

Autophagy is a lysosomal degradation process that plays an important role in normal tissue homeostasis, but its role in developmental processes is still largely unknown. Impairment of the autophagy process has been observed in many human genetic disorders, as has been demonstrated in cells and tissues isolated from lysosomal storage disease (LSD) models.1 Notably, LSD patients often display defective bone development and growth. These observations prompted us to study the role of autophagy during bone growth. By analyzing bone development in mice lacking the essential autophagy related gene 7 (Atg7) in chondrocytes (Atg7 chKO) we demonstrated that autophagy controls the secretion of type II collagen (Col2), one of the main components of cartilage extra cellular matrix (ECM). In the absence of autophagy, collagen molecules abnormally accumulated within the endoplasmic reticulum (ER) of chondrocytes and the levels of Col2 in the cartilaginous ECM was lower in Atg7 chKO compared to control mice.2
Collagens are synthesized as precursors (procollagens, PCs) in the ER, and undergo several post-translational modifications, which permit the folding and assembly of native triple helix PC chains that are then secreted into the ECM. PCs are large and complex molecules that can be prone to misfolding during assembly and secretion processes. Our work suggests that chondrocytes use autophagy as an ER quality control pathway (ERQC) that prevents the accumulation of misfolded PC molecules within the ER. Indeed, triple helix PCs cannot undergo ER-Associated Degradation (ERAD) due to their physical state and our data suggest that the misfolded PC2 is instead sequestered by autophagosomes. The native collagen specific chaperone heat shock protein 47 (HSP47), was absent from autophagosomes containing PC2, suggesting that autophagy recognizes PC2 molecules in their non-native conformation.
Autophagy may also influence PC secretion indirectly either by controlling ER homeostasis through a recently described FAM134B-mediated ER-phagy3 or by regulating the biogenesis of COPII coated vesicles, which are deputed to ER-Golgi trafficking of collagens.4 Thus, multiple mechanisms can explain the role of autophagy as regulator of PC secretion. Notably, recent works by the Chagin lab show that chondrocytes lacking autophagy display a higher apoptotic rate compared to control cells.5 It would be interesting to evaluate whether collagen storage and ER dysfunction in Atg7 ChKO chondrocytes account for cell death.
We observed induction of autophagy in growth plate chondrocytes during post-natal development. Notably, collagen production by chondrocytes was also increased at these ages, suggesting that the two processes could be co-regulated. To identify the potential regulatory pathways involved in chondrocyte autophagy, we screened chondrogenic molecules for their potential to induce autophagy in vitro. Fibroblast growth factor 18 (FGF18) was the only molecule that significantly increased autophagy flux. FGF signaling is among the most studied pathways in bone due to its involvement in many forms of skeletal disorders.6 FGF18 plays a key role in skeletal growth and development in mice, regulating multiple aspects of chondrogenesis in the growth plate,6 including ECM composition.
Mice lacking FGF18 exhibited very low levels of autophagy in chondrocytes compared to control mice, pointing to FGF18 as a physiological regulator of chondrocyte autophagy. Among the different FGF receptors, our in vitro and in vivo data suggest that FGF18 acts mainly through FGF receptor 4 (FGFR4), and to a lesser extent through FGFR3, to regulate autophagy. Fgf18+/− and Fgfr4−/− mice failed to induce chondrocyte autophagy during post-natal bone development and as a consequence displayed PC2 storage in the ER of chondrocytes and decreased Col2 levels in the ECM. Biochemical studies suggested that FGF18 induces autophagosome biogenesis through the autophagy initiation complex VPS34-Beclin1. When active, this complex produces phosphatidylinositol-3-phosphate (PtdIns(3)P) which is essential for autophagosome biogenesis. Consistent with this model, we rescued autophagy in the growth plates of Fgf18+/− mice by using TAT-Beclin1, a peptide able to enhance the activity of endogenous Beclin1 protein. Notably, TAT-Beclin1 restored ECM matrix defects in Fgf18+/− and Fgfr4−/− mice, indicating autophagy as a physiological regulator of FGF-mediated bone growth. Thus, our study has identified a novel mechanism by which FGF signaling regulates post-natal bone development (Fig. 1).
Figure 1.
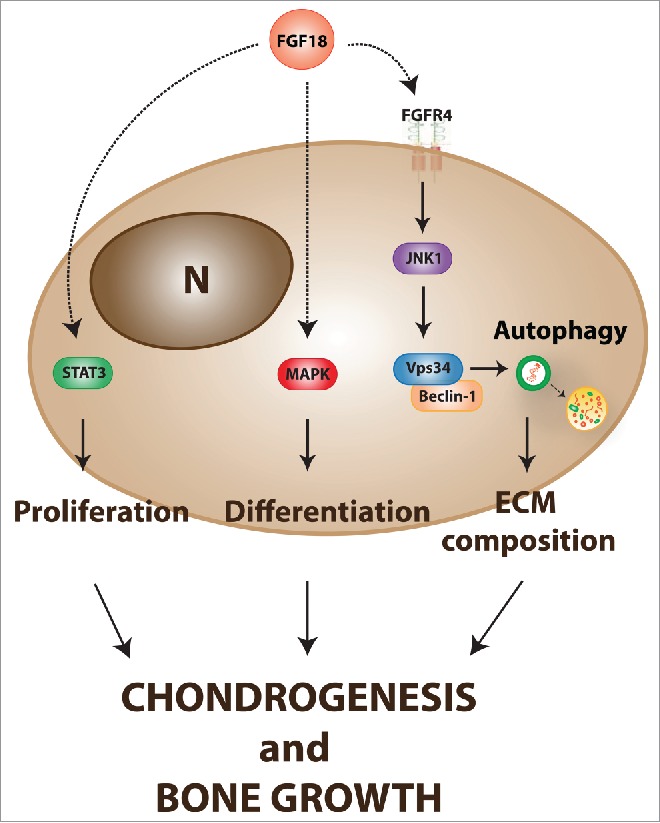
Proposed role of FGF-mediated induction of autophagy during bone growth. FGF signaling modulates multiple aspects of chondrogenesis and bone growth through the regulation of different intracellular pathways. During post-natal bone development, FGF18 induces chondrocyte autophagy through the activation of the Beclin1-Vps34 complex via FGFR4 and c-Jun N-terminal kinases (JNK). The FGF-mediated induction of autophagy controls collagen homeostasis and ECM composition during postnatal bone growth. (N = nucleus).
A fundamental question is now to understand whether autophagy dysfunction is involved in the pathogenesis of FGF-related skeletal disorders. The most common form of dwarfism in humans, achondroplasia, is caused by activating mutations in FGFR3.6 A recent report from Wang et al. showed defective autophagy in the growth plates of an achondroplasia mouse model (FGFRach), suggesting that this could be the case.7 We have in our hands all of the tools required to explore autophagy as a new therapeutic option for the treatment of achondroplasia.
References
- [1].Settembre C. Genes Dev 2008; 22:2645-50; PMID:18832069; http://dx.doi.org/ 10.1101/gad.1711308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cinque L. Nature 2015; 528:272-5; PMID:26595272; http://dx.doi.org/ 10.1038/nature16063 [DOI] [PubMed] [Google Scholar]
- [3].Khaminets A. Nature 2015; 522:354-358; PMID:26040720; http://dx.doi.org/10.1038/nature14498 [DOI] [PubMed] [Google Scholar]
- [4].Stadel D. Mol Cell 2015; 60:89-104; PMID:26431026; http://dx.doi.org/ 10.1016/j.molcel.2015.09.010 [DOI] [PubMed] [Google Scholar]
- [5].Vuppalapati KK, et al.. J Bone Miner Res 2015; 30:2249-61; PMID:26077727; http://dx.doi.org/ 10.1002/jbmr.2575 [DOI] [PubMed] [Google Scholar]
- [6].Ornitz DM, et al.. Genes Dev 2002; 16:1446-1465; PMID:12080084; http://dx.doi.org/ 10.1101/gad.990702 [DOI] [PubMed] [Google Scholar]
- [7].Wang X, et al.. Autophagy 2015; 11:1998-2013; PMID:26491898; http://dx.doi.org/ 10.1080/15548627.2015.1091551 [DOI] [PMC free article] [PubMed] [Google Scholar]