

ABSTRACT
Activation and proliferation of T cells are tightly regulated during the immune response. We show here that kinetics of proliferation of PHA activated T cells follows the expression of cMyc. Expression of p53 is also elevated and remains high several days after activation. To investigate the role of p53 in activated T cells, its expression was further elevated with nultin-3 treatment, a small molecule that dissociates the E3 ubiquitin protein ligase MDM2 from p53. Concomitantly, cMyc expression and proliferation decreased. At the other end of the cMyc-p53 axis, inhibition of cMyc with 10058-F4 led to down regulation of p53, likely through the lower level of cMyc induced p14ARF, which is also known to dissociate the p53-MDM2 complex. Both compounds induced cell cycle arrest and apoptosis. We conclude that the feedback regulation between cMyc and p53 is important for the T cell homeostasis. We also show that the two compounds modulating p53 and cMyc levels inhibited proliferation without abolishing the cytotoxic function, thus demonstrating the dichotomy between proliferation and cytotoxicity in activated T cells.
KEYWORDS: apoptosis, cMyc, nutlin-3, p53, T cells, 10058-F4
Introduction
T cells are central in immune response. In the absence of an antigen, T cells are in the ‘resting state’. They are activated by ligation of specific T cell receptor (TCR) with peptide antigen loaded major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs). Subsequently, proliferation of antigen specific T cells is initiated accompanying their differentiation determined functions.1,2
The dynamics of cellular homeostasis in the acute immune response is tightly regulated. Cessation of proliferation and the return of the T cells to a resting state is essential. A part of the T cells that have gone through this initial proliferation are eliminated by apoptosis – activation induced cell death (AICD). We have previously shown that the tumor suppressor p53 contributes to this process through the induction of the pro-apoptotic protein SAP.3,4
p53 regulates cell cycle progression and contributes to the maintenance of genome integrity.5 Recent studies have also indicated that p53 suppresses cellular senescence through the inhibition of mTOR pathway.6,7 Cellular metabolism can also be altered by p53 via PTEN-mTOR pathway.8 Loss of p53 function, either by mutation in p53 itself, overexpression of the p53 antagonist MDM2, or expression of viral proteins such as the Human Papilloma Virus (HPV) E6 protein, may allow evasion from apoptosis or senescence in response to oncogenic stress.9 Mutations in p53 occur in more than 50% of common human malignancies.10 p53 knockout mice are prone to develop T cell lymphomas in spite of apparently normal T cell development and immune response,11,12 underlining the role of p53 in regulating T cell proliferation.
cMyc activity is essential for the expansion of T cell clones. cMyc is a transcription factor regulating diverse sets of genes involved in proliferation, metabolism, apoptosis, senescence and differentiation.13 The induction of different target genes, thus execution of the different roles of cMyc depends upon the cellular context.14 Deregulation of cMyc expression by chromosomal translocation or gene amplification contributes to the genesis of several malignancies, such as Burkitt lymphoma.15 The role of cMyc in the generation of T cell lymphomas was studied in transgenic mice. Targeted expression of cMyc in T cells (CD2-myc) induced a low incidence of spontaneous T cell lymphomas.16 However, expression of CD2-myc showed synergy with the p53 deficient genotype, as mice developed thymic lymphomas with dramatically increased frequency and reduced latency compared to both parental groups.17
cMyc induces p53 through the involvement of p14ARF.18 Competitive binding of cMyc induced p14ARF to MDM2 inhibits p53 degradation which is otherwise mediated through the formation of a MDM2-p53 complex and MDM2-mediated p53 ubiquitination.19 In turn, it has also been shown that p53 can suppress cMyc transcription.20 Thus, cMyc and p53 can regulate each other at several levels to control cell growth and survival. This motivated us to study the cMyc-p53 network in a model where the cMyc and p53 pathways were unaltered. To this end we activated primary T cells with PHA as model for proliferating lymphocytes. To our knowledge, the roles of cMyc and p53 in T cell homeostasis have not been studied yet. Here we show that both cMyc and p53 are induced upon T cell activation. Further induction of p53 by nutlin-3 leads to reduced expression of cMyc, while inhibition of cMyc results in lower levels of p53. We conclude that the cMyc-p53 feedback mechanism contributes to the cessation of T cell proliferation in the immune response.
Results
cMyc and p14ARF expression are induced in activated T cells
We have previously reported that p53 is expressed in activated T cells where it induces the pro-apoptotic protein SAP that contributes to T cell homeostasis.3,4 Here we continued to study parameters related to the expression of p53 in proliferating T cells. Following activation with PHA, proliferation and cMyc expression were induced (Fig. 1A), reaching a peak on day 3 after which both started to decline. In accordance with cMyc induction, its target p14ARF was also upregulated in the activated T cells (Fig. 1B). In line with our previous results, p53 was induced and further upregulated at later timepoints in activated T cells (Fig. 1B). While p53 levels remained high, the levels of p21 mRNA and protein gradually decreased (Fig. 1B & E). The results were confirmed at the mRNA level also for the other 3 genes. cMyc mRNA expression was high only on the first day (Fig. 1C), while p14ARF mRNA gradually increased (Fig. 1F). Similarly, p53 mRNA gradually increased until day 3, thereafter declined (Fig. 1D). The high level of cMyc accompanied by high p53 levels, suggested the possibility that, like in other cell types, expression of cMyc in activated T cells may lead to p53 induction through the p14ARF-MDM2 pathway.
Figure 1.
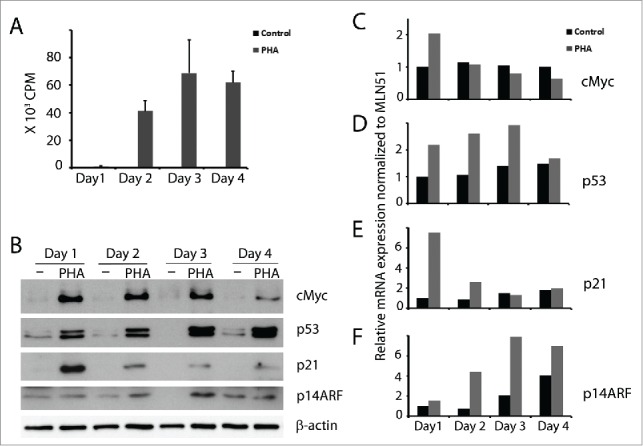
cMyc, p53, p21 and p14ARF expression in activated T cells. T cells were isolated from buffy coat and cultured without and with PHA (1 µg/ml). (A) 3H-thymidine incorporation of 105 cells cultured from day 0 and pulsed with 3H-thymidine at the indicated time points for 16 h. (B) cMyc, p53, p21, p14ARF were detected by immunoblot. Kinetics of (C) cMyc, (D) p53, (E) p21 and (F) p14ARF mRNA expression were measured by real time RT-PCR at the indicated time points (black bars - control T cells; gray bars - PHA activated T cells).
High p53 levels downregulate cMyc expression in activated T cells
We have reported earlier that nutlin-3 treatment potentiates p53 and p21 expression and inhibits cell proliferation in activated T cells.4 Elevation of p53 has been shown to suppress cMyc transcription in different cell lines and by that cause cell cycle arrest.20 Based on these findings, we asked whether p53 would suppress cMyc expression in activated T cells. To this end, we treated activated T cells with nutlin-3 for 24 and 48 hours. This led to increased p53 level (Fig. 2B) and decreased proliferation (Fig. 2A).
Figure 2.

Nutlin-3 induced p53 downregulated cMyc expression in activated T cells. (A) 105 T cells were activated with PHA (1 µg/ml) and cultured without and with nutlin-3 (5 µM). Activated T cells were pulsed with 3H-thymidine at the indicated time point for 16 h (black bars - activated T cells cultured with the solvent control DMSO; gray bars – activated T cells cultured with nutlin-3). (B) cMyc, p53, p21 and p14ARF were detected at the indicated time points in T cells cultured with and without PHA (1 µg/ml) and with and without nutlin-3 (5 µM). (C) mRNA expression of cMyc, p21 and p53 measured by real time RT-PCR in T cells activated with PHA for 24 h and treated with or without nutlin-3 (5 µM) for the last 8 h (black bars - activated T cells cultured with the solvent control DMSO; gray bars - activated T cells cultured with nutlin-3). Values represent average of 3 experiments with SD.
Levels of p21, a bona fide p53 target, did not change in cells cultured with nutlin-3 for 24 h, but after 48 h it was elevated (Fig. 2B). Importantly, in activated T cells upregulation of p53 by nutlin-3 was accompanied by downregulation of cMyc expression. As a consequence of lower cMyc levels, p14ARF was also downregulated at 24 h (Fig. 2B). Thus, p53 induction had a negative effect on cMyc expression in activated T cells. In addition, p53s pro-apoptotic transcriptional target BAX21 was upregulated at 48 h in activated T cell cultures treated with nutlin-3.
We confirmed p53s inhibitory effect on cMyc at transcriptional level as well. Short term treatment with nutlin-3 (last 8 hours) of T cells activated for 24 h led to decreased cMyc mRNA expression, suggesting that p53 can suppress cMyc transcription (Fig. 2C). As control, we confirmed upregulation of p21 mRNA levels (Fig. 2C) and we found no changes in p53 mRNA levels, consistent with the notion that nutlin-3 elevates p53 protein levels by inhibiting MDM2-mediated p53 degradation in the proteasome.22
Pharmacological inhibition of Notch or cMyc inhibits proliferation and expression of p14ARF and p53 in activated T cells
The Notch pathway is involved in cMyc induction following TCR stimulation, thus promoting proliferation.23 In order to further study the regulatory interplay between cMyc and p53, we tested the effect of Notch inhibition by DAPT24 (γ secretase inhibitor) in activated T cells. This treatment lead to decreased T cell proliferation in a dose-dependent manner (Fig. 3A), decreased cMyc expression (Fig. 3B), and correspondingly lower levels of p14ARF, but also lower levels of p53 after 24 h (Fig. 3B). These results suggested that p53 induction in activated T cells is downstream of Notch activation, possibly mediated by cMyc-p14ARF. For a more direct proof, we tested whether specific inhibition of cMyc would affect p53 expression. T cells were activated with PHA and cultured with the cMyc inhibitor (10058-F4, at a concentration of 50 μM).25 As expected, treatment with the cMyc inhibitor suppressed proliferation of PHA-activated T cells (Fig. 3C) and resulted in lower cMyc protein expression (Fig. 3D) and in lower p14ARF levels after 48 h. In parallel, expression of p53 was decreased (Fig. 3D), confirming the involvement of cMyc/p14ARF in its induction. When nutin-3 was in culture, p14ARF levels were also low in the activated T cells, consistent with the lower levels of cMyc (Fig. 3D).
Figure 3.
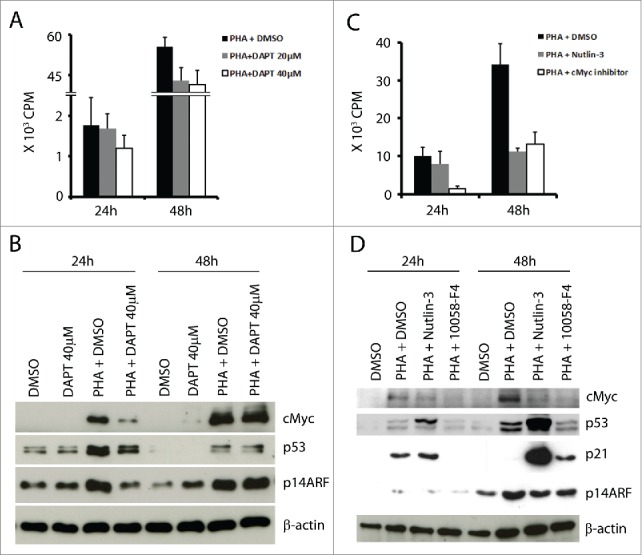
Inhibition of Notch (with DAPT) or of cMyc (with 10058-F4) reduced T cell proliferation and mediated downregulation of cMyc, p53 and p14ARF in activated T cells. (A) 105 T cells were activated and cultured with or without Notch inhibitor DAPT (20 and 40 µM) for 24 h and 48 h and pulsed with 3H-thymidine for the last 16 h. (B) cMyc, p53, and p14ARF were detected at the indicated time points in T cells cultured with or without PHA (1 µg/ml) and with or without DAPT (40 µM). (C) 105 T cells were activated and cultured with or without 10058-F4 (50 µM) from day 0 and pulsed with 3H-thymidine at the indicated time point for 16 h. (D) cMyc, p53, p21 and p14ARF were detected at the indicated time points in T cells cultured with or without PHA (1 µg/ml), nutlin-3 (5 µM) and 10058-F4 (50 µM).
Pharmacological induction of p53 or inhibition of cMyc leads to cell cycle arrest and apoptosis in PHA-activated T cells
As a consequence of p53 induction, activation of downstream target genes can promote cell cycle arrest and/or apoptosis. On the other hand, cMyc is pivotal for cell proliferation. In line with these functions, treatments with nutlin-3 or cMyc inhibitor repressed proliferation of activated T cells. Next, we studied to what extent cell cycle arrest and apoptosis are responsible for this effect. We examined cell cycle distribution by BrdU incorporation and PI staining, and apoptosis by Annexin V and propidium iodide (PI) staining. Both nutlin-3 and 10058-F4 induced cell cycle arrest and apoptosis. A considerably lower fraction of activated T cells treated with nutlin-3 or cMyc inhibitor were found in S phase, which decreased to 0.35 and 0.27, respectively, as compared to control cells. At the same time, a higher proportion of cells were in the sub-G1 phase, which increased by 2.8 and 1.6-fold, respectively (Fig. 4A). The Annexin V positive apoptotic cell population increased by 1.8-fold in nutlin-3 treated cells and 1.5-fold in 10058-F4 treated cells, consistent with the increased sub-G1 population (Fig. 4B).
Figure 4.
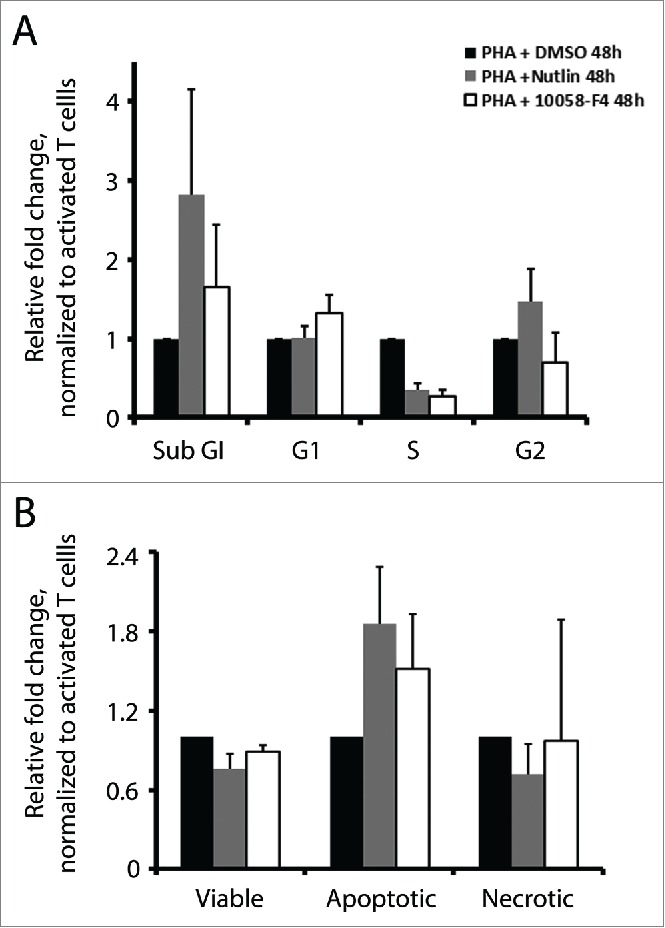
Nutlin-3 and 10058-F4 induced cell cycle arrest and apoptosis. (A) Cell cycle analysis by BrdU incorporation and PI staining in activated T cells cultured with or without nutlin-3 (5 µM) or 10058-F4 (50 µM) for 48 h. The values represent average of 2 experiments with SD. (B) Apoptosis in activated T cells cultured with or without nutlin-3 (5 µM) or 10058-F4 (50 µM) for 48 h was measured by flow cytometry upon staining cells with Annexin V and PtdIns. The values represent average of 2 experiments with SD.
Induction of p53 by nutlin-3 or inhibition of cMyc by 10058-F4 inhibits T cell proliferation but not T cell cytotoxic function
Interference with p53 or cMyc in activated T cells leads to a decrease in proliferation as cells enter cell cycle arrest or apoptosis. Next, we studied how nutlin-3 treatment influences T cell proliferation and cytotoxic function if the T cells are exposed to it prior to activation. We induced p53 in T cells by treating them with nutlin-3 for 48 h, after which nutlin-3 was washed off and the cells were activated with PHA. Cell proliferation assessed by 3H-thymidine incorporation showed that nutlin-3 pre-treated T cells, expressing higher levels of p53 at the time of activation, had similar proliferation rates as untreated cells (Fig. 5A).
Figure 5.
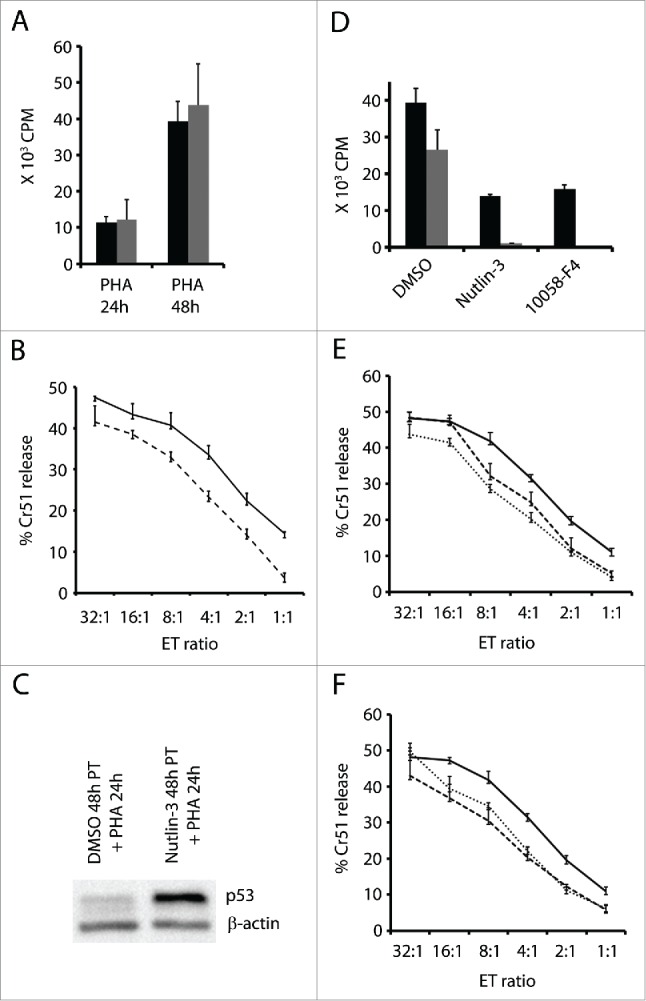
Nutlin-3 or 10058-F4 do not significantly affect the cytotoxic function of activated T cells. (A) 105 T cells pre-treated with nutlin-3, 5 µM (gray bars) or DMSO-solvent control (black bars) for 48 h were washed and then activated with PHA (1 µg/ml) and pulsed with 3H-thymidine for 16 h at the indicated time points. (B) Cytotoxic assay – Killing of chromium-51 labeled allogeneic LCL by T cells activated for 24 h following a 48 h pre-treatment with nutlin-3, 5 µM (broken line) or DMSO-solvent control (solid line). (C) p53 expression in activated T cells pre-treated with nutlin-3 (5 µM) or DMSO for 48 h, washed and activated by by PHA (1 µg/ml) for 24 h. (D) 105 T cells were activated for 48 h and cultured with nutlin-3 (5 µM) or 10058-F4 (50 µM) simultaneously (black bars) or alternatively cells were cultured with each compound only for the last 24 h (gray bars). Cells treated with DMSO served as control. (E) Cytotoxic assay - Killing of chromium-51 labeled allogeneic LCL by activated T cells treated with nutlin-3 5 µM (broken line) or 10058-F4, 50 µM (dotted line) in parallel with PHA and (F) for the last 24 h of the 48 h activation. Cells treated with DMSO served as control.
We also tested the cytotoxic activity of these T cells in a Cr51 release assay following 24 h of activation. Allogeneic LCLs were used as target cells. Activated T cells pre-treated with nutlin-3, and therefore expressing higher levels of p53 (Fig. 5C), exhibited a marginally decreased ability to kill when compared to activated T cells without nutlin-3 pre-treatment (Fig. 5B). For example, at an E:T ratio of 32:1, nutlin-3 pre-treated T cells killed 41.5% whereas the control T cells killed 47.5% of the target cells.
Next, we studied the effect of cMyc inhibition and p53 upregulation on cytotoxicity of T cells that had already gone through the initial step of activation. T cells were activated with PHA for 48 h, and cultured with nutlin-3 or cMyc inhibitor for the last 24 h. In another experimental setup, T cells were activated and simultaneously cultured with or without nutlin-3/cMyc inhibitor for 48 h. 3H-thymidine incorporation in the above cultures showed that nutlin-3 and cMyc inhibitor suppressed proliferation even more extensively if they were added after activation occurred (24 h after PHA) (Fig. 5D). However, the cytotoxic function of the cells to which nutlin-3 or cMyc inhibitor was added simultaneously or after the initial phase of activation, was only marginally affected (Fig. 5E&F). No significant difference in surface expression of MHC class II was detected in activated T cells cultured with or without nutlin-3/cMyc inhibitor (data not shown).
Discussion
Engagement of TCR induces proliferation of T cells. As one of the first steps in this process, Notch signaling induces cMyc.23,26 Induction of cMyc is essential for entering the cell cycle. Activated T cells undergo rapid cell division, with each cell dividing in vivo once every 2 h27 and each activated T cell undergoing about 15 divisions.28 At such a high rate of proliferation, largely driven by cMyc, it is critical to keep its expression under tight control during and after the immune response in order to prevent the establishment of a sustained and deregulated T cell proliferation.
We have shown earlier that p53 is induced in PHA activated T cells and contributes to T cell homeostasis, at least in part, through the induction of the proapoptotic protein SAP.3,4 Here we have studied a mechanism for the p53 induction in these cells, and the consequences of high p53 levels. Our results demonstrate that the kinetics of cMyc expression and its downstream target p14ARF parallel T cell proliferation. It is well known that expression of cMyc is upregulated after ligation of TCR-CD3 complexes and that this is essential for the expansion of lymphocytes during an immune response.23 Notch receptors are involved in T cell activation and have been shown to induce cMyc.23 Similarly, Notch1 has been shown to promote proliferation of T cell lymphoblastic leukemia cells by directly inducing cMyc expression.26 Deregulated cMyc expression is associated with uncontrolled cell division thereby contributing to genomic instability and development of cancer.13
The cMyc-p14ARF axis is well established as one of the pathways that can induce p53.18,29,30 Disruption of this pathway can contribute to the genesis of lymphomas.31 In HTLV-1 associated T cell lymphomas the transactivation function of p53 is inhibited by the binding of viral Tax protein to the N terminal of p53.32,33 In vivo study in a mouse model of T-ALL reported that tumor cells express low levels of p53. In this model, the aberrant Notch signaling results in low p19ARF protein levels, thus allowing unperturbed formation of the MDM2-p53 complex and degradation of p53.34 Thus, similar to the development of other cancer types, deregulation/inactivation of p53 pathway seems to be crucial in T cell lymphomagenesis. Expansion of CTLs was more efficient from p53−/− mice than from p53+/+ mice,35 suggesting a role for p53 in regulating T cell proliferation and survival. A study in a p53 silenced humanized mouse model showed normal T cell development and normal immune response to antigen stimulation. However, upon prolonged antigen stimulation, p53 silenced T cells showed a significant growth advantage over T cells expressing p53.36 Based on these studies we hypothesized that in normal T cells activated by PHA, upregulated cMyc induces p14ARF, which in turn binds to MDM2 and inhibits its negative regulatory effect on p53, thus leading to elevated p53 levels. To test if this is a plausible mechanism for p53 induction in activated T cells, we first modulated cMyc expression through the indirect inhibition of Notch by DAPT, a γ-secretase inhibitor.37,24 Treatment with DAPT lowered cMyc levels and proliferation of activated T cells.23 The cMyc target p14ARF was also downregulated in these cells. As a consequence of the lower levels of cMyc and p14ARF, p53 expression was also decreased. These results imply that initiation of T cell proliferation can itself be one of the contributing factors for p53 induction in activated T cells. Along with p53, its transcriptional target p21 is also induced following T cell activation, but p21 declines when more vigorous proliferation starts. It is possible that p21 is downregulated by cMyc, as described in tumor lines.38 It has also been shown that p21 expression is suppressed in T cells by the zinc finger protein Znf131, thus controlling proliferation of T cells.39
To further ascertain that p53 is induced by the cMyc–p14ARF pathway, we inhibited cMyc using 10058-F4,25 a small-molecule inhibitor that interferes with cMyc/Max heterodimerization, blocking cMyc-mediated transactivation.40 10058-F4 suppressed cMyc protein expression in activated T cells and also inhibited T cell proliferation. cMyc downregulation was again accompanied by lower p14ARF and p53. The lower level of p53 is presumably a direct consequence of downregulation of p14ARF, although more indirect mechanisms cannot be excluded. In addition to the cMyc-p14ARF pathway, elevated ROS levels in activated T cells41,42 may also contribute to upregulation of p53.
We also studied the consequence of further upregulation of p53 by nutlin-3 in activated T cells. Small molecules that disrupt MDM2-p53 binding and thus elevate p53 levels, are already in clinical trials for cancer treatment.43 Therefore, it is of interest to examine the effects of systemic upregulation of p53 on T cell function, as T cell mediated immune surveillance contributes to the control of tumors. In line with a previous study showing that nutlin-3 induces apoptosis in T cell leukemia cell lines,44 we found that nutlin-3 induced apoptosis in activated T cells, and a corresponding increase in the expression of the pro-apoptotic protein Bax. In addition, and as shown here, nutlin-3 induced a massive increase in p21 and decrease in cMyc, changes that would contribute to cell cycle arrest. It is conceivable that the decrease in cMyc mRNA levels was a result of transcriptional suppression by p53 in activated T cells treated with nutlin-3. This is in line with the previous finding that p53-mediated cMyc suppression is essential for cell cycle arrest.20 Also, in a recent study it was observed that nutlin-3 induced p53 suppressed cMyc at the transcriptional level in M2 macrophages.45
The decrease in 3H-Thymidine incorporation of activated T cells simultaneously treated with nutlin-3 can be attributed to cell cycle arrest and cell death. However, these T cells retained the cytotoxic function, although with a marginally decreased efficiency. The effects were similar when T cells were treated with nutlin-3 after the activation was already initiated.
The fate of activated T cells that exited the cell cycle may be influenced by the p53-mTOR crosstalk. Recent studies in cell lines showed that through its target TSC2, p53 can inhibit the mTOR pathway, thus favoring reversible quiescence over senescence.7,46,47 By integrating immunological and metabolic signals, the mTOR pathway plays an important role in T cell activation and differentiation.48-50 Considering the p53-mTOR relationship, additional studies are needed to clarify whether p53 can modulate mTOR activity in T cells and thus influence their function and senescence.
To test whether proliferation is influenced by high levels of p53 present in T cells prior to activation, we assessed the proliferative capacity of nutlin-3 pretreated T cells. This experiment corresponds to a possible clinical situation in which activation of T cells occurs after/during anticancer therapy with a p53-MDM2 inhibitor. As a result, T cells will express high levels of p53 before undergoing the process of activation. Although p53 rapidly decreases upon nutlin-3 removal in tumor cells lines,51 this seems not to be the case in normal T cells. A substantial amount of p53 is still observed after nutlin-3 was washed off, which is even further increased following activation. 3H-thymidinde incorporation showed that in spite of the elevated p53 levels, proliferation of the cells did not decrease. This is remarkable as T cells cultured with nutlin-3 at the time of activation show impaired proliferation. This finding indicates that high levels of p53 in T cells do not necessarily inhibit proliferation and that the effect possibly depends on the cellular context, on the exact timing of different signaling events. Further studies are required to elucidate the exact role of p53 in T cell activation and proliferation.
Nutlin-3 pretreatment did not influence MHC class II expression, a marker of T cell activation. Although studies from p53 knockout mice have shown that p53 loss has no significant influence on the T cell function, consequence of p53 expression and its role in regulating genes involved in the immune response is yet to be explored in details. p53 has been already described to facilitate immune cell function in macrophages where it regulates IL-6 production.52 This suggests the plausible role of p53 in regulating T cell immune response not only by regulating proliferation but also other genes involved in T cell response.
We also tested the effect of the c-Myc inhibitor 10058-F4 on the cytotoxic function of activated T cells. Despite potent inhibition of proliferation, the cytotoxic function was retained with only a marginally decreased efficiency. Like nutlin-3, the cMyc inhibitor did not alter expression of the activation marker MHC-class II. However, it is possible that other functions are altered, e.g. an earlier study reported that cMyc inhibition by 10058-F4 affected specific cytokine production of T cells: it repressed IL-4 and IL-17 production by Th2 and Th17 cells, but not IL-2 and INF-γ by Th17 and Th1 cells.53
By using these 2 compounds that target cMyc and p53, we have thus been able to demonstrate a dichotomy between proliferation and cytotoxic function in activated T cells. Our results suggest that cancer therapy with small molecules that disrupt p53-MDM2 binding and/or inhibit cMyc expression may have deleterious effects on proliferation of activated T cells but not on their cytotoxic function.
Based on our results, we propose a model for a feedback loop mechanism of cMyc-p53 regulation in activated T cells (Fig. 6). This model suggests that cMyc induced following activation of T cells contributes, at least in part, to upregulation of p53, which in turn at later time points will downregulate cMyc levels. This will contribute to cessation of T cell proliferation and to T cell homeostasis.
Figure 6.
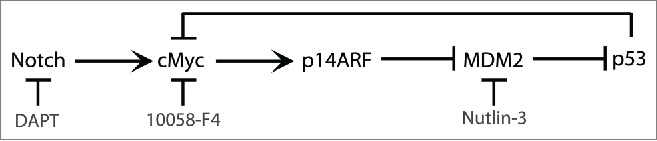
Schematic representation of the cMyc-p53 feedback regulation in activated T cells.
Materials and methods
Separation, activation and treatment of T cell
Peripheral blood mononuclear cells (PBMCs) were separated from buffy coats of healthy donors by Ficoll-Paque (GE Health Care, 17-1440-02). T cells from PBMCs were obtained by negative selection. T cells were plated at a 0.5 × 106 cells/ml density in complete RPMI with or without 1 µg/ml PHA (Sigma, L8902). When indicated 5 µM of nutlin-3 (Sigma, N6287), 50 µM 10058-F4 (Sigma, F3680) or DAPT (Sigma, D5942) were added to the medium. DMSO was added to the control cultures.
For proliferation, 105 T cells were plated in day 0 in a 96 well plate and cultured in complete RPMI medium. To parallel cultures, 1 µg/ml PHA (Sigma, L8902) was added. At specific time points, one µCi 3H-thymidine (Perkin Elmer, NET02250UC) was added to each well and incubated at 37°C in 5% CO2 for the last 16 h. Cells were harvested on a glass fiber filter and the radioactivity was measured in a liquid scintillation counter (Perkin Elmer, Microbeta 1450). In specific experiments, cells were treated with 5 µM of nutlin-3 (Sigma, N6287), 50 µM 10058-F4 (Sigma, F3680) or DAPT (Sigma, D5942). DMSO was added to the control cultures.
Immunoblotting
The cells were lysed in SDS and 2-mercaptoethanol containing loading buffer and aliquots corresponding to 1.5 × 105 cells were loaded in each lane in a SDS-PAGE gel. The following antibodies were used to detect the respective proteins; p53 (Santa Cruz, sc-126), cMyc (Life Technologies, 13-2500), p21 (BD Transduction Laboratories, 610233), p14ARF (Santa Cruz, sc-73434), Bax (Cell Signaling, 2772S). As a control for equal amounts of protein loaded β-actin (Sigma, A1978) was detected.
RNA isolation, cDNA synthesis and real time PCR
Total cellular RNA was extracted from cells using the Quick-RNA MiniPrep kit (Zymo Research), and then reverse transcribed using the SuperScript VILO cDNA synthesis kit (Invitrogen), according to the manufacturer's instructions. The relative level of cMyc, p21, p14 and p53 mRNA transcripts were determined with the LightCycler FastStart DNA Master SYBR Green I kit (Roche) kit in a LightCycler 1.2 instrument (Roche) using the standard curve method. Each PCR mixture was initially denatured at 95°C for 10 min and then cycled 40 times at 95°C for 8 s, 60°C for 5 s, and 72°C for 8 s. Target genes were measured and normalized simultaneously with the endogenous control MLN51. The following primer sequences were used: p53 forward primer: 5′-CCCAACAACACCAGCTCCT-3′, reverse primer: 5′- CCTGGGCATCCTTGAGTTC-3′; cMyc forward primer: 5′-CACCACCAGCAGCGACTCT-3′, reverse primer: 5′-GCTGTGAGGAGGTTTGCTGT-3′; p21 forward primer: 5′-GCAGACCAGCATGACAGATTT-3′, reverse primer: 5′-GGATTAGGGCTTCCTCTTGGA-3′; p14/ARF forward primer: 5′-CCTCGTGCTGATGCTACTGA-3′, reverse primer: 5′-CTGCCCATCATCATGACCT-3′ and MLN51 forward primer: 5′-CAAGGAAGGTCGTGCTGGTT-3′, reverse primer: 5′-ACCAGACCGGCCACCAT-3′.
Apoptosis detection
FITC-conjugated Annexin V reagent (BD PharMingen) was used to detect apoptosis according to the manufacturer's instructions.
Cell cycle analysis
BrdU (BD PharMingen) was used to label cells to study cell cycle according to the manufacturer's instructions. Mouse-anti BrdU antibody (BD PharMingen, 543580) and rabbit anti-mouse FITC conjugated (Dako, F0232) secondary antibody were used for detection.
Cytotoxicity assay
T cells isolated as described above were activated with 1 µg/ml PHA (Sigma, L8902) in complete RPMI for 2 d along with the mentioned treatments with the compounds. For analysis of cytotoxicity, 10 thousand 51Cr-labeled allogeneic LCL cells were cultured with serially diluted activated T cells in triplicates for each dilution for 4 h at 37°C in 5% CO2. Radioactivity in the supernatant of cultures was measured in a gamma counter.
Disclosure of potential conflicts of interest
K.G.W. is co-founder, shareholder and board member of Aprea AB, a company that develops p53-based cancer therapy.
Acknowledgments
This work was supported by the grants from the Swedish Cancer Society (Cancerfonden) and Karolinska Institutet. H.M., D.S. and N.N. are recipients of cancer research fellowships from the Cancer Research Institute (New York) and Concern Foundation (Los Angeles).
References
- [1].Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A 2008; 73:975-83; PMID:18785267; http://dx.doi.org/ 10.1002/cyto.a.20643 [DOI] [PubMed] [Google Scholar]
- [2].Purnama C CX, Larbi . An Overview of T Cell Subsets and Their Potential use as markers of immunological aging. International Trends in Immunity 2013; 1(4)21-32. [Google Scholar]
- [3].Nagy N, Matskova L, Kis LL, Hellman U, Klein G, Klein E. The proapoptotic function of SAP provides a clue to the clinical picture of X-linked lymphoproliferative disease. Proc Natl Acad Sci U S A 2009; 106:11966-71; PMID:19570996; http://dx.doi.org/ 10.1073/pnas.0905691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Madapura HS, Salamon D, Wiman KG, Lain S, Klein G, Klein E, Nagy N. p53 contributes to T cell homeostasis through the induction of pro-apoptotic SAP. Cell Cycle 2012; 11:4563-9; PMID:23165210; http://dx.doi.org/ 10.4161/cc.22810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].DP L. p53, guardian of the genome. Nature 1992; 358:2; PMID:16145231614523 [Google Scholar]
- [6].Dulic V. Be quiet and you'll keep young: does mTOR underlie p53 action in protecting against senescence by favoring quiescence? Aging (Albany NY) 2011; 3:3-4; PMID:21248373; http://dx.doi.org/ 10.18632/aging.100257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Christy B, Demaria M, Campisi J, Huang J, Jones D, Dodds SG, Williams C, Hubbard G, Livi CB, Gao X, et al.. p53 and rapamycin are additive. Oncotarget 2015; 6:15802-13; PMID:26158292; http://dx.doi.org/ 10.18632/oncotarget.4602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Levine AJ, Harris CR, Puzio-Kuter AM. The interfaces between signal transduction pathways: IGF-1/mTor, p53 and the Parkinson Disease pathway. Oncotarget 2012; 3:1301-7; PMID:23211569; http://dx.doi.org/ 10.18632/oncotarget.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/ 10.1016/j.cell.2009.04.037 [DOI] [PubMed] [Google Scholar]
- [10].Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al.. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502:333-9; PMID:24132290; http://dx.doi.org/ 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Donehower LA HM, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356:7; http://dx.doi.org/ 10.1038/356215a0 [DOI] [PubMed] [Google Scholar]
- [12].Donehower LA HM, Vogel H, McArthur MJ, Montgomery CA Jr, Park SH, Thompson T, Ford RJ, Bradley A. Effects of Genetic Background on Tumorigenesis in p53 deficient mice. Mol CArcinog 1995; 14:16-22; PMID:7546219; http://dx.doi.org/ 10.1002/mc.2940140105 [DOI] [PubMed] [Google Scholar]
- [13].Dang C. c-Myc Target Genes Involved in Cell MINIREVIEW c-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol Cell Biol 1999; 19:1-11; PMID:9858526; http://dx.doi.org/ 10.1128/MCB.19.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Link JM, Hurlin PJ. The activities of MYC, MNT and the MAX-interactome in lymphocyte proliferation and oncogenesis. Biochim Biophys Acta 2015; 1849:554-62; PMID:24731854; http://dx.doi.org/ 10.1016/j.bbagrm.2014.04.004 [DOI] [PubMed] [Google Scholar]
- [15].Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Translocation of the C-Myc Gene into the Immunoglobulin Heavy-Chain Locus in Human Burkitt-Lymphoma and Murine Plasmacytoma Cells. P Natl Acad Sci-Biol 1982; 79:7837-41; PMID:NOT_FOUND; http://dx.doi.org/ 10.1073/pnas.79.24.7837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stewart M, Cameron E, Campbell M, McFarlane R, Toth S, Lang K, Onions D, Neil JC. Conditional expression and oncogenicity of c-myc linked to a CD2 gene dominant control region. Int J Cancer 1993; 53:1023-30; PMID:8473043; http://dx.doi.org/ 10.1002/ijc.2910530628 [DOI] [PubMed] [Google Scholar]
- [17].Blyth K, Terry A, Ohara M, Baxter EW, Campbell M, Stewart M, Donehower LA, Onions DE, Neil JC, Cameron ER. Synergy between a Human C-Myc Transgene and P53 Null Genotype in Murine Thymic Lymphomas - Contrasting Effects of Homozygous and Heterozygous P53 Loss. Oncogene 1995; 10:1717-23; PMID:7753548 [PubMed] [Google Scholar]
- [18].Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev 1998; 12:2424-33; PMID:9694806; http://dx.doi.org/ 10.1101/gad.12.15.2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wu XW, Bayle JH, Olson D, Levine AJ. The P53 Mdm-2 Autoregulatory Feedback Loop. Genes Dev 1993; 7:1126-32; PMID:8319905; http://dx.doi.org/ 10.1101/gad.7.7a.1126 [DOI] [PubMed] [Google Scholar]
- [20].Ho JS, Ma W, Mao DY, Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol 2005; 25:7423-31; PMID:16107691; http://dx.doi.org/ 10.1128/MCB.25.17.7423-7431.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Miyashita T, Reed JC. Tumor-Suppressor P53 Is a Direct Transcriptional Activator of the Human Bax Gene. Cell 1995; 80:293-9; PMID:7834749; http://dx.doi.org/ 10.1016/0092-8674(95)90513-8 [DOI] [PubMed] [Google Scholar]
- [22].Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al.. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844-8; PMID:14704432; http://dx.doi.org/ 10.1126/science.1092472 [DOI] [PubMed] [Google Scholar]
- [23].Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, Zhang H, Huppa JB, Tsai YH, Lobry C, et al.. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol 2013; 14:262-70; PMID:23377202; http://dx.doi.org/ 10.1038/ni.2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Geling A, Steiner H, Willem M, Bally-Cuif L, Haass C. A gamma-secretase inhibitor blocks Notch signaling in vivo and causes a severe neurogenic phenotype in zebrafish. EMBO Rep 2002; 3:688-94; PMID:12101103; http://dx.doi.org/ 10.1093/embo-reports/kvf124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mustata G, Follis AV, Hammoudeh DI, Metallo SJ, Wang HB, Prochownik EV, Lazo JS, Bahar I. Discovery of Novel Myc-Max Heterodimer Disruptors with a Three-Dimensional Pharmacophore Model. J Med Chem 2009; 52:1247-50; PMID:19215087; http://dx.doi.org/ 10.1021/jm801278g [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O'Neil J, Neuberg D, Weng AP, et al.. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A 2006; 103:18261-6; PMID:17114293; http://dx.doi.org/ 10.1073/pnas.0606108103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yoon H, Kim TS, Braciale TJ. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One 2010; 5:e15423; PMID:21079741; http://dx.doi.org/ 10.1371/journal.pone.0015423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Butz EA, Bevan MJ. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity 1998; 8:167-75; PMID:9491998; http://dx.doi.org/ 10.1016/S1074-7613(00)80469-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U S A 1998; 95:8292-7; PMID:9653180; http://dx.doi.org/ 10.1073/pnas.95.14.8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang YP, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998; 92:725-34; PMID:9529249; http://dx.doi.org/ 10.1016/S0092-8674(00)81401-4 [DOI] [PubMed] [Google Scholar]
- [31].Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 1999; 13:2658-69; PMID:10541552; http://dx.doi.org/ 10.1101/gad.13.20.2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pise-Masison CA, Choi KS, Radonovich M, Dittmer J, Kim SJ, Brady JN. Inhibition of p53 transactivation function by the human T-cell lymphotropic virus type 1 Tax protein. J Virol 1998; 72:1165-70; PMID:9445014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hatta Y, Koeffler HP. Role of tumor suppressor genes in the development of adult T cell leukemia/lymphoma (ATLL). Leukemia 2002; 16:1069-85; PMID:12040438; http://dx.doi.org/ 10.1038/sj.leu.2402458 [DOI] [PubMed] [Google Scholar]
- [34].Beverly LJ, Felsher DW, Capobianco AJ. Suppression of p53 by Notch in lymphomagenesis: implications for initiation and regression. Cancer Res 2005; 65:7159-68; PMID:16103066; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1664 [DOI] [PubMed] [Google Scholar]
- [35].Zhou XZ, Wong S, Walter J, Jacks T, Eisen HN. Increased generation of CD8(+) T cell clones in p53 mutant mice. J Immunol 1999; 162:3957-60; PMID:10201915 [PubMed] [Google Scholar]
- [36].Gimeno R, Weijer K, Voordouw A, Uittenbogaart CH, Legrand N, Alves NL, Wijnands E, Blom B, Spits H. Monitoring the effect of gene silencing by RNA interference in human CD34+ cells injected into newborn RAG2−/− gammac−/− mice: functional inactivation of p53 in developing T cells. Blood 2004; 104:3886-93; PMID:15319293; http://dx.doi.org/ 10.1182/blood-2004-02-0656 [DOI] [PubMed] [Google Scholar]
- [37].Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, et al.. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem 2001; 76:173-81; PMID:11145990; http://dx.doi.org/ 10.1046/j.1471-4159.2001.00012.x [DOI] [PubMed] [Google Scholar]
- [38].Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 2002; 419:729-34; PMID:12384701; http://dx.doi.org/ 10.1038/nature01119 [DOI] [PubMed] [Google Scholar]
- [39].Iguchi T, Aoki K, Ikawa T, Taoka M, Taya C, Yoshitani H, Toma-Hirano M, Koiwai O, Isobe T, Kawamoto H, et al.. BTB-ZF Protein Znf131 Regulates Cell Growth of Developing and Mature T Cells. J Immunol 2015; 195:982-93; PMID:26136427; http://dx.doi.org/ 10.4049/jimmunol.1500602 [DOI] [PubMed] [Google Scholar]
- [40].Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003; 22:6151-9; PMID:13679853; http://dx.doi.org/ 10.1038/sj.onc.1206641 [DOI] [PubMed] [Google Scholar]
- [41].Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, Marrack PC. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity 1999; 10:735-44; PMID:10403648; http://dx.doi.org/ 10.1016/S1074-7613(00)80072-2 [DOI] [PubMed] [Google Scholar]
- [42].Tripathi P, Hildeman D. Sensitization of T cells to apoptosis–a role for ROS? Apoptosis 2004; 9:515-23; PMID:15314279; http://dx.doi.org/ 10.1023/B:APPT.0000038033.14925.02 [DOI] [PubMed] [Google Scholar]
- [43].Sahin U, Kariko K, Tureci O. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov 2014; 13:759-80; PMID:25233993; http://dx.doi.org/ 10.1038/nrd4278 [DOI] [PubMed] [Google Scholar]
- [44].Hasegawa H, Yamada Y, Iha H, Tsukasaki K, Nagai K, Atogami S, Sugahara K, Tsuruda K, Ishizaki A, Kamihira S. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia 2009; 23:2090-101; PMID:19710698; http://dx.doi.org/ 10.1038/leu.2009.171 [DOI] [PubMed] [Google Scholar]
- [45].Li L, Ng DS, Mah WC, Almeida FF, Rahmat SA, Rao VK, Leow SC, Laudisi F, Peh MT, Goh AM, et al.. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ 2015; 22:1081-93; PMID:25526089; http://dx.doi.org/ 10.1038/cdd.2014.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344-52; PMID:20606252; http://dx.doi.org/ 10.18632/aging.100160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY) 2010; 2:535-7; PMID:20876940; http://dx.doi.org/ 10.18632/aging.100202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 2012; 12:325-38; PMID:22517423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 2008; 205:565-74; PMID:18283119; http://dx.doi.org/ 10.1084/jem.20071477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, Guertin DA, Lamb RF, Chi H. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity 2013; 39:1043-56; PMID:24315998; http://dx.doi.org/ 10.1016/j.immuni.2013.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].van Leeuwen IMM, Higgins M, Campbell J, Brown CJ, McCarthy AR, Pirrie L, Westwood NJ, Lain S. Mechanism-specific signatures for small-molecule p53 activators. Cell Cycle 2011; 10:1590-8; PMID:21490429; http://dx.doi.org/ 10.4161/cc.10.10.15519 [DOI] [PubMed] [Google Scholar]
- [52].Lowe JM, Menendez D, Bushel PR, Shatz M, Kirk EL, Troester MA, Garantziotis S, Fessler MB, Resnick MA. p53 and NF-kappaB coregulate proinflammatory gene responses in human macrophages. Cancer Res 2014; 74:2182-92; PMID:24737129; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bandukwala HS, Gagnon J, Togher S, Greenbaum JA, Lamperti ED, Parr NJ, Molesworth AMH, Smithers N, Lee K, Witherington J, et al.. Selective inhibition of CD4(+) T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci USA 2012; 109:14532-7; PMID:22912406; http://dx.doi.org/ 10.1073/pnas.1212264109 [DOI] [PMC free article] [PubMed] [Google Scholar]