

ABSTRACT
Testicular teratomas result from anomalies in embryonic germ cell development. In the 129 family of inbred mouse strains, teratomas arise during the same developmental period that male germ cells normally enter G1/G0 mitotic arrest and female germ cells initiate meiosis (the mitotic:meiotic switch). Dysregulation of this switch associates with teratoma susceptibility and involves three germ cell developmental abnormalities seemingly critical for tumor initiation: delayed G1/G0 mitotic arrest, retention of pluripotency, and misexpression of genes normally restricted to embryonic female and adult male germ cells. One misexpressed gene, cyclin D1 (Ccnd1), is a known regulator of cell cycle progression and an oncogene in many tissues. Here, we investigated whether Ccnd1 misexpression in embryonic germ cells is a determinant of teratoma susceptibility in mice. We found that CCND1 localizes to teratoma-susceptible germ cells that fail to enter G1/G0 arrest during the mitotic:meiotic switch and is the only D-type cyclin misexpressed during this critical developmental time frame. We discovered that Ccnd1 deficiency in teratoma-susceptible mice significantly reduced teratoma incidence and suppressed the germ cell proliferation and pluripotency abnormalities associated with tumor initiation. Importantly, Ccnd1 expression was dispensable for somatic cell development and male germ cell specification and maturation in tumor-susceptible mice, implying that the mechanisms by which Ccnd1 deficiency reduced teratoma incidence were germ cell autonomous and specific to tumorigenesis. We conclude that misexpression of Ccnd1 in male germ cells is a key component of a larger pro-proliferative program that disrupts the mitotic:meiotic switch and predisposes 129 inbred mice to testicular teratocarcinogenesis.
KEYWORDS: Cyclin D1 (Ccnd1), development, germ cells, pluripotency, teratomas
Introduction
Germ cells arise during embryogenesis as pluripotent primordial germ cells (PGCs) that differentiate into mature gametes and ultimately the cells and tissues of an adult organism.1,2 Defects during embryonic male germ cell development can lead to the formation of testicular germ cell tumors (TGCTs), which are classified as teratomas, non-seminomas, or seminomas.3-6 The 129 family of inbred mice has proven to be an important model system for studying the embryonic origins and genetic risk factors of human TGCTs.7,8 Spontaneous TGCTs occur at a measurable incidence (˜8%) only in 129 inbred strains and are first evident at embryonic day (E)15.5 as foci of highly proliferative and pluripotent tumor stem cells (embryonal carcinoma cells or EC cells).9,10 Mouse EC cells differentiate to form teratomas, disorganized cell masses consisting of embryonic tissue types at various stages of differentiation.9,11,12 Tumor pathogenesis and genetic susceptibility is highly similar between teratomas in 129 mice and human Type I pediatric teratomas and adolescent/adult Type II non-seminomas.6,9-12 In fact, several genes (e.g. Kitl, Dmrt1, and Pten) associated with human TGCT risk in genome-wide association studies were first shown to influence teratoma incidence when disrupted in 129 mice.13-18 Although these mutations, and several others, have distinct phenotypic effects regardless of genetic background, they are dependent on the 129 genetic background to modify teratoma incidence.7,14,15,19-24 Thus, a complex array of unknown genetic variants (i.e. susceptibilty mutations) and downstream transcriptional networks specific to the 129 inbred background are required for teratoma initation in mice.7
In mice, testicular teratoma initiation coincides with germ cell sex specification from E13.5 to E15.5.3,11,19,25 XX and XY PGCs are morphologically and functionally indistinguishable until E13.5 when signals from somatic cells in the nascent gonad induce sex-specific differentiation.26-29 One of the first signs of sexual dimorphism is germ cell entry into meiosis or mitotic arrest (the mitotic:meiotic switch).27 In the ovary, retinoic acid acts in concert with genes such as Dazl to activate Stra8 expression and induce female germ cell (oogonia) entry into prophase I of meiosis.30-33 In the testis, signals from somatic cells, including FGF9, Activins, and prostaglandin D2, induce male germ cell (gonocyte) lineage commitment, which is accompanied by G1/G0 mitotic arrest.34-36 Gonocytes remain mitotically quiescent until after birth when they form the spermatogonial lineage.28 Concomitant with sex-specific differentiation and the mitotic:meiotic switch, germ cells of both sexes downregulate the core regulators of pluripotency, Oct4, Sox2, and Nanog, which are critical for the initial specification and maintenance of PGCs.37-39
Gonocytes activate the G1-S phase cell cycle checkpoint in a gradual and unsynchronized manner due to a shift in the expression of positive and negative regulators of the G1-S phase transition. Prior to becoming mitotically quiescent, gonocytes express cyclins E1 and E2 (CCNE1/2) and cyclin D3 (CCND3), which form complexes with cyclin dependent kinases 2 (CDK2) and 4 or 6 (CDK4/6), respectively.40 These cyclin-CDK complexes hyperphosphorylate (inactivate) retinoblastoma protein 1 (pRB1), leading to derepression of E2F transcription factors and activation of genes required for progression into S phase.40-42 Cyclin D-CDK4/6 complexes also promote the G1-S transition through the sequestration of cyclin E-CDK2 inhibitors p27Kip1 (CDKN1B) and p21Cip1(CDKN1A).42 Starting around E13.5, decreases in expression of CCNE1/2 and CCND3 and increases in expression of p27Kip1and cyclin D-CDK4/6 inhibitors p15INK4B (CDKN2B) and p16INK4A(CDKN2A) result in hypophosphorylation (activation) of pRB1, suppression of E2F transcriptional activity, and gonocyte transition into G1/G0 arrest.40
Disruption of the mitotic:meiotic switch in gonocytes appears to be critical for teratoma initiation. Using mouse strains with low versus high teratoma incidence (129 vs. the chromosome substitution strain 129-Chr19MOLF/Ei),43 and resistant to teratoma formation (FVB), we previously found that teratoma-susceptible gonocytes continued to proliferate and retain pluripotency during the mitotic:meiotic switch.44 Studies of several genetic modifiers of teratoma incidence in 129 mice have similarly found that gonocyte proliferation and pluripotency after E13.5 directly correlate with tumor susceptibility.14,15,19,20,45 Importantly, the number of aberrantly proliferating and pluripotent gonocytes at E15.5 increased with tumor incidence. Because EC cells first appear at E15.5,10,11 retention of proliferative and pluripotent capacity through this time point is most likely necessary to establish a proliferative and pluripotent tumor stem cell population.
Curiously, from E13.5 to E15.5, teratoma-susceptible gonocytes also misexpress genes normally restricted to pre-meiotic oogonia and post-natal spermatogonia.20,44 As observed with proliferation and pluripotency, expression of these genes through E15.5 increases with tumor risk, indicating that they also contribute to the transformation of gonocytes into EC cells. Which of these aberrantly expressed genes contribute to teratoma risk remains largely unknown. To date, only misexpression of Stra8 has been shown to affect teratoma incidence.44 However, the influence of one additional misexpressed gene, cyclin D1 (Ccnd1), on G1-S cell cycle progression suggests that it may also be a driver of testicular teratocarcinogenesis.
In the male germ cell lineage, CCND1 is normally expressed in post-natal and adult spermatogonia but not embryonic germ cells.40,46 However, in teratoma-susceptible mice, gonocytes aberrantly express CCND1 from E13.5 to E15.5, with the number of gonocytes expressing CCND1 at E15.5 increasing with teratoma incidence.44 Importantly, CCND1 expression continues after gonocyte transformation into EC cells.44 D-type cyclins are potent oncogenes, whose overexpression overwhelms negative regulators of the G1-S transition, promotes growth factor independent proliferation, and induces tumorigenesis.42 Specifically, CCND1 is located within one of the most frequently amplified loci in the human cancer genome and its overexpression in the absence of any genomic alterations is also a common occurrence in many cancers. 42,47-49 Studies in genetic mouse models have shown that Ccnd1 is essential for both tumor formation and maintenance in several tissues. For example, in female mice bearing Erb2-driven mammary adenocarcinomas, Ccnd1 deficiency suppresses tumor cell proliferation and induces senescence, which together inhibit tumor initiation and growth.50 Importantly, tumor cells appear to be uniquely dependent on the expression of individual D-type cyclins for their proliferation. Mice deficient for individual D-type cyclins are viable, fertile, and present with relatively minor phenotypes, demonstrating that these proteins are non-essential and likely have redundant functions in most normal cell lineages.51,52
In the present study, we examine the contributions of Ccnd1 misexpression in mouse gonocytes to teratoma susceptibility and the developmental abnormalities associated with tumor initiation. We demonstrate that Ccnd1 is aberrantly expressed in teratoma-susceptible gonocytes that fail to enter G1/G0 arrest during the mitotic:meiotic switch and that Ccnd1 is the only D-type cyclin misexpressed from E13.5 to E15.5. Using Ccnd1 knockout mice, we show that Ccnd1 expression significantly contributes to tumor incidence in teratoma susceptible mice without being necessary for normal germ cell or testis development. Importantly, we demonstrate that Ccnd1 deficiency suppresses both the aberrant proliferation and retention of pluripotency phenotypes associated with gonocyte transformation into pluripotent EC cells. Based on these findings, we propose that misexpression of Ccnd1 in gonocytes during the mitotic:meiotic switch is a key component of a 129 inbred background-dependent, pro-proliferative program that drives the developmental abnormalities necessary for gonocyte transformation into EC cells.
Results
Dysregulation of the G1-S phase checkpoint in gonocytes increases with teratoma risk
We previously reported that teratoma-susceptible gonocytes delay entry into G0 arrest during the mitotic:meiotic switch (E13.5 to E15.5) and that teratoma risk increased with the incidence of gonocyte proliferation at E15.5. To test whether delayed entry into mitotic arrest is caused by dysregulation of the G1-S phase checkpoint, we examined pRB1 phosphorylation (inactivation) in gonocytes of a teratoma-resistant strain, FVB/NJ (FVB), and 2 teratoma-susceptible strains, the 129-Chr19MOLF/Ei chromosome substitution strain (M19) and the 129/SvImJ (129) inbred strain. M19 mice, in which both copies of chromosome 19 are derived from the MOLF/Ei inbred strain, have a high risk of developing teratomas (80% of males affected).43 In contrast, 129 inbred mice have a low risk of developing teratomas (8% of males affected).11 Using these 3 strains, gonocyte abnormalities associated with increasing teratoma risk can be identified.44 Furthermore, because most M19 and 129 germ cells develop normally, the developmental characteristics of teratoma-susceptible gonocytes that do not transform into EC cells can also be studied.11,43,44
To identify phospho-pRB1-positive gonocytes, we immunostained embryonic testes from FVB, 129, and M19 embryos harboring a germ cell-specific GFP transgene (Oct4::GFP)21,53 (Fig. 1A and B; Fig. S1). At E13.5, the percentage of phospho-pRB1-positive gonocytes was similar in the testes of all strains. At E14.5, the percentage of phospho-pRB1-positive gonocytes decreased in all strains compared to E13.5, but the incidence of phospho-pRB1 staining was significantly higher in teratoma-susceptible testes (8% positive in FVB versus 24-29% positive in 129 and M19). Interestingly, less than 1% of FVB and 129 gonocytes remained phospho-pRB1-positive at E15.5. In contrast, 14% of E15.5 M19 gonocytes remained phospho-pRB1-positive. Together, these results demonstrate that activation of G1-S phase checkpoint is delayed in a sub-population of teratoma-susceptible gonocytes during the mitotic:meiotic switch and that tumor incidence increases with the occurrence of phospho-pRB1-positive germ cells at E15.5.
Figure 1.
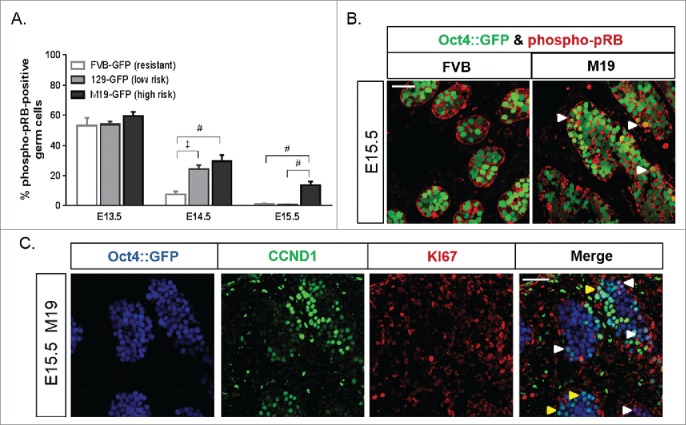
The G1-S phase transition is perturbed in teratoma-susceptible gonocytes. Dysregulation of the G1-S phase checkpoint in gonocytes increased with teratoma risk. (A) Oct4::GFP transgenic FVB (teratoma-resistant), 129 (low teratoma risk), and M19 (high teratoma risk) embryonic testes were sectioned and immunostained for phospho-pRB1 (E13.5, E14.5, E15.5). GFP-positive germ cells, positive and negative for phospho-pRB1, were counted. Data are plotted as the percentage of germ cells positive for phospho-pRB1 ± SEM (n = 7-11). ‡, P < 0.001; #, P < 0.0001. (B) Confocal microscopy images of E15.5 testes immunostained for phospho-pRB1. At E15.5, FVB germ cells are phospho-pRB1-negative, but some M19 germ cells remain phospho-pRB1-positive. Small groups of phospho-pRB1-positive germ cells are noted (arrowheads). Scale bar: 50 μm. (C) Confocal microscopy images of an E15.5 M19 testis sectioned and immunostained for CCND1 and the proliferative marker KI67. All KI67-positive gonocytes were also positive for CCND1; small groups are noted (white arrowheads). However, several CCND1-positive gonocytes were KI67-negative; small groups are noted (yellow arrowheads). Scale bar: 50 μm.
Previous studies have revealed that aberrant expression of Ccnd1 in teratoma-susceptible gonocytes initiates at E13.5, becomes restricted to a continually smaller sub-population of germ cells through E15.5, and is ultimately expressed only in the few cells predisposed to transformation into EC cells.44 Based on these observations and the role of Ccnd1 in promoting cell cycle progression, we hypothesized that aberrant Ccnd1 expression may contribute to the delayed mitotic arrest of teratoma-susceptible gonocytes during the mitotic:meiotic switch. To determine whether gonocytes aberrantly expressing Ccnd1 are the same sub-population of cells delaying entry into G0 arrest, we co-immunolabeled Oct4::GFP transgenic E15.5 M19 testes for CCND1 and the proliferation marker KI67, which distinguishes cells in any stage of the active cell cycle from those arrested in G0. Importantly, all KI67-positive gonocytes were also positive for CCND1 (Fig. 1C, noted with white arrowheads), directly linking aberrant CCND1 expression in gonocytes to delayed G0 arrest. However, several CCND1-positive gonocytes were KI67-negative (Fig. 1C, noted with yellow arrowheads). Thus, expression of negative regulators of the G1-S phase transition appears to be sufficient to induce G0 arrest in some gonocytes even in the presence of aberrant CCND1 expression.
Cyclin D1 is the only D-type cyclin misexpressed in teratoma-susceptible gonocytes during the mitotic:meiotic switch
Next, we measured cyclin D2 (Ccnd2) and D3 (Ccnd3) expression in FVB, 129, and M19 germ cells isolated by fluorescence-activated cell sorting (FACS) with the Oct4::GFP transgene (Fig. 2A and B) to test whether cyclin D1 is the only D-type cyclin aberrantly expressed in teratoma-susceptible gonocytes. Previous studies have established that embryonic male germ cells predominantly express Ccnd3 prior to and after mitotic arrest, with expression decreasing from E13.5 to E15.5.40,46 In the same studies, Ccnd2 transcript and protein levels were found to be low and unchanged in E13.5 to E15.5 gonocytes.40,46 Similar to these reports, we found that expression of Ccnd3 in gonocytes of all strains decreased from E13.5 to 15.5 and that Ccnd2 expression levels were unchanged during this time period (Fig. 2A and B). Importantly, no significant differences in Ccnd2 or Ccnd3 expression were observed between teratoma-susceptible and resistant gonocytes. Thus, Ccnd1 is the only D-type cyclin misexpressed in teratoma-susceptible gonocytes during the mitotic:meiotic switch.
Figure 2.
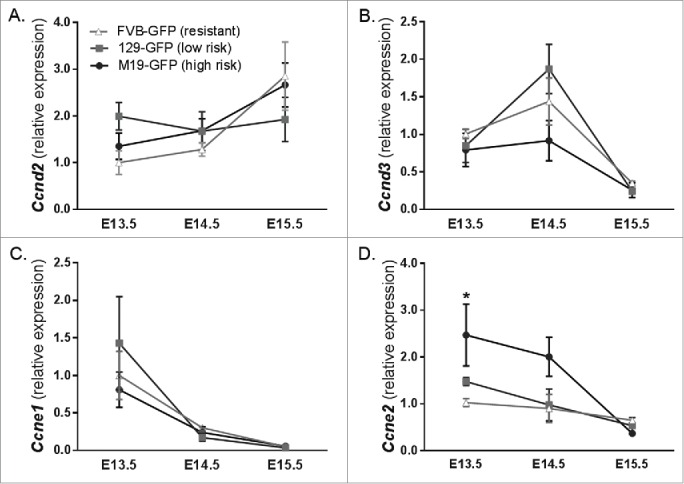
Cyclin D1 is the only D-type cyclin misexpressed in teratoma-susceptible gonocytes during the mitotic:meiotic switch. (A-D) Oct4::GFP transgenic FVB, 129 and M19 gonocytes from E13.5-E15.5 gonads were isolated by FACS and expression of G1-S phase cyclins Ccnd2, Ccnd3, Ccne1, and Ccne2, respectively, was analyzed by qPCR (n = 4-23). Gene expression at all embryonic time points is plotted relative to expression in E13.5 FVB male germ cells. *, P < 0.05.
In addition, we measured gonocyte expression of the E-type cyclins Ccne1 and Ccne2. Similar to previous reports,40 we found that gonocyte expression of Ccne1 and Ccne2 decreased from E13.5 to 15.5 in all strains (Fig. 2C and D). At each time point, expression of Ccne1 was statistically similar between gonocytes of all strains (Fig. 2C). In contrast, gonocyte expression of Ccne2 was significantly higher in M19 testes compared to FVB testes at E13.5 and remained elevated, but not significantly, in M19 through E14.5 (Fig. 2D). By E15.5, Ccne2 expression in M19 gonocytes decreased to levels observed in FVB gonocytes. Curiously, gonocyte expression of Ccne2 in low teratoma risk 129 testes was not significantly different from FVB at any time points assayed (Fig. 2D). Thus, retention of Ccne2 expression may contribute to delayed entry of M19 gonocytes into G0 arrest at the beginning of the mitotic:meiotic switch. However, because Ccne2 expression was similar between all strains at E15.5, the critical time point at which EC cells first appear, it is uncertain whether misexpression of Ccne2 at E14.5 contributes to teratoma initiation in M19 mice. Together, the results of our D- and E-type cyclin expression analyses suggest that cyclin D1 is the key G1-S cyclin contributing to the gonocyte proliferation defect that precedes teratoma initiation.
Ccnd1 deficiency reduces teratoma incidence
Cyclin D1 is a well-known oncogene and its overexpression has been previously shown to disrupt the G1-S checkpoint, promote aberrant proliferation, and contribute to tumor initiation in several tissues.42 Therefore, we next wanted to test whether Ccnd1 deficiency suppresses teratoma susceptibility. However, because of the inherently low teratoma incidence in 129 males, large numbers of mice would need to be screened to test whether Ccnd1 deficiency reduces susceptibility. To enable a more statistically powerful test, the Ccnd1tm1Wbg knockout allele (referred to as Ccnd1KO)54 was bred onto the M19 background. Because the M19 strain has a teratoma incidence of ˜80%, tests for reduced incidence are relatively easy in a modest number of mice.43,45
Previous studies in teratoma-resistant C57BL/6J mice demonstrated that Ccnd1 deficient mice are smaller and often die after 4 weeks of age, but those that reach sexual maturation are fertile.54 Thus, Ccnd1 is dispensable for male germ cell specification and maturation. Importantly, a similar phenotypes was observed in M19 mice homozygous for the Ccnd1 knockout allele (Ccnd1KO/KO). Ccnd1 was not required for spermatogenesis to occur. Histologically, Ccnd1 deficient testes appeared normal with spermatozoa present in the seminiferous tubules of males that reached sexual maturity (Fig. S2A).
To verify that the spermatogonial stem cell population was appropriately established in Ccnd1 deficient males, we analyzed the testes of post-natal day 8 (P8), M19-Ccnd1KO/KO and control mice transgenic for Oct4::GFP, which is expressed in the male germ cell lineage through post-natal stages.53 Both Hematoxylin and Eosin staining and analysis of GFP expression patterns in P8 testis sections verified that spermatogonial stem cells had migrated to the basement membrane of the seminiferous tubule in Ccnd1 deficient mice (Fig. S3A & B). Furthermore, several GFP-positive spermatogonia had begun to express c-KIT, a marker of differentiated spermatogonia,55 indicating that the first wave of spermatogenesis had initiated in M19-Ccnd1KO/KOtestes (Fig. S3B).56 Together, our analysis of post-natal and adult testes show that Ccnd1 is not required for the development of the male germ cell lineage in teratoma-susceptible mice.
Intercrosses between M19 mice heterozygous for the Ccnd1 deletion (Ccnd1KO/+) generated experimental mice heterozygous (Ccnd1KO/+) or homozygous for the Ccnd1 deletion and control wild-type littermates (Ccnd1+/+) for surveys of tumor incidence. M19-Ccnd1+/+ male offspring derived from the M19-Ccnd1KO/+intercross had a teratoma incidence of 79% (37 of 47 with a tumor), which is consistent with previously published data for M19.43 Importantly, of the 34 M19-Ccnd1KO/KO mice examined, only 10 (29%) had a teratoma, which represented a 63% reduction in teratoma incidence compared to wild-type controls (P<5E-4, Table 1). Histopathology verified that the few tumors that formed in M19-Ccnd1KO/KO mice were teratomas (Fig. S2B). Interestingly, tumor surveys revealed that only 72 of 110 (65%) M19-Ccnd1KO/+ males had a teratoma. Although the 18% reduction in teratoma incidence in M19-Ccnd1KO/+ compared to wild-type M19 controls was not significant with the samples sizes analyzed (P=0.098, Table 1), it suggests that Ccnd1 has a gene dosage effect on teratoma susceptibility. Based on the results of our tumor survey, we conclude that Ccnd1 expression significantly contributes to teratoma incidence.
Table 1.
Teratoma incidence on the M19 background.
| Genotype | N | Tumor | % Affected* |
|---|---|---|---|
| M19-Ccnd1+/+ | 47 | 37 | 79 |
| M19-Ccnd1KO/+ | 110 | 72 | 65 |
| M19-Ccnd1KO/KO | 34 | 10 | 29 |
Fisher's Exact test: M19-Ccnd1+/+ vs M19-Ccnd1KO/+
P = 0.098; M19-Ccnd1+/+ vs M19-Ccnd1KO/KO
P < 5E-4
Ccnd1 is not a 129 susceptibility gene
In segregating crosses between 129/Sv and other strains, only 1 affected male was found among more than 11,000 progeny tested, which is consistent with 129-specific genetic variants in as many as 8 susceptibility genes being necessary for teratoma formation.11,57 Because Ccnd1 deficiency reduced but did not block tumorigenesis, we hypothesized that Ccnd1 is not one of these susceptibility genes. In fact, analysis of single nucleotide polymorphisms (SNPs) at the Ccnd1 locus in 129S1/SvImJ and several other teratoma-resistant inbred strains of mice failed to identify potential coding or regulatory SNP variants specific to 129 (Table S1). Furthermore, 129 and FVB, the teratoma-resistant strain in which Ccnd1 is not expressed in gonocytes,44 had the same variant for each SNP tested. Thus, although Ccnd1 misexpression significantly contributes to teratoma incidence, it appears to not be a 129-derived teratoma susceptibility gene. Instead, other genetic variants in the 129 genome must influence teratoma incidence, in part, by inducing Ccnd1 expression in gonocytes.
Ccnd1 deficiency does not inhibit Sertoli or Leydig cell specification
Somatic cell regulation of the testis microenvironment is critical to male germ cell development and, therefore, has the potential to influence tumorigenesis. To determine whether Ccnd1 deficiency altered somatic cell lineage development through the mitotic:meiotic switch, we measured expression of markers specific to pre-Sertoli cells (Sox9, Dhh) and developing Leydig cells (Cyp11a1) in FACS-enriched, Oct4::GFP-negative somatic cells from E14.5 and E15.5, M19-Ccnd1+/+ and M19-Ccnd1KO/KO testes. Importantly, Ccnd1 deficiency did not affect expression of the Sertoli or Leydig cell markers at either time point (Fig. 3). Together with our data showing that spermatogonial stem cells specify and have the capacity to differentiate into c-KIT-positive spermatogonia during the first wave of meiosis after birth and that adult sperm production occurs in Ccnd1 knockout adult males (Fig. S2A and S3), these results suggest that Ccnd1 deficiency does not adversely affect the specification of these 2 critical somatic cell lineages. Thus, the mechanisms by which Ccnd1 deficiency reduces teratoma incidence are most likely germ cell autonomous.
Figure 3.
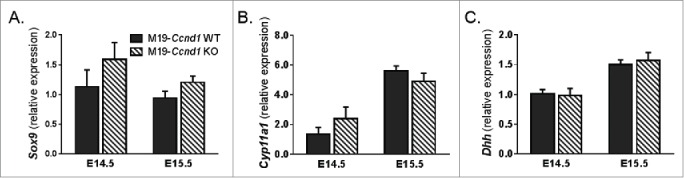
Sertoli and Leydig cell development is unaltered in Ccnd1 deficient gonads through the mitotic:meiotic switch. Ccnd1 deficient somatic cells have unaltered expression of (A & B) pre-Sertoli and (C) developing Leydig cell markers. Oct4::GFP-negative somatic cells were isolated by FACS from E14.5, E15.5 M19-Ccnd1+/+ (M19-Ccnd1 WT) and M19-Ccnd1KO/KO (M19-Ccnd1 KO) gonads, and gene expression was analyzed by qPCR (n = 3-5). Gene expression at all embryonic time points is plotted relative to expression in E14.5 M19-Ccnd1 WT male somatic cells.
Ccnd1 deficiency suppresses the aberrant proliferation of teratoma-susceptible gonocytes
Because of the close association between CCND1 misexpression in gonocytes and their delayed entry into G0 arrest (Fig. 1C), we hypothesized that Ccnd1 deficiency reduced teratoma incidence in M19 mice by suppressing aberrant germ cell proliferation. To test this hypothesis, we first employed the Oct4::GFP transgene to investigate the influence of Ccnd1 deficiency on total germ cell number in M19 testes from E13.5 to E15.5 (Fig. 4). Aberrant expression of Ccnd1 does not initiate until E13.5.44 Thus, it was not surprising to discover that Ccnd1 deficiency did not alter total germ cell number up to E13.5. Additionally, at E14.5 and E15.5, total germ cell number was unaffected by Ccnd1. Together with our findings that spermatogonial stem cell specification and spermatogenesis occur in the absence of Ccnd1 in teratoma susceptible neonates and adults, respectively (Fig. S2A and S3), these results demonstrate that Ccnd1 is dispensable for male germ cell specification and maturation. Thus, the mechanisms by which Ccnd1 deficiency reduced teratoma susceptibility appear to be specific to the process of tumor initiation.
Figure 4.
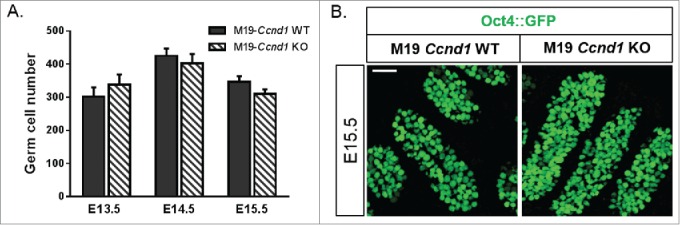
Ccnd1 deficiency does not affect germ cell number. (A) Oct4::GFP transgenic M19 wild-type (WT) and M19 Ccnd1 homozygous knockout (KO) embryonic testes were sectioned and Oct4::GFP-positive germ cells were counted (E13.5, E14.5, E15.5). Data are plotted as the number of Oct4::GFP-positive germ cells ± SEM (n = 4-14). (B) Confocal microscopy images of E15.5 Oct4::GFP transgenic testes from M19-Ccnd1 WT and KO embryos. Scale bar: 50 μm.
Next, we immunostained Oct4::GFP transgenic M19-Ccnd1+/+ and M19-Ccnd1KO/KOembryonic testes for phospho-pRB1 to test whether Ccnd1 deficiency activated the G1-S phase checkpoint in teratoma-susceptible gonocytes (Fig. 5A and B, Fig. S4). At E13.5, the percentage of phospho-pRB1-positive gonocytes was similar in both Ccnd1-deficient and wild-type M19 testes (˜58% positive). However, at E14.5, the incidence of phospho-pRB1-positive gonocytes was significantly reduced in Ccnd1-deficient testes (22% positive in M19-Ccnd1KO/KO vs. 40% positive in M19-Ccnd1+/+). A significant difference in phospho-pRB1 staining was also observed at E15.5, at which time only 2% of M19-Ccnd1KO/KOgonocytes remained phospho-pRB1-positive compared to 14% of M19-Ccnd1+/+gonocytes.
Figure 5.
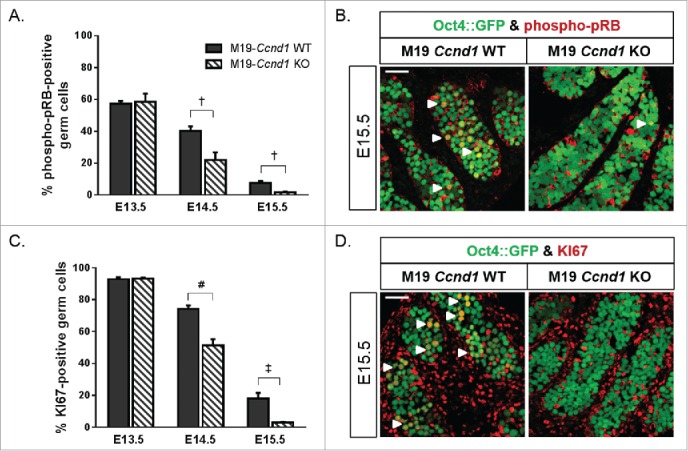
Ccnd1 deficiency suppresses G1-S checkpoint dysregulation and aberrant proliferation of teratoma-susceptible gonocytes. Oct4::GFP transgenic M19-Ccnd1 WT and KO embryonic testes were sectioned and immunostained for (A) phospho-pRB1 or (C) KI67 (E13.5, E14.5, E15.5). GFP-positive germ cells, positive and negative for phospho-pRB1 or KI67-positive, were counted. Data are plotted as the percentage of germ cells positive for phospho-pRB1 or KI67 ± SEM (n = 4-14). †, P < 0.01; ‡, P < 0.001; #, P < 0.0001. (B, D) Confocal microscopy images of E15.5 Oct4::GFP transgenic M19-Ccnd1 WT and KO gonads immunostained for (B) phospho-pRB1 or (D) KI67. Small groups of phospho-pRB1 or KI67-positive germ cells are noted (arrowheads). Scale bar: 50 μm.
We also immunolabelled Oct4::GFP transgenic, M19-Ccnd1+/+ and M19-Ccnd1KO/KOembryonic testes for KI67 to test whether Ccnd1 deficiency induced gonocyte entry into G0 arrest after E13.5 (Fig. 5C and D; Fig. S5). We previously reported that 80-90% of E13.5 gonocytes in teratoma-resistant and susceptible testes were KI67-positive.44 It was not until E14.5 that retention of KI67 staining was observed in teratoma-susceptible germ cells. Ccnd1 deficiency did not affect the percentage of KI67-positive gonocytes (93% positive in M19-Ccnd1KO/KO and M19-Ccnd1+/+) at E13.5. However, at E14.5 and continuing through E15.5, Ccnd1 deficiency significantly reduced the percentage of M19 gonocytes positive for KI67 so that by E15.5 only 3% of M19-Ccnd1KO/KOgonocytes remained KI67-positive compared to 18% of M19-Ccnd1+/+gonocytes. Collectively, these results demonstrate that aberrant Ccnd1 expression is critical for delaying G0 arrest in gonocytes after E13.5 and inducing the aberrant proliferation phenotype associated with teratoma initiation in mice.
Ccnd1 deficiency suppresses retention of pluripotency by teratoma-susceptible gonocyte
In addition to delaying entry into G0 arrest, teratoma-susceptible gonocytes retain pluripotent capacity during the mitotic:meiotic switch, which appears to be necessary for transformation into pluripotent EC cells starting at E15.5.44 Curiously, reprogramming of pluripotency and differentiation of several stem cells, including germ cells, embryonic stem (ES) cells, and EC cells, occurs during G0 arrest.37-39,58-60 Therefore, aberrant cyclin D1 expression in gonocytes during the mitotic:meiotic switch and the resulting delay in G0 arrest may cause those same cells to remain pluripotent. To test whether Ccnd1 misexpression promotes gonocyte pluripotency, we immunostained Oct4::GFP transgenic M19-Ccnd1+/+ and M19-Ccnd1KO/KOtestes for NANOG (Fig. 6; Fig. S6). We previously showed that 90% of E13.5 gonocytes in teratoma-resistant and susceptible testes were NANOG-positive.44 It was not until after E14.5 that retention of NANOG expression was observed in tumor-susceptible gonocytes. At E13.5, the percentage of M19 gonocytes positive for NANOG was not affected by Ccnd1 deficiency (84% positive in M19-Ccnd1KO/KO and 85% positive in M19-Ccnd1+/+). However, at E14.5, the incidence of NANOG-positive gonocytes significantly decreased in Ccnd1-deficient testes compared to wild-type controls (40% positive in M19-Ccnd1KO/KO and 62% positive in M19-Ccnd1+/+). A significant difference in NANOG staining was also observed at E15.5, at which time only 7% of M19-Ccnd1KO/KOgonocytes remained NANOG-positive compared to 23% of M19-Ccnd1+/+gonocytes. Interestingly, in Ccnd1 deficient mice, the reduction in the percentage of NANOG-positive germ cells (Fig. 6A) closely mirrored the decrease in the percentage of KI67-positive germ cells (Fig. 5C). Together with previous evidence that rapid transition from G1 into S phase promotes pluripotency,61-63 these data suggest that Ccnd1 deficiency suppresses the pluripotency defect in teratoma-susceptible germ cells as a direct consequence of its effect on proliferation.
Figure 6.
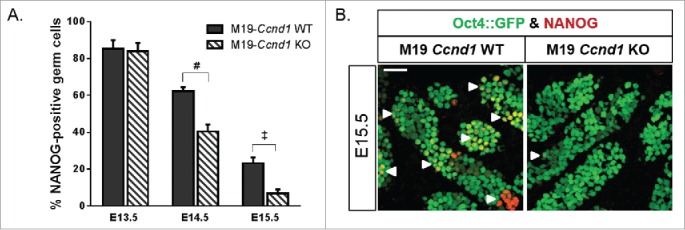
Ccnd1 deficiency suppresses aberrant pluripotency in teratoma-susceptible gonocytes. (A) Oct4::GFP transgenic M19-Ccnd1 WT and KO embryonic testes were sectioned and immunostained for NANOG (E13.5, E14.5, E15.5). GFP-positive germ cells, positive and negative for NANOG, were counted. Data are plotted as the percentage of germ cells positive for NANOG ± SEM (n = 4-13). ‡, P < 0.001; #, P < 0.0001. (B) Confocal microscopy images of E15.5 Oct4::GFP transgenic M19-Ccnd1 WT and KO gonads immunostained for NANOG. Small groups of NANOG-positive germ cells are noted (arrowheads). Scale bar: 50 μm.
Discussion
For several decades, it has been hypothesized that testicular teratomas arise in 129 inbred mice from gonocytes that remain mitotically active and pluripotent following the mitotic:meiotic switch.3,11,19,25 However, despite considerable effort, the 129-specific genetic variants and transcriptional networks required for teratoma initiation in mice remained undefined. We previously reported that 129 and M19 teratoma-susceptible gonocytes aberrantly express Ccnd1 during the mitotic:meiotic switch.44 Given its known role as positive regulator of G1-S cell cycle progression and as an oncogene when aberrantly expressed in a variety of tissues, we hypothesized that misexpression of Ccnd1 in gonocytes during the mitotic:meiotic switch disrupts the balance of positive to negative regulators of the cell cycle, delays entry into mitotic arrest, and facilitates germ cell transformation into pluripotent EC cells. In the present study we demonstrate that Ccnd1 is an important driver of both the gonocyte proliferation and pluripotency defects that precede tumor initiation in 129 inbred mice. Importantly, Ccnd1 expression was dispensable for somatic cell specification and male germ cell development and maturation, suggesting that the mechanisms by which Ccnd1 deficiency reduced teratoma incidence were germ cell autonomous and specific to tumorigenesis. However, a more in-depth analysis of Ccnd1 deficient testes will be necessary to rule out potential subtle changes in somatic and germ cell development and differentiation.
Gonocyte entry into G1/G0 mitotic arrest during the mitotic:meiotic switch is dependent on coordinated alterations in the expression of positive and negative regulators of the G1-S phase transition.40 During embryogenesis, D-type cyclin expression is primarily restricted to CCND3 in male germ cells.40,46 Curiously, even though its expression decreases during the mitotic:meiotic switch, CCND3 protein persisted in FVB gonocytes through at least E17.5, a time point at which these cells are quiescent. Thus, expression of negative regulators of the G1-S transition must be sufficient to counteract residual D-type cyclin expression to induce G1/S mitotic arrest.46 A sufficient increase in the ratio of positive to negative regulators of G1-S cell cycle progression during the mitotic:meiotic switch might tip the balance toward proliferation rather than G1/G0 arrest. We had previously demonstrated that Ccnd1 expression levels are significantly higher in teratoma-susceptible gonocytes.44 Here, our expression analyses of Ccnd2 and Ccnd3, and the E-type cyclins, Ccne1 and Ccne2, revealed that Ccnd1 is the only G1-S phase cyclin upregulated in teratoma-susceptible gonocytes at E15.5, the time point at which EC cells first form. In fact, a transient increase in Ccne2 expression at E13.5 in M19 gonocytes was the only other significant change in G1-S phase cyclin expression observed. Thus, cyclin D1 appears to be the G1-S phase cyclin delaying G1/G0 arrest of teratoma-susceptible gonocytes. Supporting this conclusion, our phospho-pRB1 and KI67 immunostaining of teratoma-susceptible gonocyte showed that Ccnd1 deficiency induced G1/G0 mitotic arrest in a more developmental stage-appropriate manner.
Interestingly, co-staining experiments with KI67 revealed that some CCND1-positive M19 gonocytes are in G1/G0 mitotic arrest. These results are in agreement with our phospho-pRB1 staining results and our previous KI67 and CCND1 single staining experiments,44 which together revealed that there are more M19 CCND1-positive gonocytes at E14.5 and E15.5 (60% and 50% CCND1-postive) than gonocytes in the active cell cycle (46% and 25% KI67-positive; 29% and 14% phosphor-Rb1-positive). Thus, some germ cells overcome the pro-proliferative effects of Ccnd1 misexpression. The ability of teratoma-susceptible gonocytes to become quiescent in the face of aberrant Ccnd1 expression may be dependent on the ratio of Ccnd1 expression to the expression of negative regulators of the G1-S transition. Western et al.64 previously demonstrated that, even in 129 inbred mice, gonocytes are induced to express negative regulators of the G1-S transition (e.g., p27Kip1, p15INK4B, and p16INK4A) during the mitotic:meiotic switch. However, a recent microarray study revealed that the expression of one cyclin D-CDK4/6 inhibitor p15INK4B is 5-fold lower in 129 gonocytes compared to teratoma-resistant C57BL/6J gonocytes at E14.5. Therefore, the relative abundance of Ccnd1 and p15INK4B expression might determine whether a teratoma-susceptible germ cell becomes mitotically quiescent or remains proliferative.
The teratoma-forming capacity of EC cells is dependent upon their pluripotent capacity.65 Thus, retention of both proliferation and pluripotency appear to be necessary for gonocyte transformation into EC cells. Whether mitotic arrest and downregulation of pluripotency in gonocytes are linked or independently regulated is not well understood. Miles et al.35 previously demonstrated that Activin signaling from somatic cells and autocrine NODAL signaling induces gonocytes to enter into mitotic arrest and transiently maintain pluripotency, respectively. Separate studies showed that subsequent loss of NODAL expression by gonocytes facilitates suppression of pluripotency.66 Thus, aspects of gonocyte G1/G0 arrest and suppression of pluripotency are independently regulated. However, our discoveries that Ccnd1 deficiency suppressed teratoma-susceptible gonocyte pluripotency (NANOG expression) during the mitotic:meiotic switch and that this decrease was reflected by a similar reduction in KI67-positive cells suggest that retention of pluripotency is, at least in part, dependent on the misexpression of genes that promote G1-S cell cycle progression and proliferation. Importantly, a link between the regulation of the cell cycle and pluripotency is well established. In both ES and EC cells, rapid transition through G1 into S phase facilitates the maintenance of pluripotency.58,60 A short G1 and long S phase promotes the euchromatic state of chromatin and suppresses differentiation, which preferentially occurs during the G1 phase in pluripotent cells.61-63
Based on our tumor surveys of Ccnd1 knockout mice, it is evident that Ccnd1 misexpression promotes but is not necessary for testicular teratocarcinogenesis. Furthermore, although Ccnd1 deficiency suppressed the incidence of aberrant gonocyte proliferation and retention of pluripotency during the mitotic:meiotic switch, some gonocytes remained proliferative and pluripotent at E15.5. These results imply that Ccnd1 is not one of the 129-derived susceptibility genes necessary for teratoma formation. Rather, Ccnd1 is likely to be one of several downstream targets of 129 susceptibility genes that disrupt G1/G0 mitotic arrest. In agreement with these conclusions, our analysis of SNP variants at the Ccnd1 locus failed to identify SNP variants that may directly dysregulate Ccnd1 expression in 129 gonocytes. As previously mentioned, microarray expression analysis of 129 and C57BL/6J E14.5 gonocytes found that at least one negative regulator of the G1-S transition, p15INK4B, is expressed at higher levels in C57BL/6J gonocytes compared to 129 germ cells. Thus, the cumulative influences of these downstream mediators on mitotic arrest likely determine overall teratoma incidence, but are not individually required for tumorigenesis to occur. What induces this pro-proliferative transcriptional program in 129 gonocytes is not known.
The concurrent timing of Ccnd1 misexpression, the mitotic:meiotic switch, and sex specification may provide clues to the underlying cause of dysregulation at the mitotic:meiotic switch in 129 mice. We previously showed that oogonia in both teratoma-resistant and susceptible mouse strains transiently express Ccnd1 from E12.5 to E15.5, just prior to initiating meiosis.44 Thus, Ccnd1 misexpression in teratoma-susceptible gonocytes occurs at the same developmental time points that Ccnd1 is normally expressed in pre-meiotic oogonia. These observations suggest that a signal normally restricted to the developing ovary is aberrantly active in the teratoma-susceptible testis. Alternatively, a signal normally active in the testis may be absent in both the ovary and teratoma-susceptible testis. Additional studies are needed to determine the pro-proliferative signal inducing Ccnd1.
Our discovery that Ccnd1 misexpression in mouse gonocytes contributes to a mitotic:meiotic switch defect that leads to tumorigenesis is likely to inform future studies into human TGCT initiation and new treatment paradigms. Recent gene expression studies of human TGCT stem cells suggest that dysregulation of the mitotic:meiotic switch drives tumor initiation.67,68 Furthermore, CCND1 expression has been shown to predominantly localize to the EC cell compartment of human TGCTs, where it appears to be an important mediator of EC cell proliferation, survival, and resistance to cisplatin.69,70 Thus, small molecule inhibition of cyclin D associated kinase activity,71 alone or in combination with cisplatin, may be an important alternative treatment strategy for some TGCTs.
Materials and methods
Mice
129S1/SvImJ (129, JR#002448) and FVB/NJ (FVB, JR#001800) mice were obtained from the Jackson Laboratory (Bar Harbor ME). 129S1/SvImJ mice homosomic for the Chr19MOLF/Ei chromosome substitution (M19) were obtained from our research colony.43 FVB.129S2(B6)-Ccnd1tm1Wbg/J (FVB-Ccnd1KO, JR#002935) mice were obtained from the Jackson Laboratory (Bar Harbor ME). The Ccnd1KO allele was then backcrossed onto the 129 inbred background for at least 10 generations to establish a congenic strain, and then transferred with crosses to the M19 background. The Ccnd1KO allele was PCR genotyped as previously described.54 The germ cell-specific Oct4ΔPE:GFP (Oct4::GFP) transgene21,53 was previously backcrossed onto the 129, FVB, and M19 backgrounds to establish congenic lines.45 All protocols were approved by the BCM Institutional Animal Care and Use Committee.
Tumor surveys
Crosses between M19 mice heterozygous for Ccnd1KO were used to produce wild-type (M19-Ccnd1+/+), heterozygous knockout (M19-Ccnd1KO/+) and homozygous knockout (M19-Ccnd1KO/KO) male offspring to survey for teratomas. Males, 4–8 weeks of age, were necropsied prior to genotyping and testes were visually and histologically examined for tumors, which are readily detected at this age.43,72 Fisher's Exact Tests were used to assess statistical differences between the number of teratoma-affected M19 wild-type (M19-Ccnd1+/+) control and heterozygous knockout (M19-Ccnd1KO/+) or homozygous knockout (M19-Ccnd1KO/KO) experimental progeny.
Histology
Tissues were fixed in 10% buffered formalin, embedded in paraffin, sectioned (6 µm), and stained with hematoxylin and eosin. Bright field images were taken with the Zeiss Axioplan2 microscope.
SNP analysis
The Mouse Genome Informatics (http://www.informatics.jax.org/) Strain, SNP and Polymorphism analysis tool was used to search for strain-specific genetic variants in the Ccnd1 locus. 129S1/SvImJ was used as the reference strain and compared to selected testicular teratoma-resistant inbred strains C57BL/6J, FVB/NJ, A/J, C3H/HeJ, BALB/cJ, and DBA/2J. A SNP query was performed to identify SNPs within 10 kb of Ccnd1 and different between the reference 129 strain and the selected teratoma-resistant strains. SNPs in functional classes coding-nonsynonymous, intron, locus-region, mRNA-UTR, splice-site, and noncoding-transcript polymorphisms were identified. SNPs resulting in synonymous amino acid changes were excluded.
Timed-matings and gonad dissections
For immunofluorescence and fluorescence-activated cell sorting (FACS), wild-type females were bred to males homozygous for the Oct4::GFP transgene to produce FVB, 129 or M19 transgenic embryos heterozygous for Oct4::GFP. Crosses between M19-Ccnd1KO/+ females and M19-Ccnd1KO/+ males homozygous for Oct4::GFP were used to produce M19-Ccnd1+/+, M19-Ccnd1KO/+ and M19-Ccnd1KO/KO embryos and post-natal day 8 (P8) mice heterozygous for Oct4::GFP. Embryonic day 0.5 (E0.5) was assumed to be noon of the day the vaginal plug was observed. Pregnant females were euthanized by carbon dioxide followed by cervical dislocation and gonads were removed from embryos in ice-cold 1xPBS. Embryos older than E14.5 were decapitated prior to dissection. P8 mice were anesthetized and euthanized by decapitation and gonads were removed from mice in ice-cold 1xPBS. Gonad morphology identified the sex of E13.5 to E15.5 embryos and P8 mice. Tissues were collected for PCR genotyping for the Ccnd1 knockout allele as previously described. 44,54
Fluorescence-activated cell sorting (FACS)
FACS with the Oct4::GFP transgene has been previously described.45,73 Briefly, gonads were digested in 0.25% trypsin (Life Technologies, Grand Island, NY) for 15 min. at 37°C. Tissues were triturated into single-cell suspensions and filtered through a 35 µm nylon mesh cell strainer (Falcon Corning, Tewksbury MA). The mesh was washed with 2% BSA in 1xPBS to inactivate the trypsin, and the cells were kept on ice until FACS with the BD Biosciences FACSAria system. The Oct4::GFP transgene was used to sort GFP-positive germ cells from GFP-negative somatic cells from both gonads of a single embryo, which yielded similar numbers and purity as previously described.45,73
Quantitative real-time PCR expression analysis
Germ cell RNA was prepared using the RNeasy Micro Kit (Qiagen, Valencia CA). RNA was reverse transcribed with the SuperScript First-Strand Synthesis System (Life Technologies). Quantitative real-time PCR (qPCR) was performed with the StepOnePlus real-time PCR system (Life Technologies) and the FastStart Universal SYBR Green Master (ROX) (Roche Diagnostics, Indianapolis IN) using manufacturer recommended protocols. Expression was normalized to the ubiquitously expressed housekeeping gene Rpl7 as previously described.45,74 One-way ANOVA with the Bonferroni post-test for pair-wise comparisons were used to detect significant differences in expression between gonocytes from FVB (control) and 129 and M19 testes. Unpaired t-tests were used to detect significant differences in expression between somatic cells from M19-Ccnd1+/+ and M19-Ccnd1KO/KO testes. Primer sequences are available upon request.
Immunohistochemistry
Gonads were removed from E13.5 to E15.5 embryos and P8 mice and processed for sectioning and immunofluorescence as previously described.45 Sections were incubated with a 1:300 dilution of rabbit polyclonal anti-phospho-pRB1 (#9308, Cell Signaling, Danvers MA), 1:100 dilution of rabbit monoclonal anti-CCND1 (ab16663, Abcam, Cambridge MA), 1:500 dilution of rabbit polyclonal anti-KI67 (ab15580, Abcam, Cambridge MA), 1:100 dilution of rabbit polyclonal anti-NANOG (IHC-00205, Bethyl Laboratories, Montgomery TX), or 1:40 dilution of goat polyclonal anti-c-KIT (AF1356, R&D Systems, Minneapolis MN) antibody overnight at 4°C. For some experiments involving CCND1 immunostaining, sections were co-incubated with a 1:100 dilution of rat monoclonal anti-KI67 (5698, eBioscience, San Diego CA) antibody. For secondary detection by confocal microscopy, sections were incubated with a 1:500 dilution of goat anti-rabbit AlexaFluor 555 (A-21428, Life Technologies) antibody, 1:500 dilution of donkey anti-goat AlexaFluor 555 (ab150130, Abcam), or 1:500 dilutions of goat anti-rat AlexaFluor 647 (A-21247, Life Technologies) and goat anti-rabbit AlexaFluor 555 (A-21428, Life Technologies) in blocking solution for 2 hrs at room temperature. For secondary detection by fluorescence microscopy, sections were incubated with a 1:500 dilution of goat anti-rabbit AlexaFluor 594 (A-27016, Life Technologies) antibody in blocking solution for 2 hrs at room temperature. Nuclei were counter-stained with DAPI. Confocal images of sections were taken with a Nikon A1-Rs inverted Laser Scanning Microscope. Fluorescence microscopy images of tissue sections were collected with the Zeiss Axioplan2 microscope.
Cell counts
Oct4::GFP-positive germ cells positive or negative for phospoh-pRB1, KI67, or NANOG immunostaining were counted as previously described.45 One-way ANOVA with the Bonferroni post-test for pair-wise comparisons was used to test for significant differences in the percentage phosphor-pRB1-positive germ cells between FVB (control) and 129 and M19 testes. Unpaired t-tests were used to detect significant differences in the number of Oct4::GFP-positive germ cells or the percentage of phospho-pRB1, KI67, and NANOG-positive germ cells between M19-Ccnd1+/+ and M19-Ccnd1KO/KO testes.
Supplementary Material
Acknowledgments
We thank the Baylor College of Medicine Human Tissue Acquisition and Pathology, Integrated Microscopy, and Cytometry and Cell Sorting Advanced Technologies Cores for assistance with tissue processing, confocal microscopy, and FACS, respectively.
Funding
This work was supported by National Institutes of Health grant HD059945 (JDH), Cancer Prevention and Research Institute of Texas (CPRIT) grants RP150081 (JDH) and RP140102 (JMR), and the Baylor Research Advocates for Student Scientists (BRASS) Scholarship Program (EPD). Resources accessed through all cores were supported by NIH-NCI grant CA125123 to the Dan L. Duncan Cancer Center. Additionally, NIH grants HD007495 and DK56338, and a grant from the John S. Dunn Gulf Coast Consortium for Chemical Genomics supported resources accessed through the Integrated Microscopy Core.
References
- [1].Kunwar PS, Siekhaus DE, Lehmann R. In vivo migration: a germ cell perspective. AnnuRevCell DevBiol 2006; 22:237-65. [DOI] [PubMed] [Google Scholar]
- [2].Aponte PM, van Bragt MP, de Rooij DG, van Pelt AM. Spermatogonial stem cells: characteristics and experimental possibilities. APMIS 2005; 113:727-42; PMID:16480445; http://dx.doi.org/ 10.1111/j.1600-0463.2005.apm_302.x [DOI] [PubMed] [Google Scholar]
- [3].Stevens L. Development of resistance to teratocarcinogenesis by primordial germ cells in mice. J Natl Cancer Inst 1966; 37:859-67; PMID:6005945 [PubMed] [Google Scholar]
- [4].Looijenga LH, Stoop H, de Leeuw HP, de Gouveia Brazao CA, Gillis AJ, van Roozendaal KE, van Zoelen EJ, Weber RF, Wolffenbuttel KP, van DH, et al.. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res 2003; 63:2244-50; PMID:12727846 [PubMed] [Google Scholar]
- [5].Almstrup K, Hoei-Hansen CE, Wirkner U, Blake J, Schwager C, Ansorge W, Nielsen JE, Skakkebaek NE, Rajpert-de ME, Leffers H. Embryonic stem cell-like features of testicular carcinoma in situ revealed by genome-wide gene expression profiling. Cancer Res 2004; 64:4736-43; PMID:15256440; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-0679 [DOI] [PubMed] [Google Scholar]
- [6].Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. NatRevCancer 2005; 5:210-22 [DOI] [PubMed] [Google Scholar]
- [7].Heaney JD, Nadeau JH. Testicular germ cell tumors in mice: new ways to study a genetically complex trait. Methods MolBiol 2008; 450:211-31 [DOI] [PubMed] [Google Scholar]
- [8].Bustamante-Marin X, Garness JA, Capel B. Testicular teratomas: an intersection of pluripotency, differentiation and cancer biology. The International journal of developmental biology 2013; 57:201-10; PMID:23784831; http://dx.doi.org/ 10.1387/ijdb.130136bc [DOI] [PubMed] [Google Scholar]
- [9].Stevens L, Hummel K. A description of spontaneous congenital testicular teratomas in strain 129 mice. JNatlCancer Inst 1957; 18:719-47. [PubMed] [Google Scholar]
- [10].Stevens L. Testicular teratomas in fetal mice. J Natl Cancer Inst 1962; 28:247-67; PMID:13917068 [PubMed] [Google Scholar]
- [11].Stevens L. The biology of teratomas. Adv Morphog 1967; 6:1-31; PMID:4894128; http://dx.doi.org/ 10.1016/B978-1-4831-9953-5.50005-6 [DOI] [PubMed] [Google Scholar]
- [12].Stevens L. Origin of testicular teratomas from primordial germ cells in mice. JNatlCancer Inst 1967; 38:549-52. [PubMed] [Google Scholar]
- [13].Heaney JD, Lam MY, Michelson MV, Nadeau JH. Loss of the transmembrane but not the soluble kit ligand isoform increases testicular germ cell tumor susceptibility in mice. Cancer Res 2008; 68:5193-7; PMID:18593919; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kimura T, Suzuki A, Fujita Y, Yomogida K, Lomeli H, Asada N, Ikeuchi M, Nagy A, Mak TW, Nakano T. Conditional loss of PTEN leads to testicular teratoma and enhances embryonic germ cell production. Development 2003; 130:1691-700; PMID:12620992; http://dx.doi.org/ 10.1242/dev.00392 [DOI] [PubMed] [Google Scholar]
- [15].Krentz AD, Murphy MW, Kim S, Cook MS, Capel B, Zhu R, Matin A, Sarver AL, Parker KL, Griswold MD, et al.. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. ProcNatlAcadSciUSA 2009; 106:22323-8; http://dx.doi.org/ 10.1073/pnas.0905431106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kanetsky PA, Mitra N, Vardhanabhuti S, Li M, Vaughn DJ, Letrero R, Ciosek SL, Doody DR, Smith LM, Weaver J, et al.. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. NatGenet 2009; 41(7):811-5; PMID:19483682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Turnbull C, Rapley EA, Seal S, Pernet D, Renwick A, Hughes D, Ricketts M, Linger R, Nsengimana J, Deloukas P, et al.. Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. NatGenet 2010; 42:604-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Andreassen KE, Kristiansen W, Karlsson R, Aschim EL, Dahl O, Fossa SD, Adami HO, Wiklund F, Haugen TB, Grotmol T. Genetic variation in AKT1, PTEN and the 8q24 locus, and the risk of testicular germ cell tumor. HumReprod 2013; 28:1995-2002 [DOI] [PubMed] [Google Scholar]
- [19].Noguchi T, STEVENS LC. Primordial germ cell proliferation in fetal testes in mouse strains with high and low incidences of congenital testicular teratomas. JNatlCancer Inst 1982; 69:907-13 [PubMed] [Google Scholar]
- [20].Cook MS, Munger SC, Nadeau JH, Capel B. Regulation of male germ cell cycle arrest and differentiation by DND1 is modulated by genetic background. Development 2011; 138:23-32; PMID:21115610; http://dx.doi.org/ 10.1242/dev.057000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Youngren KK, Coveney D, Peng X, Bhattacharya C, Schmidt LS, Nickerson ML, Lamb BT, Deng JM, Behringer RR, Capel B, et al.. The Ter mutation in the dead end gene causes germ cell loss and testicular germ cell tumours. Nature 2005; 435:360-4; PMID:15902260; http://dx.doi.org/ 10.1038/nature03595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Harvey M, McArthur MJ, Montgomery CA Jr., Bradley A, Donehower LA. Genetic background alters the spectrum of tumors that develop in p53-deficient mice. FASEB J 1993; 7:938-43; PMID:8344491 [DOI] [PubMed] [Google Scholar]
- [23].Krentz AD, Murphy MW, Zhang T, Sarver AL, Jain S, Griswold MD, Bardwell VJ, Zarkower D. Interaction between DMRT1 function and genetic background modulates signaling and pluripotency to control tumor susceptibility in the fetal germ line. DevBiol 2013; 377:67-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schemmer J, Arauzo-Bravo MJ, Haas N, Schafer S, Weber SN, Becker A, Eckert D, Zimmer A, Nettersheim D, Schorle H. Transcription factor TFAP2C regulates major programs required for murine fetal germ cell maintenance and haploinsufficiency predisposes to teratomas in male mice. PloS one 2013; 8:e71113; PMID:23967156; http://dx.doi.org/ 10.1371/journal.pone.0071113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Matin A, Collin GB, Varnum DS, Nadeau JH. Testicular teratocarcinogenesis in mice-a review. APMIS 1998; 106:174-82; PMID:9524576; http://dx.doi.org/ 10.1111/j.1699-0463.1998.tb01333.x [DOI] [PubMed] [Google Scholar]
- [26].Park SY, Jameson JL. Minireview: transcriptional regulation of gonadal development and differentiation. Endocrinology 2005; 146:1035-42; PMID:15604204; http://dx.doi.org/ 10.1210/en.2004-1454 [DOI] [PubMed] [Google Scholar]
- [27].McLaren A. Germ and somatic cell lineages in the developing gonad. MolCell Endocrinol 2000; 163:3-9; http://dx.doi.org/ 10.1016/S0303-7207(99)00234-8 [DOI] [PubMed] [Google Scholar]
- [28].McLaren A. Meiosis and differentiation of mouse germ cells. SympSocExpBiol 1984; 38:7-23. [PubMed] [Google Scholar]
- [29].Adams IR, McLaren A. Sexually dimorphic development of mouse primordial germ cells: switching from oogenesis to spermatogenesis. Development 2002; 129:1155-64; PMID:11874911 [DOI] [PubMed] [Google Scholar]
- [30].Koubova J, Menke DB, Zhou Q, Capel B, Griswold MD, Page DC. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. ProcNatlAcadSciUSA 2006; 103:2474-9; http://dx.doi.org/ 10.1073/pnas.0510813103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bowles J, Knight D, Smith C, Wilhelm D, Richman J, Mamiya S, Yashiro K, Chawengsaksophak K, Wilson MJ, Rossant J, et al.. Retinoid signaling determines germ cell fate in mice. Science 2006; 312:596-600; PMID:16574820; http://dx.doi.org/ 10.1126/science.1125691 [DOI] [PubMed] [Google Scholar]
- [32].Lin Y, Gill ME, Koubova J, Page DC. Germ cell-intrinsic and -extrinsic factors govern meiotic initiation in mouse embryos. Science 2008; 322:1685-7; PMID:19074348; http://dx.doi.org/ 10.1126/science.1166340 [DOI] [PubMed] [Google Scholar]
- [33].Bowles J, Koopman P. Retinoic acid, meiosis and germ cell fate in mammals. Development 2007; 134:3401-11; PMID:17715177; http://dx.doi.org/ 10.1242/dev.001107 [DOI] [PubMed] [Google Scholar]
- [34].Bowles J, Feng CW, Spiller C, Davidson TL, Jackson A, Koopman P. FGF9 suppresses meiosis and promotes male germ cell fate in mice. DevCell 2010; 19:440-9 [DOI] [PubMed] [Google Scholar]
- [35].Miles DC, Wakeling SI, Stringer JM, van den Bergen JA, Wilhelm D, Sinclair AH, Western PS. Signaling through the TGF beta-activin receptors ALK4/5/7 regulates testis formation and male germ cell development. PloS one 2013; 8:e54606; PMID:23342175; http://dx.doi.org/ 10.1371/journal.pone.0054606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Moniot B, Ujjan S, Champagne J, Hirai H, Aritake K, Nagata K, Dubois E, Nidelet S, Nakamura M, Urade Y, et al.. Prostaglandin D2 acts through the Dp2 receptor to influence male germ cell differentiation in the foetal mouse testis. Development 2014; 141:3561-71; PMID:25142465; http://dx.doi.org/ 10.1242/dev.103408 [DOI] [PubMed] [Google Scholar]
- [37].Yamaguchi S, Kimura H, Tada M, Nakatsuji N, Tada T. Nanog expression in mouse germ cell development. Gene ExprPatterns 2005; 5:639-46 [DOI] [PubMed] [Google Scholar]
- [38].Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 2003; 17:126-40; PMID:12514105; http://dx.doi.org/ 10.1101/gad.224503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pesce M, Wang X, Wolgemuth DJ, Scholer H. Differential expression of the Oct-4 transcription factor during mouse germ cell differentiation. MechDev 1998; 71:89-98 [DOI] [PubMed] [Google Scholar]
- [40].Western PS, Miles DC, van den Bergen JA, Burton M, Sinclair AH. Dynamic regulation of mitotic arrest in fetal male germ cells. Stem Cells 2008; 26:339-47; PMID:18024419; http://dx.doi.org/ 10.1634/stemcells.2007-0622 [DOI] [PubMed] [Google Scholar]
- [41].Spiller CM, Wilhelm D, Koopman P. Retinoblastoma 1 protein modulates XY germ cell entry into G1/G0 arrest during fetal development in mice. Biology of reproduction 2010; 82:433-43; PMID:19864318; http://dx.doi.org/ 10.1095/biolreprod.109.078691 [DOI] [PubMed] [Google Scholar]
- [42].Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene 2005; 24:2909-15; PMID:15838524; http://dx.doi.org/ 10.1038/sj.onc.1208618 [DOI] [PubMed] [Google Scholar]
- [43].Matin A, Collin GB, Asada Y, Varnum D, Nadeau JH. Susceptibility to testicular germ-cell tumours in a 129.MOLF-Chr 19 chromosome substitution strain. NatGenet 1999; 23:237-40. [DOI] [PubMed] [Google Scholar]
- [44].Heaney JD, Anderson EL, Michelson MV, Zechel JL, Conrad PA, Page DC, Nadeau JH. Germ cell pluripotency, premature differentiation and susceptibility to testicular teratomas in mice. Development 2012; 139:1577-86; PMID:22438569; http://dx.doi.org/ 10.1242/dev.076851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Heaney JD, Michelson MV, Youngren KK, Lam MY, Nadeau JH. Deletion of eIF2beta suppresses testicular cancer incidence and causes recessive lethality in agouti-yellow mice. HumMolGenet 2009; 18:1395-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Beumer TL, Roepers-Gajadien HL, Gademan IS, Kal HB, de Rooij DG. Involvement of the D-type cyclins in germ cell proliferation and differentiation in the mouse. BiolReprod 2000; 63:1893-8 [DOI] [PubMed] [Google Scholar]
- [47].Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al.. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463:899-905; PMID:20164920; http://dx.doi.org/ 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology 2004; 145:5439-47; PMID:15331580; http://dx.doi.org/ 10.1210/en.2004-0959 [DOI] [PubMed] [Google Scholar]
- [49].Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nature reviews Cancer 2011; 11:558-72; PMID:21734724; http://dx.doi.org/ 10.1038/nrc3090 [DOI] [PubMed] [Google Scholar]
- [50].Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001; 411:1017-21; PMID:11429595; http://dx.doi.org/ 10.1038/35082500 [DOI] [PubMed] [Google Scholar]
- [51].Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004; 18:2699-711; PMID:15545627; http://dx.doi.org/ 10.1101/gad.1256504 [DOI] [PubMed] [Google Scholar]
- [52].Wolgemuth DJ, Roberts SS. Regulating mitosis and meiosis in the male germ line: critical functions for cyclins. PhilosTransRSocLond B BiolSci 2010; 365:1653-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yoshimizu T, Sugiyama N, De Felice M, Yeom YI, Ohbo K, Masuko K, Obinata M, Abe K, Scholer HR, Matsui Y. Germline-specific expression of the Oct-4/green fluorescent protein (GFP) transgene in mice. DevGrowth Differ 1999; 41:675-84; http://dx.doi.org/ 10.1046/j.1440-169x.1999.00474.x [DOI] [PubMed] [Google Scholar]
- [54].Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 1995; 82:621-30; PMID:7664341; http://dx.doi.org/ 10.1016/0092-8674(95)90034-9 [DOI] [PubMed] [Google Scholar]
- [55].Schrans-Stassen BH, van de Kant HJ, de Rooij DG, van Pelt AM. Differential expression of c-kit in mouse undifferentiated and differentiating type A spermatogonia. Endocrinology 1999; 140:5894-900; PMID:10579355; http://dx.doi.org/ 10.1210/endo.140.12.7172 [DOI] [PubMed] [Google Scholar]
- [56].Nebel BR, Amarose AP, Hacket EM. Calendar of gametogenic development in the prepuberal male mouse. Science 1961; 134:832-3; PMID:13728067; http://dx.doi.org/ 10.1126/science.134.3482.832 [DOI] [PubMed] [Google Scholar]
- [57].Stevens L, Mackensen JA. Genetic and environmental influences on teratocarcinogenesis in mice. JNatlCancer Inst 1961; 27:443-53 [Google Scholar]
- [58].Singh AM, Dalton S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell 2009; 5:141-9; PMID:19664987; http://dx.doi.org/ 10.1016/j.stem.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang Y, Blelloch R. Cell cycle regulation by MicroRNAs in embryonic stem cells. Cancer Res 2009; 69:4093-6; PMID:19435891; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Filipczyk AA, Laslett AL, Mummery C, Pera MF. Differentiation is coupled to changes in the cell cycle regulatory apparatus of human embryonic stem cells. Stem Cell Res 2007; 1:45-60; PMID:19383386; http://dx.doi.org/ 10.1016/j.scr.2007.09.002 [DOI] [PubMed] [Google Scholar]
- [61].Herrera RE, Chen F, Weinberg RA. Increased histone H1 phosphorylation and relaxed chromatin structure in Rb-deficient fibroblasts. ProcNatlAcadSciUSA 1996; 93:11510-5; http://dx.doi.org/ 10.1073/pnas.93.21.11510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mummery CL, van Rooijen MA, van den Brink SE, de Laat SW. Cell cycle analysis during retinoic acid induced differentiation of a human embryonal carcinoma-derived cell line. Cell Differ 1987; 20:153-60; PMID:3568136; http://dx.doi.org/ 10.1016/0045-6039(87)90429-5 [DOI] [PubMed] [Google Scholar]
- [63].Jonk LJ, de Jonge ME, Kruyt FA, Mummery CL, van der Saag PT, Kruijer W. Aggregation and cell cycle dependent retinoic acid receptor mRNA expression in P19 embryonal carcinoma cells. MechDev 1992; 36:165-72. [DOI] [PubMed] [Google Scholar]
- [64].Western PS, Ralli RA, Wakeling SI, Lo C, van den Bergen JA, Miles DC, Sinclair AH. Mitotic arrest in teratoma susceptible fetal male germ cells. PLoSONE 2011; 6:e20736; http://dx.doi.org/ 10.1371/journal.pone.0020736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gidekel S, Pizov G, Bergman Y, Pikarsky E. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell 2003; 4:361-70; PMID:14667503; http://dx.doi.org/ 10.1016/S1535-6108(03)00270-8 [DOI] [PubMed] [Google Scholar]
- [66].Spiller CM, Feng CW, Jackson A, Gillis AJ, Rolland AD, Looijenga LH, Koopman P, Bowles J. Endogenous Nodal signaling regulates germ cell potency during mammalian testis development. Development 2012; 139:4123-32; PMID:23034635; http://dx.doi.org/ 10.1242/dev.083006 [DOI] [PubMed] [Google Scholar]
- [67].Jorgensen A, Nielsen JE, Almstrup K, Toft BG, Petersen BL, Rajpert-De Meyts E. Dysregulation of the mitosis-meiosis switch in testicular carcinoma in situ. The Journal of pathology 2013; 229:588-98; PMID:23303528; http://dx.doi.org/ 10.1002/path.4154 [DOI] [PubMed] [Google Scholar]
- [68].Adamah DJ, Gokhale PJ, Eastwood DJ, Rajpert De-Meyts E, Goepel J, Walsh JR, Moore HD, Andrews PW. Dysfunction of the mitotic:meiotic switch as a potential cause of neoplastic conversion of primordial germ cells. IntJAndrol 2006; 29:219-27. [DOI] [PubMed] [Google Scholar]
- [69].Noel EE, Yeste-Velasco M, Mao X, Perry J, Kudahetti SC, Li NF, Sharp S, Chaplin T, Xue L, McIntyre A, et al.. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. AmJPathol 2010; 176:2607-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Freemantle SJ, Vaseva AV, Ewings KE, Bee T, Krizan KA, Kelley MR, Hattab EM, Memoli VA, Black CC, Spinella MJ, et al.. Repression of cyclin D1 as a target for germ cell tumors. IntJOncol 2007; 30:333-40. [PubMed] [Google Scholar]
- [71].Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von BH, et al.. The requirement for cyclin D function in tumor maintenance. Cancer Cell 2012; 22:438-51; PMID:23079655; http://dx.doi.org/ 10.1016/j.ccr.2012.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lam MY, Youngren KK, Nadeau JH. Enhancers and suppressors of testicular cancer susceptibility in single- and double-mutant mice. Genetics 2004; 166:925-33; PMID:15020477; http://dx.doi.org/ 10.1534/genetics.166.2.925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Molyneaux KA, Schaible K, Wylie C. GP130, the shared receptor for the LIF/IL6 cytokine family in the mouse, is not required for early germ cell differentiation, but is required cell-autonomously in oocytes for ovulation. Development 2003; 130:4287-94; PMID:12900446; http://dx.doi.org/ 10.1242/dev.00650 [DOI] [PubMed] [Google Scholar]
- [74].Jeong YJ, Choi HW, Shin HS, Cui XS, Kim NH, Gerton GL, Jun JH. Optimization of real time RT-PCR methods for the analysis of gene expression in mouse eggs and preimplantation embryos. MolReprodDev 2005; 71:284-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.