent kinase (Cdk)-mediated targeting of repair components. However, the growing number of regulators and targets identified in this response suggests that several levels of regulation exist, and that we have just started scratching the surface in our understanding of this complex pathway.
Proteins in the SUMO-targeted ubiquitin ligase (STUbL) family are among the emerging regulators of DNA damage response. STUbLs typically target poly-SUMOylated repair components for ubiquitination and proteasome-mediated degradation, affecting both repair pathway choice and repair progression. A key target of the human STUbL RNF4 is the chromatin component KAP1, particularly in its phosphorylated form (S824pKAP1, or pKAP1).1 KAP1 is required for NHEJ,2 raising the question of what prevents RNF4-dependent degradation of KAP1 during G1, when NHEJ is needed the most.
According to a new study by Kuo et al., this regulation operates at the level of RNF4 stability.3 RNF4 protein levels are maintained low in G1 via proteasome-mediated degradation, and dramatically increase during G1-S transition3 (Fig. 1). Interestingly, RNF4 stabilization is Cdk2-dependent.3 This is consistent with the recent discovery that RNF4 is a target of Cdk2,4 and suggests the interesting possibility that Cdk2 regulates RNF4 by affecting its turnover.
Figure 1.
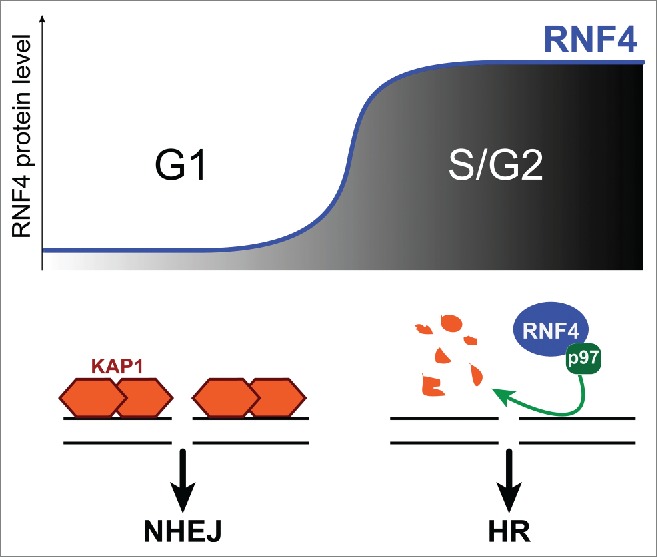
Model for how RNF4 levels influence repair pathway choice during the cell cycle. In G1, low RNF4 levels enable pKAP1 accumulation at repair sites and NHEJ. S-phase entrance results in RNF4 accumulation. RNF4 and p97 induce pKAP1 degradation, thus promoting HR repair.
Notably, this study also reveals an example where regulation of protein stability is not just the end goal for STUbL proteins, but also the means by which STUbL proteins are themselves regulated, providing a 2-way role for proteasome-mediated degradation in STUbL activities. It would be interesting to determine whether this is a general feature of STUbL proteins, and whether STUbL degradation depends on its own Ub-ligase activity through a negative feedback loop. Such a mechanism would maintain a high turnover of STUbLs, enabling quick activation and deactivation of these proteins, as would be expected of a protein acting as a switch.
Even more importantly, this study suggests RNF4 stabilization as a key transition required to promote HR over NHEJ when cells enter S-phase.3 Accordingly, RNF4 accumulation in S/G2 inversely correlates with the presence of pKAP1 at repair sites.3 Because pKAP1 degradation depends on RNF4,1,3 and Kap1 loss promotes HR repair,3 it is reasonable to speculate that local degradation of pKAP1 by RNF4 provides one of the ‘licensing signals’ for channeling DSB repair toward HR in S/G2.
The ubiquitin-selective segregase p97 is also emerging as an important ‘partner in crime’ for RNF4.3 p97 has been recently identified for its role in the extraction of the Fanconi protein complex from chromatin for RNF4-dependent degradation.5 Kuo et al. now find that p97 colocalizes with RNF4-pKAP1 foci and is required for pKAP1 degradation,3 suggesting that p97 promotes pKAP1 turnover by facilitating its extraction from chromatin.
How KAP1 facilitates NHEJ over HR is still unclear. KAP1 might directly mediate 53BP1 recruitment6 and/or interfere with the ability of HP1 to recruit Brca1 (as discussed in ref.3). However, KAP1 is also a chromatin component interacting with several histone modifiers, and can influence repair pathway choice indirectly, by changing the chromatin state. Understanding how KAP1 regulates repair progression and how post-translational modifications and chromatin responses influence KAP1 functions are among the most important open questions in the field. How these effects crosstalk with KAP1-independent functions of RNF4 during repair is also a topic of intense investigation.
Finally, the Drosophila RNF4 homolog Dgrn is required for anchoring heterochromatic DSBs to the nuclear periphery, enabling ‘safe’ progression of HR repair of highly repeated sequences.7 We cannot but wonder whether in this context the STUbL Dgrn also promotes HR progression via local destabilization of a chromatin component like KAP1, and whether STUbL levels influence this regulation across the cell cycle. More studies are also required to establish whether STUbLs are required for the spatial and temporal regulation of HR repair in mammalian heterochromatin, similar to their role in Drosophila. Furthermore, it is still unclear whether RNF4 and KAP1 function the same way in heterochromatin and euchromatin. The tools seem in place for new exciting discoveries about the role of RNF4 and its homologs in DSB repair, including their specific targets in different chromatin contexts and cell cycle phases, and their impact on chromatin and nuclear dynamics.
We apologize to our colleagues whose studies are not cited here due to space limitations.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
I. C. is supported by NIH R01GM117376, The Rose Hills Foundation and the Edward Mallinckrodt Jr. Foundation.
References
- [1].Kuo CY, et al.. J Biol Chem 2014; 289:20757-72; PMID:24907272; http://dx.doi.org/ 10.1074/jbc.M114.555672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liu J, et al.. Cell Cycle 2012; 11:2643-9; PMID:22732494; http://dx.doi.org/ 10.4161/cc.20957 [DOI] [PubMed] [Google Scholar]
- [3].Kuo CY, et al.. Cell Cycle 2016; PMID:2676649226766492 [Google Scholar]
- [4].Luo K, et al.. Nucleic Acids Res 2015; 43:5465-75; PMID:25948581; http://dx.doi.org/ 10.1093/nar/gkv434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gibbs-Seymour I, et al.. Mol Cell 2015; 57:150-64; PMID:25557546; http://dx.doi.org/ 10.1016/j.molcel.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lin YH, et al.. PLoS ONE 2015; 10:e0123935; PMID:25905708; http://dx.doi.org/ 10.1371/journal.pone.0123935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ryu T, et al.. Nat Cell Biol 2015; 17:1401-11; PMID:26502056; http://dx.doi.org/ 10.1038/ncb3258 [DOI] [PMC free article] [PubMed] [Google Scholar]