

ABSTRACT
p27Kip1 was first discovered as a key regulator of cell proliferation. The canonical function of p27Kip1 is inhibition of cyclin-dependent kinase (CDK) activity. In addition to its initial identification as a CDK inhibitor, p27Kip1 has also emerged as an intrinsically unstructured, multifunctional protein with numerous non-canonical, CDK-independent functions that exert influence on key processes such as cell cycle regulation, cytoskeletal dynamics and cellular plasticity, cell migration, and stem-cell proliferation and differentiation. Many of these non-canonical functions, depending on the cell-specific contexts such as oncogenic activation of signaling pathways, have the ability to turn pro-oncogenic in nature and even contribute to tumor-aggressiveness and metastasis. This review discusses the various non-canonical, CDK-independent mechanisms by which p27Kip1 functions either as a tumor-suppressor or tumor-promoter.
KEYWORDS: cell cycle, cell migration, cyclin-dependent kinases, metastasis, non-canonical functions, p27Kip1, stem-cell biology
Introduction
p27Kip1 (hereafter p27) is an intrinsically unstructured, multifunctional protein that influences diverse biological processes ranging from cell cycle regulation to cellular migration and transcriptional regulation. The canonical function of p27 is inhibition of cyclin-dependent kinase (CDK) activity. p27 was first discovered and characterized in 1993–94 as an inhibitor of cyclin E/CDK2 complex in transforming growth factor β (TGF-β)-arrested, contact-inhibited, or lovastatin-blocked cells.1-5 The other targets of p27 are cyclin D-CDK4/6, cyclin A-CDK2/1, and cyclin B-CDK1.6-8 p27 belongs to the Cip/Kip family of CDK inhibitors, and inhibits cyclin/CDK complex activities by binding both cyclin and CDK subunits via the highly conserved cyclin- and CDK-binding domains within the N-terminal region of p27 (Fig. 1).9
Figure 1.
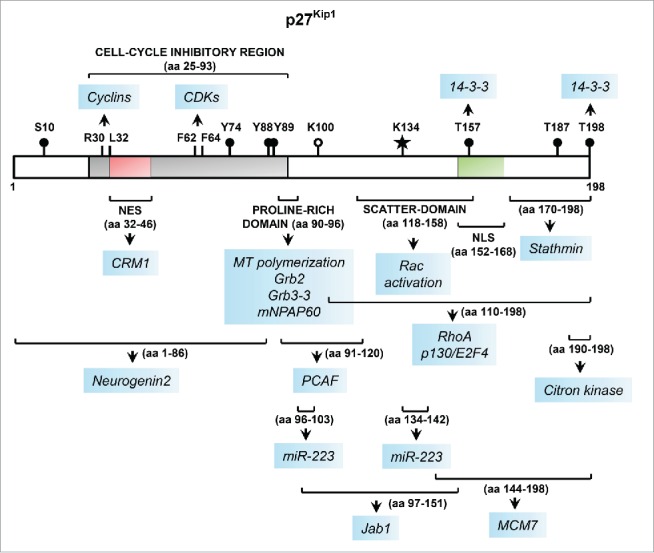
Linear representation of the key functional domains of p27 along with sites of post-translational modifications and protein-protein interactions. The p27 protein, encoded by CDKN1B, is 198-amino acids (aa) long in humans. The N-terminal portion of p27 harbors the cell-cycle inhibitory region that contains the cyclin- and CDK-binding domains comprising of aa 25–93 (shown in gray). The critical residues within this region required for binding to cyclins and CDKs are indicated. This region also harbors a nuclear export signal (NES), shown in red. The location of the nuclear localization signal (NLS) in the C-terminal portion is shown in green. The major sites of phosphorylation are denoted by black circles. The recently discovered sites of SUMOylation103 and acetylation are denoted by a star and open circle, respectively. The approximate sites of protein-protein interactions and other functional domains are denoted by brackets, below which each arrow points to the protein/microRNA partners of p27 interacting with that specific region (highlighted in blue); the interactions with CRM1104 and mNPAP60105,106, and post-translational modification by SUMOylation, included here for completeness, have not been discussed in the text.
Consistent with its CDK inhibitory role, p27 is a cell cycle-regulated protein. In response to growth inhibitory signals p27 translation and protein stability are maximal, resulting in its accumulation in G0/G1.10,11 When conditions become favorable for proliferation, p27 protein levels rapidly decrease permitting activation of cyclin E-CDK2 and cyclin A-CDK2 complexes, and G1/S transition.12 This decrease is largely accomplished at the posttranslational level by ubiquitin-dependent proteolysis. Two ubiquitin E3 ligase systems predominantly target p27 for degradation in the proteasome: KPC (Kip1 ubiquitination-promoting complex) in the cytoplasm during G0-G1 transition and SCFSkp2 (SCF: Skp1-Cul1-F box) in the nucleus in S and G2.13-16
The importance of p27 in the regulation of exit from and entry into the cell cycle was revealed by the generation of p27-deficient mice, which display features of gigantism with approximately 30% increase in body size, multiple organ hyperplasia, and retinal dysplasia, all of which are attributed to an overall increase in cellular proliferation.17-19 The p27 knockout (KO) females are infertile with impaired luteal cell differentiation and a defective uterine environment. p27 nullizygous mice spontaneously develop adenomas of the intermediate lobe of the pituitary, and p27 heterozygous mice are predisposed to increased tumorigenesis in multiple tissues when challenged with γ-irradiation or chemical mutagens.20 In addition, p27 deficiency also exacerbates the tumor-prone phenotypes caused by oncogenic activation of Ras21,22 or loss of tumor suppressors, such as p18INK4C, p16INK4A, RB, or Pten.23-26 In humans, like in rats, germline nonsense mutation of the p27 gene (CDKN1B) can predispose individuals to the syndrome of multiple endocrine neoplasia.27-30 Cell cycle inhibition is a function of nuclear p27. Accordingly, nuclear p27 is frequently reduced or undetectable in multiple human cancers, including breast, lung, colon, prostate, lymphomas, and gliomas.31,32 Recently, CDKN1B was reported to be one among the most significantly mutated genes in luminal breast cancer and aggressive primary prostate cancer.33-35 In hairy cell leukemia, it happens to be the second most common mutated gene.36 Collectively, these findings underscore the tumor-suppressive properties of p27.
Nevertheless, p27 is an atypical tumor suppressor. Inactivating mutations and loss of heterozygosity of CDKN1B are observed at very low frequencies in most human cancers.20,37-40 Thus, unlike TP53 and RB, p27 does not appear to fulfill Knudson's two-mutation criterion for a tumor suppressor gene.20 Even in hairy cell leukemia and luminal breast cancers, only a handful of all the CDKN1B mutations reported reside in the N-terminal region with deleterious effects on, or potential to hamper, its CDK-inhibitory function.33,34,36 Furthermore, in many human tumors in addition to the loss of nuclear p27, gain of cytoplasmic p27 has been reported that commonly correlates with tumor aggressiveness and adverse patient outcome.41-46 Notably, multiple oncogene-activated pathways target p27 to promote its cytoplasmic translocation, either through active nuclear export or cytoplasmic retention.47 This cytoplasmic translocation constitutes more than just inactivation of its nuclear cell cycle inhibitory functions. Numerous studies in the last 2 decades have revealed unexpected CDK-independent, non-canonical functions for p27; most of these are pro-oncogenic in nature, while others, along with its canonical functions appear to be critical for inhibition of G1-S transition. In this review, we present an overview of the current knowledge of the non-canonical functions of p27 both in normal and tumor biology.
p27 in cellular migration
Cell migration is a highly integrated, multi-step process that orchestrates morphogenesis throughout embryonic development; is vital to the process of tissue repair and regeneration, and contributes to progression of several important pathological processes, including cancer.48 Central to migration is the ability of the cell to quickly integrate external migration-promoting signals with the help of intracellular signaling “integrator” molecules and translate it into rapid changes in the dynamics of the actin and microtubule cytoskeleton. Key among these integrator molecules are members of the Rho-family GTPases.49 That CDK inhibitors could function to regulate cytoskeletal rearrangements, independent of their cell-cycle inhibitory effects, by modulating Rho-GTPases was first demonstrated by studies in yeast. In Saccharomyces cerevisiae, activation of Cdc42 (a Rho-GTPase) by its guanine-nucleotide exchange factor Cdc24 is required to orientate the actin cytoskeleton toward the pheromone-secreting partner during mating or the incipient bud site during division. In G1 phase, the CDK inhibitor Far1 binds to and sequesters Cdc24 in the nucleus. In response to mating pheromones, Far1-Cdc24 complex is exported out of the nucleus, and Far1 aids in targeting Cdc24 to the site of receptor-associated heterotrimeric G-protein activation at the plasma membrane by directly binding to Gβ subunit.50 On the other hand, bud emergence and polarized growth in G1 involves activation of Cln/Cdc28 which in turn induces phosphorylation and degradation of Far1, thereby releasing Cdc24 to be exported from the nucleus to the bud site.51 Thus, by regulating the subcellular localization of Cdc24, Far1 plays an important role in the local activation of Cdc42 required for actin cytoskeletal reorganization in response to extracellular signals and during vegetative growth. Far1 is functionally analogous to p27; although the 2 proteins are distantly related with limited sequence identity, the Cdc24-binding motif present within the C-terminal region of Far1 bears a loose sequence similarity to the “scatter-domain” (discussed below) in the C-terminal region of p27.
p27 regulates actomyosin reorganization
That cytoplasmic p27 modulates cellular motility by regulating actomyosin remodeling was first reported by Dowdy and colleagues. They demonstrated that introduction of transducible p27 protein (TAT-p27) is sufficient for actin cytoskeletal rearrangement, filopodium formation and migration in human hepatocellular carcinoma cells.52,53 Importantly, this role of p27 is independent of its CDK-inhibitory activity and requires cytoplasmic localization of p27 bearing an intact “scatter-domain” in the C-terminal region comprising of residues between 118 and 158 (Fig. 1). In these cells, while p27 translocates to the cytoplasm and colocalizes with actin in response to HGF signaling, and cell migration depends on Rac-activation and, in part, on PI-3K activation, the precise mechanism of p27-induced migration is still not completely understood. Another mechanism by which p27 regulates cell migration is through prevention of RhoA activation.54 Notably, p27-null fibroblasts have elevated levels of RhoA-GTP (active form) with increased number of actin stress fibers and focal adhesions, and exhibit a dramatic decrease in motility compared to wild-type cells. This migration defect can be rescued by re-expression of either wild-type p27 or mutant p27 (p27CK) that cannot bind cyclins and CDKs. Overexpression experiments revealed that the C-terminal half of p27 directly binds RhoA (Figs. 1 and 2), which prevents RhoA interaction with and activation by Rho-GEFs. Similarly, ectopic overexpression of a mutant p27, lacking the nuclear localization signal, that localizes predominantly in the cytoplasm in MCF-7 human breast cancer cell line results in downregulation of RhoA activity together with enhanced cell motility and survival.55 The p27-mediated RhoA regulation was also found to be critical in the cyclin D1-stimulated migration of mammary epithelial cells.56 Cyclin D1 stabilizes p27 by repressing the Skp2 component of the SCFSkp2 ubiquitin ligase complex, and physically colocalizes with p27 in the activated Rho-complex. Thus, inhibition of RhoA activation requires cytoplasmic localization of p27. Multiple mitogenic signaling pathways target p27 for phosphorylation resulting in its cytoplasmic mislocalization.47 For example, RSK1, a downstream effector of PI-3K and MAPK pathways, phosphorylates p27 at T198 (Fig. 1) which in turn mislocalizes p27 to the cytoplasm and promotes p27-RhoA interaction.57 The resulting inhibition of RhoA-ROCK pathway leads to loss of actomyosin stability. Of note, both T198 phosphorylation of p27 and p27-bound RhoA increase in early G1; the resulting reduction in actin cytoskeletal stability in G1 might further contribute to the G1-S transition and prepare cells for the shape changes that must occur during the subsequent phases of the cell cycle, although the mechanism involved is less understood. It is also plausible that in human cancers, constitutive RSK1 activation induced by oncogenic activation of MAPK and PI-3K pathways might target p27 for cytoplasmic mislocalization and contribute to enhanced cellular motility and migration.47,57
The modulation of RhoA-ROCK pathway by p27 also plays a critical role in neuronal migration during corticogenesis.58,59 Ablation of p27 in post-mitotic neurons results in relatively rounded cells with thin processes, indicating abnormal regulation of actin cytoskeleton.58 In these cells, cytoplasmic p27 was found to directly interact with CDK5, predominantly in the perinuclear region and the process tips, where the latter targets p27 for phosphorylation (at S10; Fig. 1) and stabilization. The stabilized cytoplasmic p27 then inhibits the RhoA-ROCK pathway to activate an F-actin severing protein, cofilin (by repressing its phosphorylation at S3), to promote actin reorganization and neuronal migration. p27-tuned actomyosin contractions are also vital during interneuron migration. p27-null cortical interneurons exhibit defective nucleokinesis and neurite branching resulting in delayed tangential migration.60 By inhibiting of RhoA-ROCK pathway, p27 represses myosin-II light chain phosphorylation to control actomyosin contractions during interneuron migration to drive both forward translocation of the nucleus and growth cone splitting.
Intriguingly, the promigratory effect of p27 is not seen in all cell types. In certain cell types, including vascular smooth muscle, mesangial, umbilical vein endothelial, fibrosarcoma, and glioblastoma cells, p27 inhibits migration.61-64 It has been postulated that these differences might arise from cell type-specific variations in the relative balance between Rac and Rho activity. For example, in a cell type that might have this balance tilted toward Rho activation, cytoplasmic p27 could decrease Rho activity to promote migration. Conversely, in cell types where the balance is tilted toward Rac activation, inhibition of Rho activity by p27 might block migration. Furthermore, in yet other cell types, as already described for hepatocellular carcinoma cells, p27 might function upstream of Rac, to directly impinge on Rac activation to promote migration (Fig. 2). In addition to RhoA and Rac, p27 could also regulate other critical pathways that affect cell migration depending on the cell type-specific context, as discussed below.
Figure 2.
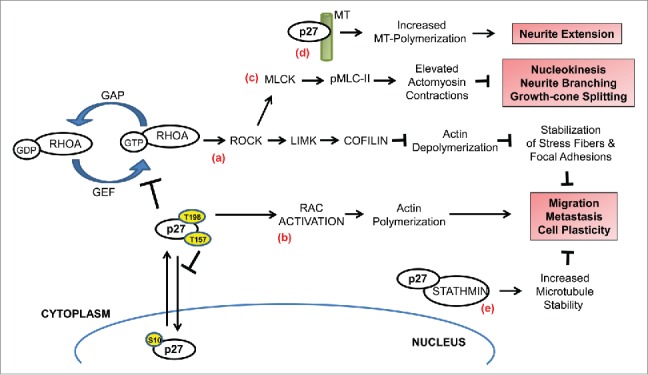
p27 regulates cell migration. Depending on the cell-specific context, p27 can either promote or inhibit cell migration in a CDK-independent manner. (a) RhoA functions as a molecular switch that cycles between inactive GDP-bound and active GTP-bound states that are promoted by GTPase-activating proteins (GAPs) and guanine–nucleotide exchange factors (GEFs), respectively. Upon stimulation, GEFs activate RhoA which in turn activates ROCKs to phosphorylate LIMKs. Thus activated, LIMK then phosphorylates cofilin to inhibit its actin-depolymerizing function to stabilize actin stress fibers and focal adhesions. Cytoplasmic p27 inhibits this pathway by directly interacting with RhoA and preventing its activation by the Rho-GEFs, resulting in increased cell motility. Mitogenic signaling pathways and oncogenic kinases target p27 for cytoplasmic relocalization through phosphorylation of specific residues (indicated by yellow circles)—phosphorylation of S10 promotes nuclear export while phosphorylation of T157 and/or T198 impairs nuclear import. (b) In certain cell types, cytoplasmic p27 can promote Rac activation to stimulate filopodium formation and cell migration (c) In migrating cortical interneurons, sustained activation of RhoA in the absence of p27 results in elevated actomyosin contractions due to increased phosphorylation of myosin-II light chain (pMLC-II) by activated MLC kinase (MLCK). This deregulation contributes to defective nucleokinesis, neurite branching, and tangential migration. Cytoplasmic p27 fine-tunes the actomyosin contractions by inhibiting RhoA activation to correct this defect. (d) p27 is a microtubule (MT)-associated protein and it promotes MT-polymerization, thereby contributing to neurite extension during interneuron migration. (e) p27 controls MT dynamics in a stathmin-dependent manner. Sequestration of stathmin, a MT-destabilizing protein, by p27 blocks the tubulin-sequestration activity of the former resulting in increased MT polymerization and inhibition of migration.
p27 regulates microtubule dynamics
Expression of p27 in HT-1080 human fibrosarcoma cells inhibits cell migration.63 Baldassarre et al. reported that this effect depends on cell to extracellular matrix (ECM) contact that induces cytoplasmic localization of p27 where the latter sequesters stathmin, a major microtubule-destabilizing protein, by direct binding via the C-terminal 28 amino acids of p27 (Fig. 1).63 Stathmin sequestration blocks its tubulin-sequestration activity, leading to increased microtubule polymerization and inhibition of migration (Fig. 2). Thus, p27 affects microtubule stability independent of its cyclin-CDK inhibitory function. Notably, in human sarcoma cell lines the p27-modulated migration appears to depend on the cellular p27 to stathmin expression ratio. A high p27/stathmin ratio correlates with reduced motility in vitro and primary sarcoma tumors in vivo, while a low p27/stathmin ratio correlates with increased motility in vitro and metastatic tumors in vivo.63
The p27-stathmin axis also appears to modulate cellular plasticity in cells migrating through 3-dimensional matrices. Mouse embryonic fibroblasts (MEFs) lacking p27 that have been immortalized or subjected to v-Src-induced transformation when challenged with 3D-ECM undergo mesenchymal to amoeboid transition (MAT) compared to cells harboring wild-type p27.65,66 The amoeboid-like morphology in these cells is due to reduced microtubule stability and increased RhoA activation resulting from loss of p27 and presumably higher stathmin activity, and is associated with not just higher motility in vitro and but also higher invasive ability in vivo.66 Incidentally, activation of c-Src, or other oncogenic kinases such as Lyn and BCR-ABL, is known to accelerate p27 degradation through phosphorylation of p27 at Y88 and Y74.67,68 Thus these kinases might tilt the balance toward low p27/stathmin ratio and favor amoeboid-like morphology in tumor-cells in which they are oncogenically activated. Furthermore, p27-modulated mechanisms such as this might come into play as an adaptive response to varying tissue microenvironments, allowing disseminating tumor-cells to utilize mesenchymal or amoeboid modes of movement that are most favorable to the conditions that they encounter in their journey away from the primary tumor.
p27 also controls microtubule dynamics in a stathmin-independent manner. In cortical interneurons, p27 is a microtubule associated protein; multiple microtubule-binding sites in both the N- and C-terminal regions of p27 appear to mediate this interaction.60 Microtubule-associated p27 promotes microtubule polymerization both in vitro and in vivo in interneurons during corticogenesis. This function requires the integrity of the proline-rich domain (residues 90–96; Figs. 1 and 2) of p27 and is important for neurite extension during interneuron migration.60
p27 in metastasis and stem cell biology
The cytoplasmic mislocalization of p27 and its ability to modulate cellular motility is likely to turn pro-oncogenic and even contribute to metastasis depending on the cell-specific contexts. Indeed, an oncogenic, CDK-independent role for cytoplasmic p27 was shown in melanoma cells. Targeted cytoplasmic expression of wild-type p27 or p27CK at subphysiologic levels in a low-metastatic B16F10 melanoma cell induces a dramatic increase in cell motility in vitro and numerous metastases to lymph nodes, lung, and peritoneum in vivo.42 This finding is further corroborated by the observation that ∼70% of invasive and metastatic human melanomas exhibit cytoplasmic p27, whereas the non-invasive, melanoma in situ shows no cytoplasmic p27.42 In a separate study, analyses of melanoma tissue microarrays in a large series of melanoma patients have identified gain of cytoplasmic p27 is associated with poor 5-year survival of metastatic melanoma patients and that it is an independent prognostic factor to predict patient outcome.41
Interestingly, in mouse and human melanoma-cell models, p27 expression is required for the tumor-initiating properties of Mift-depletion induced melanoma cancer stem cells (CSCs), suggesting a role for p27 in regulating CSC biology.69 A growing body of evidence indicates that the biological traits of CSCs are central to the multi-step invasion-colonization cascade of the metastatic process. CSCs, even though cannot be directly equated to the normal stem cells, have the property of “stemness” that is associated with self-renewal and ability to spawn differentiated progeny.70 An oncogenic, CDK-independent role for p27 in regulating stem cell biology became apparent in p27CK knock-in mice.71 In these mice, p27CK functions as a dominant oncogene (as compared to p27 KO and wild-type controls) and causes hyperplastic lesions and tumors in multiple organs, including the lung, retina, pituitary, ovary, adrenals, spleen, and thymus/lymph nodes. Furthermore, the neoplastic lesions in both lung and retina are associated with amplification of the stem/progenitor cell populations in these tissues, likely arising from their deregulated proliferation and/or differentiation. Subsequent studies show that p27CK needs to be localized in the cytoplasm in order to function as an oncogene, in the absence of which it just behaves similar to a null allele.72 Thus, p27 could function as a cooperating oncogene with those that have the ability to target it for cytoplasmic localization. Indeed, p27CK cooperates with K-Ras which targets the former for cytoplasmic localization, but not with c-Myc which does not alter the nuclear localization of p27CK.72 The importance of cytoplasmic localization of p27 in its oncogenic functions has also been corroborated in p27S10A knock-in mice: these mice are tumor-resistant in response to urethane-treatment compared to p27 wild-type, heterozygous, and nullizygous mice.22 Alanine substitution at S10 renders p27 resistant to activated K-Ras-induced phosphorylation at S10 and subsequent nuclear-export of p27.
In addition to the less understood contribution of cytoplasmic p27 to stem cell expansion, nuclear p27 is also known to regulate stem cell differentiation in various cell types. A subset of these mechanisms, involves transcriptional repression of specific stem-cell genes by p27, either through direct promoter occupancy of the target gene or by indirectly influencing transcriptional activity through regulation of transcription factor stability (Fig. 3). For example, in human embryonic stem cells (hESCs) p27 protein levels are low in undifferentiated cells and increases markedly as the cells are induced to undergo differentiation to form embryoid bodies.73,74 Similar observations have been reported with murine embryonic stem cells (mESCs).75 Using overexpression of p27 protein and ShRNA-directed silencing of p27 in hESCs, Belmonte and colleagues reported that p27 associates with the Twist1 and Brachyury promoters, and might facilitate the differentiation process through direct transcriptional repression of these genes.73 Although, at present it is unclear whether this p27-mediated transcriptional repression is independent of or dependent on its CDK-inhibitory activity. Recently, however, Serrano and colleagues provided compelling evidence for p27 in the transcriptional repression of SOX2, a pluripotency gene, during the differentiation of pluripotent cells, including murine induced pluripotent stem cells (iPSCs), mESCs, and P19 embryonal carcinoma cells.76 Absence of p27 results in delayed and incomplete silencing of SOX2 in these cells; likewise, p27-null mice exhibit defective repression of SOX2 in fibroblast, lung, retina, and pituitary. Most remarkably, SOX2 heterozygosity in p27-null mice rescues the main phenotypes associated with p27-deficiency, namely, gigantism, pituitary hyperplasia, pituitary adenomas, and retinal abnormalities. The authors demonstrate that p27 functions as a transcriptional repressor by associating with the SOX2-SRR enhancer together with repressive complex, p130-E2F4-SIN3A.76 This function of p27 is likely to be independent of its cyclin-CDK inhibitory role as described in a study by Bachs and colleagues, discussed later.77 Furthermore, the ability of decreased gene dosage of SOX2 to rescue some of the phenotypes of p27 KO mice suggests that not all phenotypes observed in the latter are attributable to enhanced CDK activity. Another mechanism by which p27 regulates differentiation is seen during neuronal differentiation of cortical progenitors.59 Here, p27 is required for the stabilization of neurogenin2 protein and thereby promoting the transcription of its target genes; this activity is carried by the N-terminal of p27, independent of its cell-cycle regulatory function.
Figure 3.
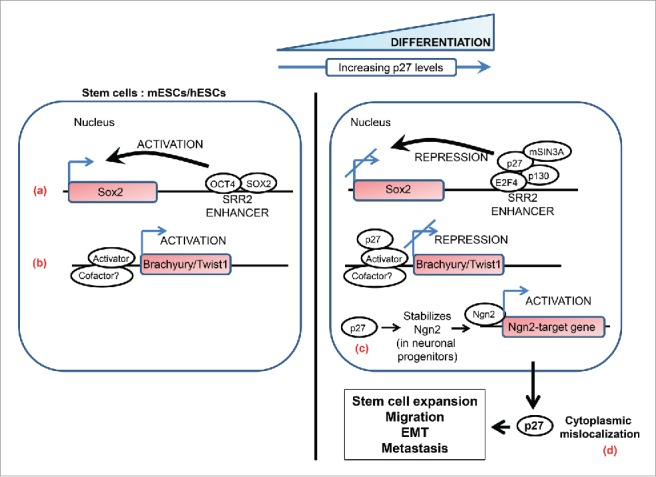
A simplified schematic highlighting the non-canonical roles of p27 in stem-cell biology and metastasis. Nuclear p27 regulates differentiation in various cell types. p27 levels are low in mouse and human embryonic stem cells (mESCs and hESCs) and it markedly increases as these cells are induced to differentiate. (a) p27 contributes to transcriptional repression of SOX2, a pluripotency gene, during differentiation of pluripotent cells and in differentiated cells by associating with the SOX2-SRR2 enhancer together with the repressive complex p130-E2F4-SIN3A. (b) In differentiating pluripotent cells p27 associates with Brachyury and Twist1 promoters to repress the transcription of these genes; the identity of the repressor complex that p27 associates with on these promoters remains to be defined. (c) Neurogenin2 (Ngn2) is a proneural basic helix-loop-helix transcription factor that plays a central role in the neuronal differentiation of cortical progenitors. p27 plays a key role in this process by stabilizing the neurogenin2 protein in a CDK-independent manner. (d) Cytoplasmic p27 contributes to tumorigenesis and metastasis by exerting influence on various processes such as expansion of the stem cell pool, cell migration and invasion, and epithelial-mesenchymal transition (see text for details).
Very recently, cytoplasmic p27 was shown to contribute to the epithelial-mesenchymal transition (EMT) process.78 EMT is a highly conserved and fundamental process that is critical for the normal embryonic development in metazoans. There is good evidence that this transdifferentiation program, driven by EMT-inducing transcription factors, is involved in the dissemination of single carcinoma cells from the sites of primary tumors.79 Recent evidence also suggests that in certain cancer types EMT might contribute to the dedifferentiation of non-CSCs resulting in the acquisition of CSC-like traits.80 Slingerland and colleagues demonstrated that ectopic overexpression of a phosphomimetic p27CK (p27CK-DD, aspartic acid residues substituted at T157 and T198) significantly enhances the mesenchymal features of a pair of human epithelial cell lines (MCF7 breast and UMUC3 bladder cancer cell lines).78 p27CK-DD binds to JAK2 and perhaps by facilitating the recruitment of STAT3 to JAK2 increases the formation of JAK2-STAT3 complexes, leading to activation of STAT3. Activated STAT3 further binds to and transcriptionally activates TWIST1 promoter resulting in increased TWIST1 expression. TWIST1 is one of the key EMT-inducing transcription factors, which markedly represses E-cadherin expression and contributes to enhanced cell migration and invasion. Importantly, overexpression of p27CK-DD causes non-transformed mammary epithelial cells to acquire invasive ability in vitro and also increases metastasis by the otherwise weakly-metastatic MDA-MB-231 and UMUC3 cell lines in vivo.78 Collectively, these results suggest a new mechanism by which cytoplasmic, C-terminally phosphorylated p27 contributes to tumor-cell invasion and metastasis.
p27 in cell-cycle regulation
By virtue of its ability to inhibit cyclin D- and cyclin E-associated activities, p27 functions as an important gatekeeper of the G1-S transition. As mentioned earlier, this is reflected in p27 KO mice that are larger than normal mice due to enhanced cellular proliferation and organ enlargement in tissues that normally express p27 at the highest levels.17-19 Conversely, overexpression of p27 in cells, including multiple cancer cell lines, holds them in G1-arrest.61,81,82 p27 might also function to serve as a gatekeeper at the G2-M transition by virtue of its ability to inhibit CDK1 activity. This latter function is evident in cells from specific tissues of the Skp2 KO mice: due to the accumulation of p27, particularly, in the S and G2 phases, these cells exhibit reduced CDK1 activity resulting in impaired ability to enter M phase, leading to endoreplication, centrosome over-duplication, polyploidy and increased apoptosis.83 Of note, the Skp2 KO mice are smaller in size compared to their littermate controls. Remarkably, the centrosome over-amplification and polyploidy phenotypes of Skp2 KO mice are rescued with concomitant ablation of p27.84 p27-deficiency is also implicated in increased genetic instability which arises from impaired G2/M arrest in response to DNA damage.85 Given these important roles in regulating the cell cycle via CDK inhibition, is it likely that p27 has also evolved to exert additional breaks on the cell cycle through non-canonical, CDK-independent mechanisms? Recent findings, discussed below, seem to answer this in the affirmative (Fig. 4).
Figure 4.
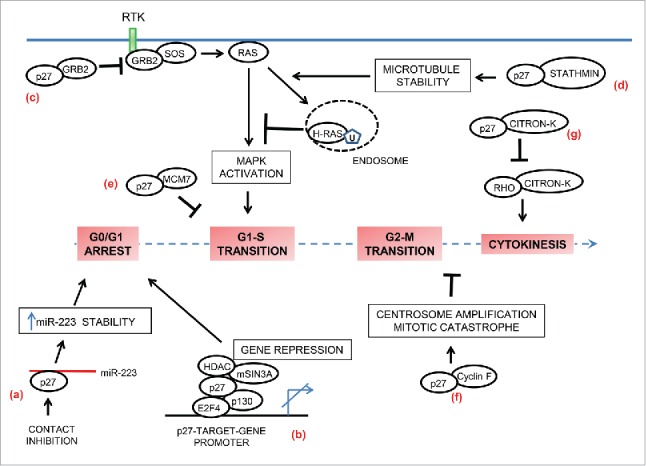
p27 in cell-cycle regulation. In addition to inhibition of cyclin-CDK activities, p27 also contributes to regulation of the cell-cycle by non-canonical means. (a) p27 is a RNA-binding protein. In contact-inhibited cells, cytoplasmic p27 directly binds and stabilizes miR-223. High levels of miR-223 then target E2F1 mRNA and thereby promote cell cycle arrest. (b) p27 also contributes to G0-G1 arrest by functioning as a transcriptional co-repressor in a CDK-independent fashion. On the promotors of specific target-genes it associates with p130/E2F4 complex and facilitates gene repression by recruiting co-repressors such as HDAC1 and mSIN3A. (c and d) p27 controls activation of MAPK pathway in a CDK-independent manner to regulate cell cycle entry. (c) In mitogen-stimulated quiescent cells, p27 is rapidly exported to the cytoplasm where it binds free GRB2 to limit GRB2-SOS complex formation and thus has the potential to attenuate MAPK activation. (d) In mitogen-stimulated cells, p27 sequesters stathmin to counteract its microtubule-destabilizing activity. The resulting enhanced microtubule-stability facilitates increased endocytic-trafficking of H-Ras and its ubiquitination, leading to attenuation of H-Ras –MAPK signaling and inhibition of cell cycle entry. (e) p27 interacts in a CDK-independent fashion with MCM7, a key component of the pre-replication complex that is assembled at the origins of replication. This interaction might prevent premature firing of the origins in late G1. (f) p27 sequesters cyclin F in a CDK-independent manner and antagonizes the cyclin F-mediated degradation of CP110 resulting in centrosome amplification and mitotic catastrophe. (g) Citron kinase (Citron-K) is essential for cytokinesis; p27 interferes with normal cytokinesis by interacting with citron-K and preventing its activation by RhoA.
That p27's tumor-suppressing ability might reside in a function in addition to CDK inhibition was first reported by Sheaff and colleagues.86 They demonstrated that the proline-rich domain (amino acids 91–96) of p27 interacts with the SH3-domain of GRB2, a key molecule in the Ras activation pathway. Upon mitogen stimulation, the resulting phosphorylation of receptor tyrosine kinases generates a binding site for the SH2-domain of GRB2. The latter via its SH3 domain then binds SOS, a guanine nucleotide exchange factor; the GRB2-SOS complex thus formed activates Ras by catalyzing exchange of GDP for GTP. In mitogen-stimulated quiescent fibroblasts, p27 is rapidly exported to the cytoplasm where it sequesters unbound GRB2 resulting in a concomitant decrease in GRB2-SOS complexes. Consistently, the p27-null MEFs maintain their SOS-GRB2 complexes significantly longer that p27+/+ cells. Despite these molecular changes, Sheaff and colleagues could not detect a prolonged activation of the Ras-MAPK pathway in p27-null cells; thus the physiological significance of the p27-GRB2 interaction still remains unclear. Contrary to this putative role in the control of MAPK pathway, an earlier study indicated GRB2-p27 interaction leads to accelerated, Jab1/CSN5-mediated p27 degradation.87
Very recently, however, Baldassarre and colleagues reported an enhanced activation of the Ras-MAPK pathway in p27-null MEFs compared to wild-type MEFs, as determined by increased phosphorylation of ERK1/2 and higher levels of MAPK target genes, such as Erg1, JunB, cFos, and cyclin D1.88 The hyperactivation of the MAPK pathway is responsible for the observed faster cell-cycle entry of p27 KO MEFs compared to control cells. Interestingly, the increased proliferation rate of the p27 KO cells is rescued by the co-ablation of stathmin.89 This is also reflected in the body-size of the p27 and stathmin double KO mice: the phenotypes of the p27-null mice such as increased body and organ weight, development of the pituitary adenomas and the outgrowth of the retinal basal layer, are all reverted in these double-KO mice.89 Together, these observed effects uncover a p27-stathmin regulatory axis in the control of Ras-MAPK pathway activation and G1-S phase transition. As discussed earlier, this aspect of p27 function impinges on the increased microtubule-stabilization resulting from direct sequestration of stathmin in a CDK-independent manner.63 Microtubule stabilization can have a profound influence on endocytosis and the endocytic vesicle trafficking processes, which in turn are strongly implicated in propagating signals originating from outside of the cell, and vice versa. Indeed, in p27-null cells, stathmin-induced microtubule destabilization hyperactivates H-Ras-MAPK signaling by reducing the endocytic trafficking of H-Ras-containing vesicles and thus diminishing mono- or bi-ubiquitylation of H-Ras.88 Re-expression of p27 or ablation of stathmin in p27-null MEFs increases H-Ras ubiquitylation resulting in decreased H-Ras activity and restoration of normal cell cycle entry kinetics.
p27 also contributes to G0-G1 arrest by functioning as a transcriptional co-repressor in a CDK-independent fashion.77 In quiescent cells p27 associates with the promoters of a number of specific target genes that are mainly involved in RNA processing and splicing, translation, cell-cycle, and mitochondrial organization. Among these, the genes that are repressed by p27 can be divided into 2 categories: repression that is dependent on inhibition of cyclin-CDK activity and those that are repressed in a CDK-independent manner. In the latter case, p27 co-localizes with p130, E2F4, and co-repressors such as histone deacetylases (HDACs) and mSIN3A on these promoters. Immunoprecipitation and ChIP-on-CHIP experiments together support a model whereby p130 first drives E2F4 to the promoters, where p27 is subsequently recruited by binding directly via its C-terminal half to p130 and E2F4. Finally, p27 recruits co-repressors such as HDAC1 and mSIN3A on these promoters culminating in transcriptional repression.77 Interestingly, the CDK-independent repression of these target genes is only achieved efficiently with either wild-type p27 or p27CK, but is inefficient with a mutant p27 lacking the first 51 residues in the N-terminal portion; importantly, loss of p27 results in the upregulation of these target genes. These results suggest that the N-terminal residues (1–51) are also needed for CDK-independent repression by p27.77 The identity of the co-repressor(s) that interact with this region to enhance the efficiency of p27-induced repression is not known at present. Of note, a significant number of these p27-target genes are overexpressed in various human tumors and correlates with reduced p27 expression therein.77,90 In light of the above findings, it is noteworthy that PCAF, a well-recognized histone acetylase and transcriptional co-activator, was identified in subsequent studies by Bachs and colleagues as a nuclear interactor of p27.91 The catalytic domain of PCAF interacts with the residues 91–120 of p27 and acetylates K100 (Fig. 1); this results in decreased stability of p27 in a Skp2-independent, but proteasome-dependent manner. It is plausible that this acetylation-induced degradation of p27 might contribute to the de-repression of the p27-target genes as cells exit G0/G1 to reenter the cell cycle.
Another unanticipated non-canonical role for p27 has emerged in the regulation of the cell cycle exit following contact inhibition where it functions as a microRNA-binding protein.92 MicroRNAs are abundant ∼21–25 nucleotide non-coding RNAs that mediate sequence-specific, post-transcriptional repression of mRNA targets. Emerging evidence suggests that microRNAs could allow fine-tune control of cell cycle progression by modulating the expression of target transcripts that encode proteins directly or indirectly involved in cell cycle progression and cellular proliferation. Baldassarre and colleagues report that p27 regulates the expression of a subset of microRNAs, including miR-223, during cell cycle exit following contact inhibition.92 It does so in 2 ways: first, it stimulates miR-223 promoter activity via the inhibition of E2F1 activity through a CDK-RB pathway; second, cytoplasmic p27 binds to mature miR-223 and prevents it from degradation in a manner independent of cyclin-CDK inhibition. Human p27 has 2 putative RNA-binding sites, one of which is highly conserved among mammals comprising of residues 90–103 and another between residues 134–142; both are required for the high affinity binding of miR-223 (Fig. 1). Thus stabilized, high levels of miR-223 then contributes to cell cycle arrest by downregulating its targets, one of which is the E2F1 mRNA. Overexpression studies in K-RasV12-transformed p27KO 3T3-fibroblasts show that miR-223 expression could strongly reduce the S-phase population of exponentially growing cells to mimic contact-inhibition. Notably, the occurrence of p27 frameshift mutations, namely, p27K134fs or p27P137fs which destroy the second RNA-binding site correlates with reduced levels of miR-223 in breast tumors.33,34,92 Likewise, the low (or absent) expression levels of p27 observed in many cancers could deregulate the p27-mediated control of miR-223 and contribute to loss of contact-inhibition. Collectively, these observations suggest a novel, CDK-independent mechanism in the regulation of G0/G1-S transition by p27 as a RNA-binding protein.
Nallamshetty et al. suggested a CDK-independent function for p27 in the inhibition of the initiation of DNA replication at the G1-S boundary.93 To meet the challenging goal of replicating the entire eukaryotic genome once, and only once, in each cell division, the origins of replication are strictly prepared and licensed only in the G1 phase when the CDK activity is low. Here the origins are referred to be in the pre-replicative state: at the molecular level it involves the sequential assembly of the origin replication complex (ORC), CDC6, CDT1, followed by the loading of the replicative helicase MCM2–7 (minichromosome maintenance proteins 2–7). The pre-replication complexes are then fired and activated, triggered by the activity of the CDKs at the G1-S boundary. Nallamshetty et al. reported that p27 co-immunoprecipitates with MCM7 in synchronized cycling NIH-3T3 cells, but not in serum-starved quiescent cells.93 This interaction was mapped to the C-terminal, 144–198 residues of p27 (Fig. 1). In cell-free, Xenopus based in vitro DNA replication assays, this region of p27 inhibits the initiation of DNA replication, suggesting a possible CDK-independent regulatory role for p27 in restraining inappropriate initiation of DNA replication prior to S phase in vivo. Curiously, however, in our recent study this CDK-independent cell cycle-inhibitory effect of p27 is not observed when p27CK is inducibly overexpressed in a U2OS-derived Tet-On cell line.81 Like parental U2OS-derived Tet-On cells, these p27CK cells traverse uninhibited through the cell cycle, under both asynchronous and synchronous cycling conditions. Intriguingly, in this model system, inducible overexpression of a mutant p27 (p27K) that can neither directly bind nor inhibit CDKs, results in a block of the G1-S phase transition; however, this effect is observed only in synchronized cycling cells following release from a nocodazole-block but not in asynchronously cycling cells.81,94 p27K differs from p27CK in that it harbors an intact cyclin-binding site (this site is mutated in p27CK by alanine substitutions at R30 and L32, along with mutation of the CDK-binding site by alanine substitutions at F62 and F64; Fig. 1). Thus, these findings point to an inhibitory role for p27 in the G1-S transition that is independent of CDK inhibition, but dependent on the presence of an intact cyclin-binding site. The identity of the molecule that interacts with this site to bring about a CDK-independent inhibition of G1-S traverse is not known at this time.
Strikingly, the block in the G1-S phase transition of synchronized cycling p27K cells is reminiscent of cyclin F KO MEFs: while fully functional cyclin F is dispensable for cell division under serum-rich conditions, cyclin F−/− MEFs when subjected to serum-starvation-induced quiescence, re-enter cell cycle with significantly delayed kinetics upon stimulation with serum.94,95 Our previous study identified cyclin F as a protein that interacts with the intact cyclin-binding site of p27K as well as that of wild-type p27.81 Whereas inducible overexpression of wild-type p27 in asynchronously growing cells results in inhibition of CDKs and consequent G1-arrest, induction of p27K leads to centrosomal over-duplication, multiple micronuclei, and mitotic catastrophe, without affecting the CDK activity. In our model system, even though the asynchronously growing p27K cells progress normally through the cell cycle, they exhibit significantly reduced proliferation rates compared to parental and p27CK cells, perhaps as a result of increased cell death due to mitotic catastrophe. Notably, centrosome over-duplication, micronuclei formation, and mitotic aberrations are also seen upon siRNA-mediated cyclin F silencing and in cyclin F-null MEFs.96 Cyclin F is an atypical cyclin that does not bind or activate any CDKs.96,97 It is the founding member of the F-box family of proteins that form substrate recognition subunits of the SCF ubiquitin ligase complexes and target specific protein substrates for ubiquitylation and consequent degradation.97,98 In the G2 phase, cyclin F physically associates with the centrosome and targets CP110 for destruction; the latter is a centrosomal protein essential for centrosome duplication.96 We demonstrated that p27K sequesters cyclin F and antagonizes the cyclin F-mediated degradation of the CP110 resulting in centrosome over-duplication.81 The physiological and/or pathological relevance of the interplay between p27 and cyclin F is not clear at this time. Interestingly, Besson and colleagues observed a phenotype of centrosome over-amplification, multinucleation, and polyploidy in fibroblasts, hepatocytes (liver), and renal proximal tubule (kidney) cells in p27CK knock-in mice compared to p27 KO mice. This study identified a CDK-independent function for p27 in disrupting the normal process of cytokinesis and abscission by interfering with the function citron kinase, a RhoA effector that is essential for cytokinesis. Citron kinase localizes at the contractile ring and mid-body during telophase and cytokinesis, where its exact role at the molecular level is less understood but is known to contribute to assembly, stability, and contraction of the contractile ring. The C-terminal 8 amino acids of p27 (Fig. 1) interact with citron kinase and prevent its activation by RhoA by interfering with citron kinase-RhoA complex formation. Of note, the centrosome amplification observed in these p27CK-expressing cells is merely a consequence of cells undergoing multiple rounds of cell cycles without undergoing complete cytokinesis, as opposed to that seen with p27K cells in our study81 where it is an outcome of misregulated centrosome replication process. Contrary to these observations, the centrosome amplification seen in Skp2 KO cells (discussed above) appears to be a consequence of endoreduplication. In all these cases, however, increased induction of cell death by mitotic catastrophe and/or apoptosis is observed. Taken together, these findings suggest a gatekeeper function for p27 at the G2-M boundary that is independent of CDK inhibition via induction of mitotic catastrophe and/or apoptosis, culminating in abrogation of cell proliferation. Alternatively, observations such as that seen with p27K might point toward a potentially pro-oncogenic function for p27 in disrupting the tight regulation of the centrosome duplication process. In this context it is important to note that over-amplification of centrosomes is known to be one of the contributing causes of aneuploidy which in turn can fuel tumorigenesis.99
Conclusions and perspectives
Almost two decades have elapsed since the first discovery of a non-canonical role for p27 in the regulation of cell motility. In the intervening years many other CDK independent functions for the nuclear and cytoplasmic p27 in the control of key physiological and pathological processes have come to fore. This multifunctional character of p27 is attributed to its intrinsically unstructured nature that allows p27 to bind and influence a variety of different proteins. Thus, depending on the cell type and cell-specific context, p27 essays the dual roles of a tumor-suppressor or tumor-promoter. The tumor-suppressor, non-canonical functions involve its ability to inhibit the cell cycle by regulating the G1-S and G2-M transitions, through influence on processes such as microtubule stability, MAPK pathway, centrosome replication, transcriptional co-repression, microRNA expression, and initiation of DNA replication. It is possible that these non-canonical functions have evolved to either complement the CDK-inhibitory, canonical “gatekeeper” functions and/or to function as additional breaks when CDK inhibition by p27 is not needed, such as in the case of RB deletion. Thus, to remove these multiple breaks, cancer cells might have to not just inactivate the cyclin-CDK-binding domain, but also disrupt these additional functions of p27, many of which are dependent on its C-terminal region. Strikingly, a large number of the reported p27 somatic mutations in prostate and luminal breast cancers, and hairy cell leukemia, reside within its C-terminal portion.33-36 Further investigations are required to clarify whether, and if so, how each of these identified mutations disrupt various aspects of p27 function.
On the other hand, the effects on tumorigenesis and metastasis appear to be largely dependent on the cytoplasmic mislocalization of p27 and modulation of the pathways involved in cell motility and invasion, cellular plasticity such as EMT and MAT, and stem cell proliferation and/or differentiation. It appears cancer cells have learnt to co-opt the ability of intrinsically unstructured p27 to interact with diverse proteins while uncoupling p27 from its nuclear inhibitory functions through predominant mislocalization to the cytoplasm. Clearly, however, there is a lot more to be learnt about the mechanisms by which p27 regulates all these processes. For example, how does cytoplasmic p27 contribute to dysregulated stem cell expansion and/or differentiation? Do other novel mechanisms exist by which cytoplasmic p27 contributes to cellular plasticity and metastasis? In addition to cellular migration and invasion, survival of and colonization by the disseminated tumor-cells are other critical aspects of the metastatic process. It is conceivable that p27 might contribute to these processes as well, both in a CDK-dependent and –independent fashion. An attractive hypothesis is that p27 might serve as a molecular switch that coordinates tumor-cell migration, dormancy, and eventual colonization of distant sites during metastasis. For instance, in carcinoma cells that have succeeded in hyperphosphorylating p27 and shuttling it to and/or retaining it in the cytoplasm, p27 might contribute to enhance EMT, motility and invasion, allowing these cells to disseminate to distant sites; however, upon arrival at distant hostile microenvironments, these cells might undergo a mesenchymal-epithelial transition, losing the hyperphosphorylated status of p27 and inducing its nuclear expression, thus facilitating an entry into dormancy. In due course, however, development of tumor-cell microenvironment and/or genetic/epigenetic conditions favorable to removing p27 from the nucleus might eventually contribute to the reawakening of these cells from dormancy to allow macrometastasis formation and further dissemination. Furthermore, these various emerging non-canonical roles could have therapeutic implications for the development of targeted anti-cancer drugs (such as GGTI-2418100, Compound-A101, and Compound #25102) that seek to indirectly or directly inhibit the degradation of p27 and thereby suppress tumor-growth, at least in part, by increasing the cellular levels of p27. It is plausible that the increased p27 in these drug-treated tumors might eventually end up turning pro-oncogenic in the event a tumor cell acquires a new oncogenic mutation that is capable of cooperating with p27. Therefore, a greater mechanistic understanding of how cytoplasmic p27 promotes tumorigenesis and/or metastasis through modulation of multiple pathways, including those involved in cell migration, EMT, MAT, and CSC biology, may ultimately permit the design of more effective combinatorial therapies in cancer-types characterized by high levels of cytoplasmic p27.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Our review is largely focused on the CDK-independent functions of p27Kip1; we apologize in advance to those authors whose contributions have been inadvertently omitted or have not received the attention they deserve.
Funding
This review is supported by funding from the Gibbs Research Foundation and SRH Foundation.
References
- [1].Koff A, Ohtsuki M, Polyak K, Roberts JM, Massague J. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science 1993; 260:536-9; PMID:8475385; http://dx.doi.org/ 10.1126/science.8475385 [DOI] [PubMed] [Google Scholar]
- [2].Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci U S A 1994; 91:5291-5; PMID:8202483; http://dx.doi.org/ 10.1073/pnas.91.12.5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev 1994; 8:9-22; PMID:8288131; http://dx.doi.org/ 10.1101/gad.8.1.9 [DOI] [PubMed] [Google Scholar]
- [4].Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massague J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994; 78:59-66; PMID:8033212; http://dx.doi.org/ 10.1016/0092-8674(94)90572-X [DOI] [PubMed] [Google Scholar]
- [5].Slingerland JM, Hengst L, Pan CH, Alexander D, Stampfer MR, Reed SI. A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor beta-arrested epithelial cells. Mol Cell Biol 1994; 14:3683-94; PMID:8196612; http://dx.doi.org/ 10.1128/MCB.14.6.3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bagui TK, Jackson RJ, Agrawal D, Pledger WJ. Analysis of cyclin D3-cdk4 complexes in fibroblasts expressing and lacking p27(kip1) and p21(cip1). Mol Cell Biol 2000; 20:8748-57; PMID:11073976; http://dx.doi.org/ 10.1128/MCB.20.23.8748-8757.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dong F, Agrawal D, Bagui T, Pledger WJ. Cyclin D3-associated kinase activity is regulated by p27kip1 in BALB/c 3T3 cells. Mol Biol Cell 1998; 9:2081-92; PMID:9693368; http://dx.doi.org/ 10.1091/mbc.9.8.2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994; 78:67-74; PMID:8033213; http://dx.doi.org/ 10.1016/0092-8674(94)90573-8 [DOI] [PubMed] [Google Scholar]
- [9].Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 1996; 382:325-31; PMID:8684460; http://dx.doi.org/ 10.1038/382325a0 [DOI] [PubMed] [Google Scholar]
- [10].Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger WJ. Repression of p27kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol Cell Biol 1996; 16:4327-36; PMID:8754833; http://dx.doi.org/ 10.1128/MCB.16.8.4327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang X, Wharton W, Donovan M, Coppola D, Croxton R, Cress WD, Pledger WJ. Density-dependent growth inhibition of fibroblasts ectopically expressing p27(kip1). Mol Biol Cell 2000; 11:2117-30; PMID:10848633; http://dx.doi.org/ 10.1091/mbc.11.6.2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bagui TK, Cui D, Roy S, Mohapatra S, Shor AC, Ma L, Pledger WJ. Inhibition of p27Kip1 gene transcription by mitogens. Cell Cycle 2009; 8:115-24; PMID:19158484; http://dx.doi.org/ 10.4161/cc.8.1.7527 [DOI] [PubMed] [Google Scholar]
- [13].Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999; 1:193-9; PMID:10559916; http://dx.doi.org/ 10.1038/12013 [DOI] [PubMed] [Google Scholar]
- [14].Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S. Degradation of p27(Kip1) at the G(0)-G(1) transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem 2001; 276:48937-43; PMID:11682478; http://dx.doi.org/ 10.1074/jbc.M107274200 [DOI] [PubMed] [Google Scholar]
- [15].Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakides TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature 2001; 413:323-7; PMID:11565035; http://dx.doi.org/ 10.1038/35095083 [DOI] [PubMed] [Google Scholar]
- [16].Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol 2004; 6:1229-35; PMID:15531880; http://dx.doi.org/ 10.1038/ncb1194 [DOI] [PubMed] [Google Scholar]
- [17].Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, et al.. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 1996; 85:733-44; PMID:8646781; http://dx.doi.org/ 10.1016/S0092-8674(00)81239-8 [DOI] [PubMed] [Google Scholar]
- [18].Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 1996; 85:721-32; PMID:8646780; http://dx.doi.org/ 10.1016/S0092-8674(00)81238-6 [DOI] [PubMed] [Google Scholar]
- [19].Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 1996; 85:707-20; PMID:8646779; http://dx.doi.org/ 10.1016/S0092-8674(00)81237-4 [DOI] [PubMed] [Google Scholar]
- [20].Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998; 396:177-80; PMID:9823898; http://dx.doi.org/ 10.1038/24179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jackson RJ, Adnane J, Coppola D, Cantor A, Sebti SM, Pledger WJ. Loss of the cell cycle inhibitors p21(Cip1) and p27(Kip1) enhances tumorigenesis in knockout mouse models. Oncogene 2002; 21:8486-97; PMID:12466968; http://dx.doi.org/ 10.1038/sj.onc.1205946 [DOI] [PubMed] [Google Scholar]
- [22].Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev 2006; 20:47-64; PMID:16391232; http://dx.doi.org/ 10.1101/gad.1384406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Franklin DS, Godfrey VL, O'Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol 2000; 20:6147-58; PMID:10913196; http://dx.doi.org/ 10.1128/MCB.20.16.6147-6158.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet 2001; 27:222-4; PMID:11175795; http://dx.doi.org/ 10.1038/84879 [DOI] [PubMed] [Google Scholar]
- [25].Park MS, Rosai J, Nguyen HT, Capodieci P, Cordon-Cardo C, Koff A. p27 and Rb are on overlapping pathways suppressing tumorigenesis in mice. Proc Natl Acad Sci U S A 1999; 96:6382-7; PMID:10339596; http://dx.doi.org/ 10.1073/pnas.96.11.6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Martin-Caballero J, Flores JM, Garcia-Palencia P, Collado M, Serrano M. Different cooperating effect of p21 or p27 deficiency in combination with INK4a/ARF deletion in mice. Oncogene 2004; 23:8231-7; PMID:15378017; http://dx.doi.org/ 10.1038/sj.onc.1207863 [DOI] [PubMed] [Google Scholar]
- [27].Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, Fend F, Graw J, Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A 2006; 103:15558-63; PMID:17030811; http://dx.doi.org/ 10.1073/pnas.0603877103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, Vierimaa O, Makinen MJ, Tuppurainen K, Paschke R, et al.. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J clin endocrinol Metab 2007; 92:3321-5; PMID:17519308; http://dx.doi.org/ 10.1210/jc.2006-2843 [DOI] [PubMed] [Google Scholar]
- [29].Molatore S, Marinoni I, Lee M, Pulz E, Ambrosio MR, degli Uberti EC, Zatelli MC, Pellegata NS. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum mutat 2010; 31:E1825-35; PMID:20824794; http://dx.doi.org/ 10.1002/humu.21354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, Ferasin S, Cetani F, Pardi E, Korbonits M, et al.. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet 2013; 9:e1003350; PMID:23555276; http://dx.doi.org/ 10.1371/journal.pgen.1003350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008; 8:253-67; PMID:18354415; http://dx.doi.org/ 10.1038/nrc2347 [DOI] [PubMed] [Google Scholar]
- [32].Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 2000; 183:10-7; PMID:10699961; http://dx.doi.org/ 10.1002/(SICI)1097-4652(200004)183:1%3c10::AID-JCP2%3e3.0.CO;2-I [DOI] [PubMed] [Google Scholar]
- [33].Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al.. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012; 486:353-60; PMID:22722193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al.. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486:400-4; PMID:22722201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al.. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012; 44:685-9; PMID:22610119; http://dx.doi.org/ 10.1038/ng.2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dietrich S, Hullein J, Lee SC, Hutter B, Gonzalez D, Jayne S, Dyer MJ, Oles M, Else M, Liu X, et al.. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood 2015; 126:1005-8; PMID:26065650; http://dx.doi.org/ 10.1182/blood-2015-04-643361 [DOI] [PubMed] [Google Scholar]
- [37].Ponce-Castaneda MV, Lee MH, Latres E, Polyak K, Lacombe L, Montgomery K, Mathew S, Krauter K, Sheinfeld J, Massague J, et al.. p27Kip1: chromosomal mapping to 12p12-12p13.1 and absence of mutations in human tumors. Cancer research 1995; 55:1211-4; PMID:7882310 [PubMed] [Google Scholar]
- [38].Kawamata N, Morosetti R, Miller CW, Park D, Spirin KS, Nakamaki T, Takeuchi S, Hatta Y, Simpson J, Wilcyznski S, et al.. Molecular analysis of the cyclin-dependent kinase inhibitor gene p27/Kip1 in human malignancies. Cancer Res 1995; 55:2266-9; PMID:7757974 [PubMed] [Google Scholar]
- [39].Spirin KS, Simpson JF, Takeuchi S, Kawamata N, Miller CW, Koeffler HP. p27/Kip1 mutation found in breast cancer. Cancer Res 1996; 56:2400-4; PMID:8625318 [PubMed] [Google Scholar]
- [40].Tanaka C, Yoshimoto K, Yang P, Kimura T, Yamada S, Moritani M, Sano T, Itakura M. Infrequent mutations of p27Kip1 gene and trisomy 12 in a subset of human pituitary adenomas. The Journal of clinical endocrinology and metabolism 1997; 82:3141-7; PMID:9284759 [DOI] [PubMed] [Google Scholar]
- [41].Chen G, Cheng Y, Zhang Z, Martinka M, Li G. Prognostic significance of cytoplasmic p27 expression in human melanoma. Cancer epidemiol biomarkers Prev 2011; 20:2212-21; PMID:21828232; http://dx.doi.org/ 10.1158/1055-9965.EPI-11-0472 [DOI] [PubMed] [Google Scholar]
- [42].Denicourt C, Saenz CC, Datnow B, Cui XS, Dowdy SF. Relocalized p27Kip1 tumor suppressor functions as a cytoplasmic metastatic oncogene in melanoma. Cancer Res 2007; 67:9238-43; PMID:17909030; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1375 [DOI] [PubMed] [Google Scholar]
- [43].Rosen DG, Yang G, Cai KQ, Bast RC Jr., Gershenson DM, Silva EG, Liu J. Subcellular localization of p27kip1 expression predicts poor prognosis in human ovarian cancer. Clin Cancer Res 2005; 11:632-7; PMID:15701850 [PubMed] [Google Scholar]
- [44].Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, et al.. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 2002; 8:1153-60; PMID:12244302; http://dx.doi.org/ 10.1038/nm761 [DOI] [PubMed] [Google Scholar]
- [45].Singh SP, Lipman J, Goldman H, Ellis FH Jr., Aizenman L, Cangi MG, Signoretti S, Chiaur DS, Pagano M, Loda M. Loss or altered subcellular localization of p27 in Barrett's associated adenocarcinoma. Cancer Res 1998; 58:1730-5; PMID:9563491 [PubMed] [Google Scholar]
- [46].Kruck S, Merseburger AS, Hennenlotter J, Scharpf M, Eyrich C, Amend B, Sievert KD, Stenzl A, Bedke J. High cytoplasmic expression of p27(Kip1) is associated with a worse cancer-specific survival in clear cell renal cell carcinoma. BJU Int 2012; 109:1565-70; PMID:21981759; http://dx.doi.org/ 10.1111/j.1464-410X.2011.10649.x [DOI] [PubMed] [Google Scholar]
- [47].Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res 2011; 17:12-8; PMID:20966355; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science 2003; 302:1704-9; PMID:14657486; http://dx.doi.org/ 10.1126/science.1092053 [DOI] [PubMed] [Google Scholar]
- [49].Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002; 420:629-35; PMID:12478284; http://dx.doi.org/ 10.1038/nature01148 [DOI] [PubMed] [Google Scholar]
- [50].Butty AC, Pryciak PM, Huang LS, Herskowitz I, Peter M. The role of Far1p in linking the heterotrimeric G protein to polarity establishment proteins during yeast mating. Science 1998; 282:1511-6; PMID:9822386; http://dx.doi.org/ 10.1126/science.282.5393.1511 [DOI] [PubMed] [Google Scholar]
- [51].Shimada Y, Gulli MP, Peter M. Nuclear sequestration of the exchange factor Cdc24 by Far1 regulates cell polarity during yeast mating. Nat Cell Biol 2000; 2:117-24; PMID:10655592; http://dx.doi.org/ 10.1038/35000073 [DOI] [PubMed] [Google Scholar]
- [52].McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol 2003; 23:216-28; PMID:12482975; http://dx.doi.org/ 10.1128/MCB.23.1.216-228.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med 1998; 4:1449-52; PMID:9846587; http://dx.doi.org/ 10.1038/4042 [DOI] [PubMed] [Google Scholar]
- [54].Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 2004; 18:862-76; PMID:15078817; http://dx.doi.org/ 10.1101/gad.1185504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wu FY, Wang SE, Sanders ME, Shin I, Rojo F, Baselga J, Arteaga CL. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res 2006; 66:2162-72; PMID:16489017; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3304 [DOI] [PubMed] [Google Scholar]
- [56].Li Z, Jiao X, Wang C, Ju X, Lu Y, Yuan L, Lisanti MP, Katiyar S, Pestell RG. Cyclin D1 induction of cellular migration requires p27(KIP1). Cancer Res 2006; 66:9986-94; PMID:17047061; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1596 [DOI] [PubMed] [Google Scholar]
- [57].Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, Smith JA, Slingerland JM. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A 2009; 106:9268-73; PMID:19470470; http://dx.doi.org/ 10.1073/pnas.0805057106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat Cell Biol 2006; 8:17-26; PMID:16341208; http://dx.doi.org/ 10.1038/ncb1338 [DOI] [PubMed] [Google Scholar]
- [59].Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, Philpott A, Roberts JM, Guillemot F. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev 2006; 20:1511-24; PMID:16705040; http://dx.doi.org/ 10.1101/gad.377106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Godin JD, Thomas N, Laguesse S, Malinouskaya L, Close P, Malaise O, Purnelle A, Raineteau O, Campbell K, Fero M, et al.. p27(Kip1) is a microtubule-associated protein that promotes microtubule polymerization during neuron migration. Dev Cell 2012; 23:729-44; PMID:23022035; http://dx.doi.org/ 10.1016/j.devcel.2012.08.006 [DOI] [PubMed] [Google Scholar]
- [61].Goukassian D, Diez-Juan A, Asahara T, Schratzberger P, Silver M, Murayama T, Isner JM, Andres V. Overexpression of p27(Kip1) by doxycycline-regulated adenoviral vectors inhibits endothelial cell proliferation and migration and impairs angiogenesis. FASEB J 2001; 15:1877-85; PMID:11532967; http://dx.doi.org/ 10.1096/fj.01-0065com [DOI] [PubMed] [Google Scholar]
- [62].Sun J, Marx SO, Chen HJ, Poon M, Marks AR, Rabbani LE. Role for p27(Kip1) in Vascular Smooth Muscle Cell Migration. Circulation 2001; 103:2967-72; PMID:11413088; http://dx.doi.org/ 10.1161/01.CIR.103.24.2967 [DOI] [PubMed] [Google Scholar]
- [63].Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P, Morrione A, Canzonieri V, Colombatti A. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell 2005; 7:51-63; PMID:15652749; http://dx.doi.org/ 10.1016/j.ccr.2004.11.025 [DOI] [PubMed] [Google Scholar]
- [64].Schiappacassi M, Lovat F, Canzonieri V, Belletti B, Berton S, Di Stefano D, Vecchione A, Colombatti A, Baldassarre G. p27Kip1 expression inhibits glioblastoma growth, invasion, and tumor-induced neoangiogenesis. Mol Cancer ther 2008; 7:1164-75; PMID:18483304; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-2154 [DOI] [PubMed] [Google Scholar]
- [65].Belletti B, Pellizzari I, Berton S, Fabris L, Wolf K, Lovat F, Schiappacassi M, D'Andrea S, Nicoloso MS, Lovisa S, et al.. p27kip1 controls cell morphology and motility by regulating microtubule-dependent lipid raft recycling. Mol Cell Biol 2010; 30:2229-40; PMID:20194624; http://dx.doi.org/ 10.1128/MCB.00723-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Berton S, Belletti B, Wolf K, Canzonieri V, Lovat F, Vecchione A, Colombatti A, Friedl P, Baldassarre G. The tumor suppressor functions of p27(kip1) include control of the mesenchymal/amoeboid transition. Mol Cell Biol 2009; 29:5031-45; PMID:19596789; http://dx.doi.org/ 10.1128/MCB.00144-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, Sun P, Tan CK, Hengst L, Slingerland J. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell 2007; 128:281-94; PMID:17254967; http://dx.doi.org/ 10.1016/j.cell.2006.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW, Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 2007; 128:269-80; PMID:17254966; http://dx.doi.org/ 10.1016/j.cell.2006.11.047 [DOI] [PubMed] [Google Scholar]
- [69].Cheli Y, Giuliano S, Botton T, Rocchi S, Hofman V, Hofman P, Bahadoran P, Bertolotto C, Ballotti R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 2011; 30:2307-18; PMID:21278797; http://dx.doi.org/ 10.1038/onc.2010.598 [DOI] [PubMed] [Google Scholar]
- [70].Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011; 331:1559-64; PMID:21436443; http://dx.doi.org/ 10.1126/science.1203543 [DOI] [PubMed] [Google Scholar]
- [71].Besson A, Hwang HC, Cicero S, Donovan SL, Gurian-West M, Johnson D, Clurman BE, Dyer MA, Roberts JM. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev 2007; 21:1731-46; PMID:17626791; http://dx.doi.org/ 10.1101/gad.1556607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Serres MP, Zlotek-Zlotkiewicz E, Concha C, Gurian-West M, Daburon V, Roberts JM, Besson A. Cytoplasmic p27 is oncogenic and cooperates with Ras both in vivo and in vitro. Oncogene 2011; 30:2846-58; PMID:21317921; http://dx.doi.org/ 10.1038/onc.2011.9 [DOI] [PubMed] [Google Scholar]
- [73].Menchon C, Edel MJ, Izpisua Belmonte JC. The cell cycle inhibitor p27(Kip)(1) controls self-renewal and pluripotency of human embryonic stem cells by regulating the cell cycle, Brachyury and Twist. Cell Cycle 2011; 10:1435-47; PMID:21478681; http://dx.doi.org/ 10.4161/cc.10.9.15421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Egozi D, Shapira M, Paor G, Ben-Izhak O, Skorecki K, Hershko DD. Regulation of the cell cycle inhibitor p27 and its ubiquitin ligase Skp2 in differentiation of human embryonic stem cells. FASEB J 2007; 21:2807-17; PMID:17475922; http://dx.doi.org/ 10.1096/fj.06-7758com [DOI] [PubMed] [Google Scholar]
- [75].Bryja V, Cajanek L, Pachernik J, Hall AC, Horvath V, Dvorak P, Hampl A. Abnormal development of mouse embryoid bodies lacking p27Kip1 cell cycle regulator. Stem Cells 2005; 23:965-74; PMID:15941856; http://dx.doi.org/ 10.1634/stemcells.2004-0174 [DOI] [PubMed] [Google Scholar]
- [76].Li H, Collado M, Villasante A, Matheu A, Lynch CJ, Canamero M, Rizzoti K, Carneiro C, Martinez G, Vidal A, et al.. p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell 2012; 11:845-52; PMID:23217425; http://dx.doi.org/ 10.1016/j.stem.2012.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Pippa R, Espinosa L, Gundem G, Garcia-Escudero R, Dominguez A, Orlando S, Gallastegui E, Saiz C, Besson A, Pujol MJ, et al.. p27Kip1 represses transcription by direct interaction with p130/E2F4 at the promoters of target genes. Oncogene 2012; 31:4207-20; PMID:22179826; http://dx.doi.org/ 10.1038/onc.2011.582 [DOI] [PubMed] [Google Scholar]
- [78].Zhao D, Besser AH, Wander SA, Sun J, Zhou W, Wang B, Ince T, Durante MA, Guo W, Mills G, et al.. Cytoplasmic p27 promotes epithelial-mesenchymal transition and tumor metastasis via STAT3-mediated Twist1 upregulation. Oncogene 2015; 34:5447-59; PMID:25684140; http://dx.doi.org/ 10.1038/onc.2014.473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2:442-54; PMID:12189386; http://dx.doi.org/ 10.1038/nrc822 [DOI] [PubMed] [Google Scholar]
- [80].Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al.. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704-15; PMID:18485877; http://dx.doi.org/ 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sharma SS, Ma L, Bagui TK, Forinash KD, Pledger WJ. A p27(Kip1) mutant that does not inhibit CDK activity promotes centrosome amplification and micronucleation. Oncogene 2011; PMID:22158041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J 1997; 16:5334-44; PMID:9311993; http://dx.doi.org/ 10.1093/emboj/16.17.5334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, et al.. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 2000; 19:2069-81; PMID:10790373; http://dx.doi.org/ 10.1093/emboj/19.9.2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004; 6:661-72; PMID:15130491; http://dx.doi.org/ 10.1016/S1534-5807(04)00131-5 [DOI] [PubMed] [Google Scholar]
- [85].Payne SR, Zhang S, Tsuchiya K, Moser R, Gurley KE, Longton G, deBoer J, Kemp CJ. p27kip1 deficiency impairs G2/M arrest in response to DNA damage, leading to an increase in genetic instability. Mol Cell Biol 2008; 28:258-68; PMID:17954563; http://dx.doi.org/ 10.1128/MCB.01536-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Moeller SJ, Head ED, Sheaff RJ. p27Kip1 inhibition of GRB2-SOS formation can regulate Ras activation. Mol Cell Biol 2003; 23:3735-52; PMID:12748278; http://dx.doi.org/ 10.1128/MCB.23.11.3735-3752.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Sugiyama Y, Tomoda K, Tanaka T, Arata Y, Yoneda-Kato N, Kato J. Direct binding of the signal-transducing adaptor Grb2 facilitates down-regulation of the cyclin-dependent kinase inhibitor p27Kip1. J Biol Chem 2001; 276:12084-90; PMID:11278754; http://dx.doi.org/ 10.1074/jbc.M010811200 [DOI] [PubMed] [Google Scholar]
- [88].Fabris L, Berton S, Pellizzari I, Segatto I, D'Andrea S, Armenia J, Bomben R, Schiappacassi M, Gattei V, Philips MR, et al.. p27kip1 controls H-Ras/MAPK activation and cell cycle entry via modulation of MT stability. Proc Natl Acad Sci U S A 2015; 112:13916-21; PMID:26512117; http://dx.doi.org/ 10.1073/pnas.1508514112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Berton S, Pellizzari I, Fabris L, D'Andrea S, Segatto I, Canzonieri V, Marconi D, Schiappacassi M, Benevol S, Gattei V, et al.. Genetic characterization of p27(kip1) and stathmin in controlling cell proliferation in vivo. Cell Cycle 2014; 13:3100-11; PMID:25486569; http://dx.doi.org/ 10.4161/15384101.2014.949512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Jeannot P, Callot C, Baer R, Duquesnes N, Guerra C, Guillermet-Guibert J, Bachs O, Besson A. Loss of p27Kip1 promotes metaplasia in the pancreas via the regulation of Sox9 expression. Oncotarget 2015; 6:35880-92; PMID:26416424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Perez-Luna M, Aguasca M, Perearnau A, Serratosa J, Martinez-Balbas M, Jesus Pujol M, Bachs O. PCAF regulates the stability of the transcriptional regulator and cyclin-dependent kinase inhibitor p27 Kip1. Nucleic Acids Res 2012; 40:6520-33; PMID:22547391; http://dx.doi.org/ 10.1093/nar/gks343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Armenia J, Fabris L, Lovat F, Berton S, Segatto I, D'Andrea S, Ivan C, Cascione L, Calin GA, Croce CM, et al.. Contact inhibition modulates intracellular levels of miR-223 in a p27kip1-dependent manner. Oncotarget 2014; 5:1185-97; PMID:24727437; http://dx.doi.org/ 10.18632/oncotarget.1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nallamshetty S, Crook M, Boehm M, Yoshimoto T, Olive M, Nabel EG. The cell cycle regulator p27Kip1 interacts with MCM7, a DNA replication licensing factor, to inhibit initiation of DNA replication. FEBS letters 2005; 579:6529-36; PMID:16289477; http://dx.doi.org/ 10.1016/j.febslet.2005.10.028 [DOI] [PubMed] [Google Scholar]
- [94].Sharma SS, Ma L, Pledger WJ. p27Kip1 inhibits the cell cycle through non-canonical G1/S phase-specific gatekeeper mechanism. Cell Cycle 2015; 14:3954-64; PMID:26697844; http://dx.doi.org/ 10.1080/15384101.2015.1100775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Tetzlaff MT, Bai C, Finegold M, Wilson J, Harper JW, Mahon KA, Elledge SJ. Cyclin F disruption compromises placental development and affects normal cell cycle execution. Mol Cell Biol 2004; 24:2487-98; PMID:14993286; http://dx.doi.org/ 10.1128/MCB.24.6.2487-2498.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].D'Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, Dynlacht B, Pagano M. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 2010; 466:138-42; PMID:20596027; http://dx.doi.org/ 10.1038/nature09140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Bai C, Richman R, Elledge SJ. Human cyclin F. EMBO J 1994; 13:6087-98; PMID:7813445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 1996; 86:263-74; PMID:8706131; http://dx.doi.org/ 10.1016/S0092-8674(00)80098-7 [DOI] [PubMed] [Google Scholar]
- [99].Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer 2002; 2:815-25; PMID:12415252; http://dx.doi.org/ 10.1038/nrc924 [DOI] [PubMed] [Google Scholar]
- [100].Kazi A, Carie A, Blaskovich MA, Bucher C, Thai V, Moulder S, Peng H, Carrico D, Pusateri E, Pledger WJ, et al.. Blockade of protein geranylgeranylation inhibits Cdk2-dependent p27Kip1 phosphorylation on Thr187 and accumulates p27Kip1 in the nucleus: implications for breast cancer therapy. Mol Cell Biol 2009; 29:2254-63; PMID:19204084; http://dx.doi.org/ 10.1128/MCB.01029-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, Corral LG, Krenitsky VP, Xu W, Moutouh-de Parseval L, et al.. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008; 111:4690-9; PMID:18305219; http://dx.doi.org/ 10.1182/blood-2007-09-112904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, et al.. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013; 154:556-68; PMID:23911321; http://dx.doi.org/ 10.1016/j.cell.2013.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lovisa S, Citro S, Sonego M, Dall'Acqua A, Ranzuglia V, Berton S, Colombatti A, Belletti B, Chiocca S, Schiappacassi M, et al.. SUMOylation regulates p27Kip1 stability and localization in response to TGFbeta. J Mol Cell Biol 2015; PMID:26450989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Connor MK, Kotchetkov R, Cariou S, Resch A, Lupetti R, Beniston RG, Melchior F, Hengst L, Slingerland JM. CRM1/Ran-mediated nuclear export of p27(Kip1) involves a nuclear export signal and links p27 export and proteolysis. Mol Biol Cell 2003; 14:201-13; PMID:12529437; http://dx.doi.org/ 10.1091/mbc.E02-06-0319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Smitherman M, Lee K, Swanger J, Kapur R, Clurman BE. Characterization and targeted disruption of murine Nup50, a p27(Kip1)-interacting component of the nuclear pore complex. Mol Cell Biol 2000; 20:5631-42; PMID:10891500; http://dx.doi.org/ 10.1128/MCB.20.15.5631-5642.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Muller D, Thieke K, Burgin A, Dickmanns A, Eilers M. Cyclin E-mediated elimination of p27 requires its interaction with the nuclear pore-associated protein mNPAP60. EMBO J 2000; 19:2168-80; PMID:10811608; http://dx.doi.org/ 10.1093/emboj/19.10.2168 [DOI] [PMC free article] [PubMed] [Google Scholar]