

ABSTRACT
RNA-Seq and gene set enrichment anylysis revealed that ovarian cancer associated fibroblasts (CAFs) are mitotically active compared with normal fibroblasts (NFs). Cellular senescence is observed in CAFs treated with H2O2 as shown by elevated SA-β-gal activity and p21 (WAF1/Cip1) protein levels. Reactive oxygen species (ROS) production and p21 (WAF1/Cip1) elevation may account for H2O2-induced CAFs cell cycle arrest in S phase. Blockage of autophagy can increase ROS production in CAFs, leading to cell cycle arrest in S phase, cell proliferation inhibition and enhanced sensitivity to H2O2-induced cell death. ROS scavenger NAC can reduce ROS production and thus restore cell viability. Lactate dehydrogenase A (LDHA), monocarboxylic acid transporter 4 (MCT4) and superoxide dismutase 2 (SOD2) were up-regulated in CAFs compared with NFs. There was relatively high lactate content in CAFs than in NFs. Blockage of autophagy decreased LDHA, MCT4 and SOD2 protein levels in CAFs that might enhance ROS production. Blockage of autophagy can sensitize CAFs to chemotherapeutic drug cisplatin, implicating that autophagy might possess clinical utility as an attractive target for ovarian cancer treatment in the future.
KEYWORDS: autophagy, CAFs, LDHA, MCT4, ovarian cancer, oxidative stress, ROS, SOD2
Introduction
Epithelial ovarian cancer has the highest mortality rate of all gynecologic cancers because most patients are detected at late-stage of tumor growth.1 Tumor growth is determined by malignant cancer cells themselves and by the tumor stromal microenvironment. Activated fibroblasts that are found in association with cancer cells are known as cancer-associated fibroblasts (CAFs), which are a key cellular component of the tumor stromal microenvironment and play important roles in cancer initiation, progression and metastasis.2,3 Surgery and platinum-based cytotoxic chemotherapy can be curative for most patients with early stage disease. However, cisplatin treatment often results in the development of chemoresistance and one of the mechanisms of cisplatin is to trigger mitochondrial outer membrane permeabilization by generating reactive oxygen species (ROS).4,5
Autophagy is a pathway by which cytoplasmic organelles or cytosolic components, including intracellular pathogens, are sequestered in a double-membrane-bound autophagosome and delivered to the lysosome for degradation. Autophagy is essential for survival, differentiation, development, and energy homeostasis, and is involved in many diseases, including cancer, neurodegeneration and microbial infection. This pathway can be stimulated by multiple forms of cellular stress, including nutrient or growth factor deprivation, hypoxia, ROS, DNA damage, protein aggregates, damaged organelles, or intracellular pathogens.6-8 Defective autophagy has been detected in different tumors, implying a tumor suppressive function of autophagy.9-12 However, autophagy also displays tumor promoting functions in many cases, which implies that the functional role of autophagy in tumorigenesis is context-dependent.13-18 ROS-induced autophagy can provide negative feedback regulation by which autophagy eliminates the source of oxidative stress and protects the cell from oxidative damage.19 By comparing CAFs isolated from ovarian cancer tissues with normal fibroblasts (NFs) that were isolated from non-cancerous prophylactic oophorectomy specimens, we show that CAFs develop mechanisms resistant to oxidative stress and that autophagy is involved in this process. Our results show that targeting autophagy might show clinical utility in ovarian cancer therapy.
Results
Ovarian CAFs are mitotically active
CAFs are the most abundant among all the ovarian cancer microenvironment cells.20 To understand the difference between NFs and CAFs will help to identify new therapeutic targets. Considering there is heterogeneity among human primary fibroblasts coming from different individuals, 2 independent NFs and 3 independent CAFs were isolated and named NF1, NF2, CAF1, CAF2 and CAF3. All of the five cell lines were fibroblast-like with elongated shapes and vimentin positive (Fig. S1). Fibroblast specific protein 1 (FSP1, aka S100A4) and α-smooth muscle actin (α-SMA) are wildly used CAF markers.21 We analyzed the abundances of FSP1 and α-SMA in our isolated NFs and CAFs. As shown in Figure 1A, the immunofluorescence intensity of FSP1 was higher in CAFs than in NFs. Protein levels of α-SMA were obviously higher in CAF2 cells and slightly higher in CAF1 cells compared with NFs cells. NF1, NF2 and CAF1 can be passaged more than 10 generations. However, CAF2 and CAF3 stopped proliferating after passage 4/5. We analyzed the cell cycle of the five cell lines at passage 4 and found that NF1, NF2 and CAF1 showed typical division cell cycle consisting of G1, S and G2/M phases, but CAF2 and CAF3 only showed G1 phase (Fig. 1B and 1C), suggesting that CAF2 and CAF3 were arrested in G1 phase. Yang et al. showed that ovarian cancer cells can transform CAFs into senescent fibroblasts in 26 of 31 ovarian carcinoma tissues.22 Since senescent cells can arrest growth in G1 phase,23 cell senescence may be the reason that CAF2 and CAF3 cannot be passaged more than 5 generations. The difference between CAF1 and the other 2 CAF cell lines implicated that heterogeneity of ovarian CAFs may contribute differently to ovarian cancer. We focused on CAF1 in our study. RNA-Seq was performed to demonstrate the difference between NF1 and CAF1. To identify the pathways that are enriched in CAF1 cells, we utilized the GSEA (gene set enrichment analysis). A total of 36 gene signatures enriched in CAF1 cells were identified (Table 1). All gene sets were with an FDR≤0.25 and ranked by their normalized enrichment scores (−2.34 ≤ NES ≤ −1.48) (Table 1). As shown in Table 1 and Figure 1D, the gene sets and the corresponding pathways enriched in CAF1 were mainly associated with mitosis and cell cycle, suggesting that CAF1 cells were mitotically active. Histone H3 is specifically phosphorylated at Ser10 during mitosis.24 We examined the levels of phospho-histone H3 in NF1 and CAF1 cells at passage 6. As shown in Figure 1E, there were more mitotic signals among CAF1 cells compared with NF1 cells. Western blot analysis showed that phospho-histone H3 was higher in CAF1 cells than in NF1 cells (Fig. 1F).
Figure 1.

CAF1 cells are mitotically active. (A) Identification of human primary ovarian NFs and CAFs. Immunostaining of FSP1 and western blot analysis of α-SMA in NFs and CAFs. Cells were stained with FSP1 (green) and counterstained with DAPI (blue). The relative intensity of α-SMA normalized to housekeeping protein was shown at the bottom. (B) Cell cycle analysis of NFs and CAFs. (C) The cell cycle phase distribution in each cell line (as in B). (D) Pathways enriched in CAF1 was revealed by GSEA indicated that CAF1 cells were mitotically active. (E) Immunostaining of phospho-histone H3 in NF1 and CAF1 cells. Cells were stained with phospho-histone H3 (green) and counterstained with DAPI (blue). (F) Western blot analysis of phospho-histone H3 in NF1 and CAF1 cells. The relative intensity of phospho-histone H3 normalized to housekeeping protein was shown at the bottom.
Table 1.
Gene sets and pathways enriched in CAF1 compared to NF1 revealed by GSEA.
| Function/pathway | Gene sets | FDR q-val |
|---|---|---|
| Mitosis | ||
| M_PHASE_OF_MITOTIC_CELL_CYCLE | 0 | |
| MITOTIC_CELL_CYCLE | 0 | |
| MITOSIS | 0.005 | |
| M_PHASE | 0.004 | |
| CHROMOSOMEPERICENTRIC_REGION | 0.046 | |
| SISTER_CHROMATID_SEGREGATION | 0.035 | |
| KINETOCHORE | 0.043 | |
| CHROMOSOME_SEGREGATION | 0.043 | |
| REGULATION_OF_MITOSIS | 0.042 | |
| MITOTIC_SISTER_CHROMATID_SEGREGATION | 0.094 | |
| MITOTIC_CELL_CYCLE_CHECKPOINT | 0.092 | |
| CHROMOSOME | 0.088 | |
| CHROMOSOMAL_PART | 0.091 | |
| SPINDLE_MICROTUBULE | 0.106 | |
| REPLICATION_FORK | 0.103 | |
| MICROTUBULE_BASED_PROCESS | 0.136 | |
| CELL_DIVISION | 0.132 | |
| Cell cycle | ||
| CELL_CYCLE_PHASE | 0.015 | |
| CELL_CYCLE_PROCESS | 0.032 | |
| CELL_CYCLE_CHECKPOINT_GO_0000075 | 0.027 | |
| REGULATION_OF_CYCLIN_DEPENDENT_ | ||
| PROTEIN_KINASE_ACTIVITY | 0.036 | |
| CELL_CYCLE_GO_0007049 | 0.135 | |
| REGULATION_OF_CELL_CYCLE | 0.128 | |
| Repair of DNA | ||
| DNA_DEPENDENT_ATPASE_ACTIVITY | 0.059 | |
| DOUBLE_STRAND_BREAK_REPAIR | 0.040 | |
| DNA_RECOMBINATION | 0.042 | |
| Cell signaling | ||
| HEMATOPOIETIN_INTERFERON_CLASSD200_ | ||
| DOMAIN_CYTOKINE_RECEPTOR_BINDING | 0.036 | |
| CHEMOKINE_ACTIVITY | 0.130 | |
| GROWTH_FACTOR_ACTIVITY | 0.136 | |
| CHEMOKINE_RECEPTOR_BINDING | 0.127 | |
| G_PROTEIN_COUPLED_RECEPTOR_BINDING | 0.227 | |
| Metastasis | ||
| METALLOPEPTIDASE_ACTIVITY | 0.048 | |
| METALLOENDOPEPTIDASE_ACTIVITY | 0.098 | |
| Protein degradation | ||
| PROTEASOME_COMPLEX | 0.046 | |
| CELLULAR_PROTEIN_CATABOLIC_PROCESS | 0.226 | |
| Organelle localization | ||
| ESTABLISHMENT_OF_ORGANELLE_ | ||
| LOCALIZATION | 0.090 | |
| ORGANELLE_LOCALIZATION | 0.122 |
Ovarian CAFs are resistant to oxidative stress
Oxidative stress can switch mitotic cells into a growth-arrested, senescence-like state in which they may survive for long periods.25 We compared the influence of exogenous oxidative stress on NFs and CAFs. When the cells were treated with H2O2 to induce oxidative stress, the viability of NF1, NF2 and CAF1 were 47%, 59% and 90%, respectively (Fig. 2A and Fig. 2B), suggesting that CAF1 cells were resistant to oxidative stress. CAF1 cells treated with H2O2 showed increased senescence-associated acidic β-galactosidase (SA-β-gal) activity as compared with untreated cells at both high cell density (Fig. 2C upper panel) and low cell density (Fig. 2C middle panel). And the morphology of low density cells treated with H2O2 became flatter and more irregularly (Figure 2C middle panel and lower panel). These results suggested that H2O2 induced senescence in CAF1 cells. Western blot analysis showed that when CAF1 cells were treated with H2O2, p21 (WAF1/Cip1) protein levels which is associated with cell cycle arrest and senescence increased (Fig. 2D). Cell cycle analysis showed that H2O2 increased mainly S phase percentage (Fig. 2E and Fig. 2F), suggesting H2O2 arrested CAF1 cells in S phase. H2O2 induced ROS production in CAF1 cells (Fig. 2G). It has been reported that both p21 (WAF1/Cip1) and ROS can induce S phase arrest.26-28 Elevated p21 (WAF1/Cip1) and ROS production may be responsible for S phase arrest of CAF1 cells treated with H2O2.
Figure 2.

Ovarian CAF1 cells are resistant to oxidative stress. (A) The effect of H2O2 on NF1 and CAF1 cells. 2 × 104 cells were seeded in 24-well plate overnight, treated with 1 mM H2O2 for 2 h. The images of cell morphology were taken under microscope. (B) Quantification of cell viability described in (A) with MTT assay (mean ± SD, n = 3). (C) SA-β-gal activity in CAF1 cells treated with or without H2O2. SA-β-gal activity of high and low density cells were shown in the upper and middle panels, respectively. Cell morphology change of low density cells was shown in the middle and lower panel. (D) Western blot analysis of protein levels of p21 and autophagosome markers in CAF1 cells treated with or without H2O2. The relative intensity of indicated proteins normalized to housekeeping protein was shown at the bottom of each panel. (E) Cell cycle analysis of CAF1 cells treated with or without H2O2. (F) The cell cycle phase distribution in CAF1 cells treated with or without H2O2 (as in E). (G) ROS measurement. CAF1 cells treated with or without H2O2 were stained with H2DCF (20 μM) for 15 min. The fluorescence intensity was determined by flow cytometry.
Autophagy protects ovarian CAFs against oxidative stress by inhibiting ROS production
ROS can induce autophagy in cells and activated autophagy can eliminate ROS.29 Autophagy may protect CAF1 against oxidative stress. Western blot analysis showed that autophagosome markers LC3-II and SQSTM1/p62 increased and decreased, respectively (Fig. 2D), suggesting autophagy was activated in CAF1 cells treated with H2O2. We investigated whether blockage of autophagy could sensitize CAF1 cells to H2O2 by knockdown of autophagy essential genes with siRNA targeting Atg5 or Beclin. As shown in Figure 3A and Figure 3B, knockdown of Atg5 or Beclin can enhance the sensitivity of CAF1 cells to H2O2. Moreover, even if there was no H2O2, knockdown of Atg5 or Beclin decreased the cell viability (Fig. 3B), suggesting that blockage of autophagy inhibited CAF1 cells proliferation. Cell cycle analysis showed that knockdown of Atg5 arrested CAF1 cells in S phase (Fig. 3C and Fig. 3D), a phenotype similar to the cells that were treated with H2O2 (Fig. 2E and Fig. 2F). Flow cytometry analysis showed that knockdown of Atg5 or Beclin increased ROS production in CAF1 cells (Fig. 3E). ROS scavenger N-acetyl-cysteine (NAC) reduced ROS production (Fig. 3F) and restored cell viability (Fig. 3G) caused by knockdown of Beclin. These results suggested that autophagy protected CAF1 cells against oxidative stress by inhibiting ROS production. Knockdown of Atg5 or Beclin can enhance the sensitivity of CAF1 cells to chemotherapeutic drug cisplatin-induced cell death (Fig. 3H), presumably by enhancing the sensitivity to cisplatin-induced oxidative stress.
Figure 3.

Autophagy protects ovarian CAF1 cells against oxidative stress by inhibiting ROS production. (A) The representative images of CAF1 cells with knockdown of indicated siRNA and treated with H2O2. Nc: negative control siRNA. (B) The effect of blockage of autophagy on cell viability and H2O2-induced cell death of CAF1 cells. CAF1 cells were reverse transfected with the indicated siRNA for 48 h in 96-well plate in triplicate, treated with or without 1 mM H2O2 for 2 h. The cell viability was measured with MTT assay (mean ± SD, n = 3). (C) Cell cycle analysis of CAF1 cells transfected with Nc or Atg5 siRNA for 48 h. (D) The cell cycle phase distribution in CAF1 cells transfected with Nc or Atg5 siRNA for 48 h (as in C). (E) ROS measurement of CAF1 cells transfected with Nc, Atg5 or Beclin siRNA for 48 h by flow cytometry. (F) NAC decreased ROS production in Beclin knockdown CAF1 cells. Cells were transfected with Nc or Beclin siRNA for 24 h, incubated with or without NAC (5 mg/ml) for 2 h, and further cultured in complete media for another 24 h. Cells were stained with H2DCF (20 μM) and lysed with RIPA buffer. The fluorescence was read with a fluorometer and normalized to protein concentration (mean ± SD, n = 4). (G) NAC restored cell viability of Beclin knockdown CAF1 cells. Cells were prepared the same way as in (F). Cell viability was measured by MTT assay (mean ± SD, n = 4). (H) The effect of blockage of autophagy on cisplatin-induced cell death of CAFs. CAF1 cells were reverse transfected with the indicated siRNA for 24 h in 96-well plate in triplicate, treated with 100 μM cisplatin for another 24 h. The Cell viability was measured with MTT assay (mean ± SD, n = 3).
MCT4 and LDHA protein levels are down-regulated and lactate is accumulated in CAFs when autophagy is inhibited
Lactate is a key metabolic player in cancer by fueling tumor cells as an energy source and contributing to immune escape and metastasis. Lactate synthesis requires lactate dehydrogenase A (LDHA), and lactate transport is mediated by monocarboxylic acid symporters: MCT1 (SLC16a1), MCT2 (SLC16a7), MCT3 (SLC16a8), and MCT4 (SLC16a3).30-32 Our RNA-Seq data showed that LDHA and MCT4 were upregulated in CAF1 compared to NF1. Both the mRNA and protein levels of LDHA and MCT4 were higher in CAF1 than in NF1 cells (Fig. 4A and Fig. 4B). Consistent with the higher protein level of LDHA, the level of lactate was higher in CAF1 cells than in NF1 cells (Fig. 4C). The intracellular pH (pHi) of NF1 cells and CAF1 cells were 6.98 ± 0.15 and 6.67 ± 0.07, respectively (p < 0.05, Fig. 4D), indicating the intracellular environment of CAF1 cells was acidic. Knockdown of Atg5 or Beclin decreased LDHA and MCT4 but not MCT2 protein levels in CAF1 cells (Fig. 4E). Down-regulation of MCT4 resulted in lactate accumulation in CAF1 cells (Fig. 4F).
Figure 4.
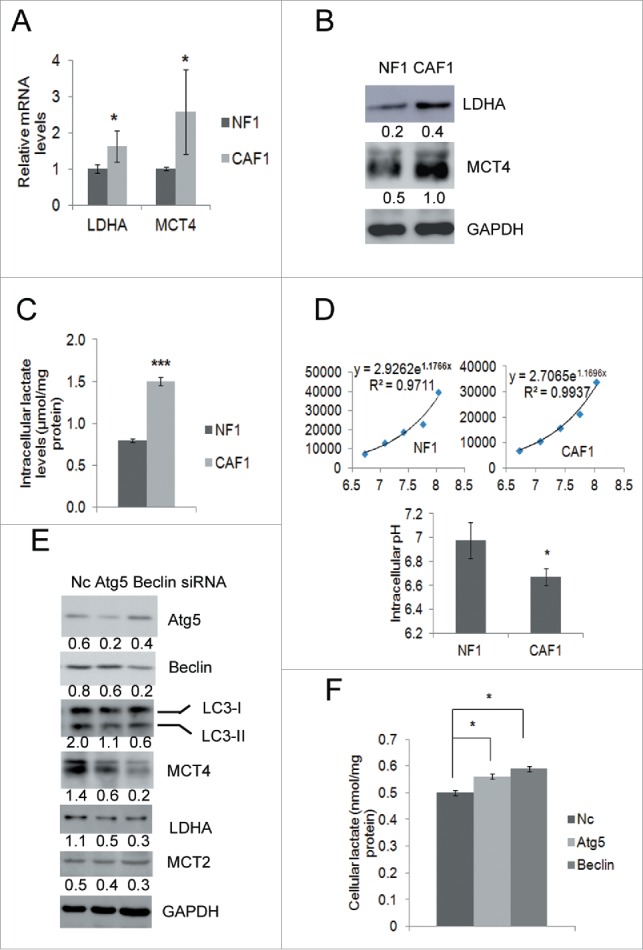
Protein levels of MCT4 and LDHA are downregulated and lactate is accumulated in CAFs when autophagy is inhibited. (A) qRT-PCR analysis of the mRNA levels of LDHA, MCT2 and MCT4 (mean ± SD, n = 3) in NF1 and CAF1 cells. (B) Western blot analysis of the protein levels of LDHA, MCT2 and MCT4 in NF1 and CAF1 cells. The relative intensity of indicated proteins normalized to housekeeping protein was shown at the bottom of each panel. (C) The lactate levels in NF1 and CAF1 cells. Cells cultured in the media without pyruvate for 24 h were lysed and assayed for lactate levels (mean ± SD, n = 3). The lactate levels were normalized to protein concentrations. (D) pHi of NF1 and CAF1 cells. 3 × 104 cells were seeded in 24-well plate overnight, stained with BCECF and lysed with RIPA buffer. The fluorescent signals were read with a fluorometer. The pHi of NF1 and CAF1 cells were calculated by the calibration curves of NF1 and CAF1 cells, respectively (mean ± SD, n = 3). The calibration curves were obtained by permeabilizing cells with nigericin at different pH values. (E) Western blot analysis of the protein levels of MCT4, LDHA and MCT2 in CAF1 cells transfected with the indicated siRNA for 48 h. The relative intensity of indicated proteins normalized to housekeeping protein was shown at the bottom of each panel. (F) The lactate level in CAF1 cells transfected with the indicated siRNA for 48 h (mean ± SD, n = 2). The lactate levels were normalized to protein concentrations. The experiment was repeated twice with similar results.
SOD2 levels increase in CAFs and can be downregulated by autophagy blockage
Intracellular ROS levels are tightly controlled by four primary antioxidant enzymes superoxide dismutase (SOD) 1 and 2, glutathione peroxidase (GPx) and catalase. They scavenge ROS and restore the redox balance.19,33 Our RNA-Seq data showed that SOD2 was enhanced in CAF1 cells. A series of anti-oxidant enzymes including SOD1, SOD2, catalase, glutathione peroxidase (Gpx) and thioredoxin reductase (TRx) were examined by RT-PCR, and only SOD2 was up-regulated in CAF1 cells (Fig. 5A). SOD2 protein levels were also higher in CAF1 than in NF1 cells (Fig. 5B). Knockdown of Atg5 or Beclin decreased mRNA levels and protein levels of SOD2 (Fig. 5C and Fig. 5D), while SOD1 protein levels were unaffected (Fig. 5D).
Figure 5.
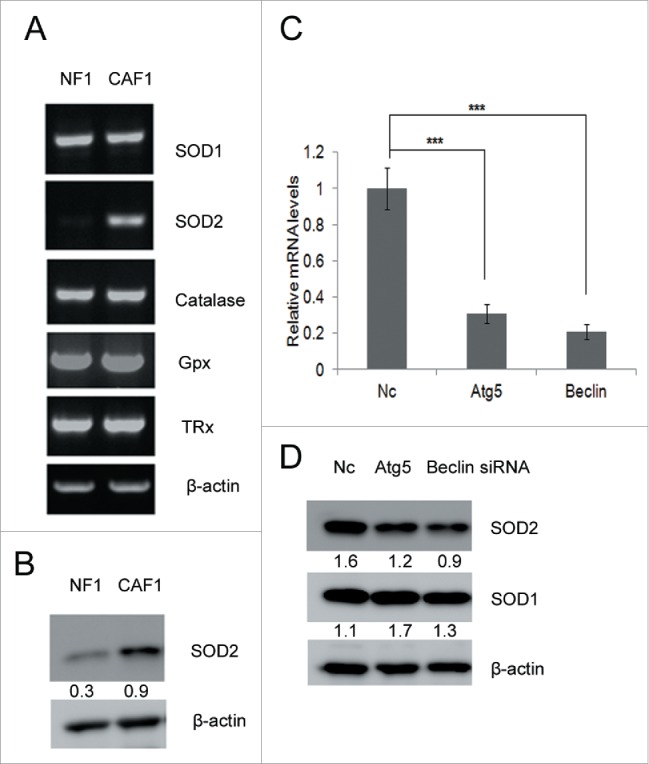
SOD2 levels increase in CAFs and can be down-regulated by autophagy blockage. (A) RT-PCR analysis of indicated antioxidant genes in NFs and CAFs. (B) Western blot analysis of SOD2 protein levels in NF1 and CAF1 cells. The relative intensity of indicated proteins normalized to housekeeping protein was shown at the bottom of each panel. (C) qRT-PCR analysis of SOD2 mRNA levels transiently transfected with the indicated siRNA for 48 h. (D) Western blot analysis of the protein levels of SOD1 and SOD2 in CAFs transiently transfected with the indicated siRNA for 48 h. The relative intensity of indicated proteins normalized to housekeeping protein was shown at the bottom of each panel.
Discussion
GSEA revealed that ovarian CAFs were mitotically active compared with NFs (Table 1 and Fig. 1D). This may reflect the fact that the tumor needs to divide rapidly. Cellular senescence is a protective state in which cells can survive the oxidative stress without proliferating.23,25 Mitotic CAFs may use this mechanism to avoid H2O2-induced cell death because elevated SA-β-gal activity and p21 (WAF1/Cip1) protein levels were observed in CAFs treated with H2O2 (Fig. 2A-D). Although overexpression of p21 (WAF1/Cip1) can result in G1, G2, or S phase arrest.27 ROS can induce S phase arrest too.26,28,34 Intracellular ROS production (Fig. 2G) and p21 (WAF1/Cip1) elevation may account for H2O2-induced CAFs cell cycle arrest in S phase (Fig. 2E and Fig. 2F).
H2O2 activated autophagy in CAFs as shown by increased LC3-II and decreased SQSTM1/p62 expression (Fig. 2D). Blockage of autophagy can increase ROS production in CAFs (Fig. 3E), leading to cell cycle arrest in S phase (Fig. 3C and Fig. 3D). Fibroblasts arrested in S phase tend to die later.35 This may be the reason that blockage of autophagy can inhibit CAFs proliferation (Fig. 3E) and enhance CAFs sensitivity to H2O2-induced cell death (Fig. 3A and Fig. 3B). ROS scavenger NAC could reduce ROS production (Fig. 3F) and restored cell viability (Fig. 3G). These results suggested that autophagy protected CAFs against oxidative stress. Moreover, blockage of autophagy could sensitize CAFs to cisplatin, implicating that autophagy play an important role in ovarian cancer chemoresistance.
It has been reported that loss of Cav-1 can cause oxidative stress, which will then induces increased MCT4 expression in stromal cells. Subsequently, stromal cells can export lactate via MCT4 to fuel cancer cells.36-38 Our RNA-Seq results showed that Cav-1 mRNA levels were similar between CAFs and NFs (data not shown). However, the proteins controlling lactate synthesis and transport, including LDHA and MCT4 (Fig. 4A and Fig. 4B), were increased in CAFs. So loss of Cav-1 cannot explain increased LDHA and MCT4 protein levels in CAFs. They were elevated probably because CAFs function as lactate reservoir to fuel cancer cells. Indeed, we showed that the lactate level was higher in CAFs than in NFs, which created an acidic intracellular environment in CAFs (Fig. 4C and Fig. 4D). Lactate is considered to have anti-oxidant function in radiotherapy.39,40 Blockage of autophagy decreased LDHA and MCT4 protein levels in CAFs (Fig. 4E), resulting in lactate accumulation in CAFs (Fig. 4F). Disturbed lactate homeostasis by autophagy blockage may facilitate ROS production.
SOD2 is localized in mitochondria and catalyzes superoxide anions to hydrogen peroxide.33 Lactate uptake increases production of mitochondrial superoxide anions.41 Up-regulation of SOD2 (Fig. 5A and Fig. 5B) may be evolved by CAFs to cope with increased superoxide anions-induced by relatively high lactate content (Fig. 4C). SOD2 has dual roles in tumorigenic progression, both as a negative modulator of cellular apoptosis and as a survival factor for cancer cells.42-44 We found that when blocking autophagy, SOD2 expression is down-regulated in CAFs (Fig. 5C). Blocking autophagy decreased expression of SOD2, LDHA and MCT4 that might contribute to enhanced ROS production, leading to low cell viability and increasing the sensitivity of CAFs to oxidative stress. In summary, ovarian CAFs utilize autophagy to eliminate intracellular ROS production when exposed to exogenous oxidative stress. Chemotherapeutic drugs commonly used in ovarian cancer including Taxol, cisplatin and cyclophosphamide can all cause oxidative stress.45-47 Autophagy might possess clinical utility as an attractive target for ovarian cancer treatment in the future.
Materials and methods
Cell culture
The primary ovarian cancer associated fibroblasts (CAFs) were isolated from ovarian cancer tissues. The normal fibroblasts (NFs) were isolated from non-cancerous prophylactic oophorectomy specimens. The clinical samples were approved by the Institutional Review Boards at Shanghai Jiaotong University, Shanghai, China. 1 cm tissue was cut into pieces and incubated in 10 ml 0.25% trypsin-EDTA (Invitrogen, Cat. 25200-072) for 15 min in a water bath set at 37°C, the solution was filtered and centrifuged to collect cells. The epithelial cells and the fibroblasts were separated by Percoll reagent (Yeasen Co. Cat. 40501ES60) as described previously.48 The cells were cultured in DMEM culture medium containing 10% fetal bovine serum at 37˚C in a humidified atmosphere containing 5% carbon dioxide.
RNA sequencing and gene set enrichment analysis
Double stranded cDNA were purified and sonicated with Diagenode Bioruptor to obtain DNA fragments of 200 to 400 bp. The fragmented DNA was then A-tailed after end repair, ligated to the adaptors and PCR amplified according to the instructions of illumina Nextera DNA Sample Prep Kit. The NGS libraries were sequenced on the illumina GA IIx (single end, 75 bp length). The sequencing depth was 29 M and 23 M reads for NFs and CAFs, respectively. After mapping to the human genome with TopHat software, the FPKM (fragments per kilobase of transcript per million fragments mapped) of NFs versus CAFs and their Log2 fold change was calculated with Cufflinks software. Then the Log2 fold change of NFs vs. CAFs was used to generate the pre-ranked gene list and loaded to do the GSEA (gene set enrichment anylysis) with the gene set database c5.all.v3.0.symbols.gmt downloaded from MSigDB.
Reverse transcription, PCR and q-PCR
Cells were lysed with RNAiso reagent (TaKaRa). Total RNA was extracted and 1 μg RNA was reverse transcribed with M-MLV Reverse Transcriptase (Promega) and subjected to PCR or quantitative PCR (q-PCR). The primers for PCR and q-PCR were list in Table S1 and Table S2, respectively. All the primers used in q-PCR were designed at http://primerdepot.nci.nih.gov.
siRNA transfection and MTT assay
The cells were transfected with 80 nM of siRNA by using DharmaFECT reagent (Thermo Fisher Scientific Inc.) according to the manufacturer's instructions. siRNA were synthesized by GenePharma Co. siRNA sequences were as follows:
Nc (negative control siRNA), sense: 5′-UUCUCCGAACGUGUCACGUTT-3′, anti-sense: 5′-ACGUGACACGUUCGGAGAATT-3′. The siRNA for Atg5 and Beclin were previously described.49 For the MTT assay, cells were incubated with 0.2 mg/ml MTT (final concentration) for 4 h and lysed with 20% sodium dodecyl sulfate-50% dimethylformamide. The OD value was read at a test and reference wavelength of 490/630 nM with a Bio-Rad ELISA reader.
Intracellular pH measurement
Cells were seeded for 24 h, then the media were replaced with a high-potassium buffers (25 mmol/L HEPES, 145 mmol/L KCl, 0.8 mmol/L CaCl2, 5.5 mmol/L glucose) at different pHs (pH 6.7, 7.1, 7.4, 7.8, and 8.0) in the presence of permeabilizing agent nigericin (10 μM) and pH fluorescent dye BCECF-AM (0.1 μM, DOJINDO) at 37°C for 30 min,50,51 lysed with RIPA buffer. The fluorescence was read at 488 nM by a fluorometer (Invision, PerkinElmer). A pH calibration curve was generated and the intracellular pH of NFs and CAFs were calculated accordingly.
Chemicals and antibodies
H2DCFDA and cisplatin were obtained from Sigma-Aldrich Co. Anti-LC3 (#3868), anti-FSP1 (#13018) and anti-LDHA (#2012) antibodies were obtained from Cell Signaling Technology. Anti-phospho-histone H3 pSer10 (PA5-17869) was obtained from Thermo Fisher Scientific. Anti-MCT2 (G-7), anti-MCT4 (H-90), HRP-conjugated goat anti-rabbit secondary antibodies were obtained from Santa Cruz Biotechnology. Anti-GAPDH, anti-vimentin and HRP-conjugated goat anti-mouse secondary antibodies were obtained from Boster Co. Anti-β-actin antibody was obtained from Transgene Biotech Co. Anti-p21 (WAF1/Cip1) (WL0362), anti-SOD1 (WL0184b) and anti-SOD2 (WL0530a) antibodies were obtained from Wanleibio Co.
Western blot analysis
Cells were lysed in RIPA buffer and quantified by bicinchoninic acid assay kit (Boster, Wuhan, China) for protein concentration. Next, 5 to 40 μg protein was loaded on to the gel and subjected to Western blot. Western blot results were quantified by Image J (NIH) software.
ROS measurement
Cells were stained with 20 μM H2DCFDA in DMEM without serum at 37°C for 15 min, and then cultured in complete DMEM for 1 h. Cells were washed with PBS and subjected to flow cytometric analysis (Beckman Coulter) or lysed in RIPA buffer and read at a wavelength of 488 nm with a fluorometer (Envision, PerkinElmer).
Cell cycle analysis
Cells were collected, washed with PBS, fixed in 70% ethanol at −20°C overnight, and then digested with 0.1 mg/ml RNase A and stained with 40 μg/ml Propidium iodide (PI). Cells were subjected to cell cycle analysis by a flow cytometer (Beckman Coulter) and MultiCycle AV software (Phoenix Flow System) was used for analyze cell cycle distribution.
Statistical analysis
Student's t- test was used to analyze the data. P values were calculated in individual assays and P < 0.05 was considered as statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Junlin Guan at Cincinnati University for critical reading and helpful discussion.
Funding
This work was supported by National Natural Science Foundation of China for young investigators (31301174), Shanghai Municipal Council for Science and Technology (No14411961500.), and State Key Laboratory of Cell Biology, Chinese Academy of Sciences, Shanghai.
References
- [1].Schauer IG, Sood AK, Mok S, Liu J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia 2011; 13:393-405; PMID:21532880; http://dx.doi.org/ 10.1593/neo.101720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005; 121:335-48; PMID:15882617; http://dx.doi.org/ 10.1016/j.cell.2005.02.034 [DOI] [PubMed] [Google Scholar]
- [3].Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth–bystanders turning into key players. Curr Opin Genet Dev 2009; 19:67-73; PMID:19211240; http://dx.doi.org/ 10.1016/j.gde.2009.01.003 [DOI] [PubMed] [Google Scholar]
- [4].Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet 2014; 384:1376-88; PMID:24767708; http://dx.doi.org/ 10.1016/S0140-6736(13)62146-7 [DOI] [PubMed] [Google Scholar]
- [5].Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P, Nephew KP, Hales DB, Stack MS. Epithelial ovarian cancer experimental models. Oncogene 2014; 33:3619-33; PMID:23934194; http://dx.doi.org/ 10.1038/onc.2013.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/ 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cicchini M, Chakrabarti R, Kongara S, Price S, Nahar R, Lozy F, Zhong H, Vazquez A, Kang Y, Karantza V. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy 2014; 10:2036-52; PMID:25483966; http://dx.doi.org/ 10.4161/auto.34398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al.. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013; 27:1447-61; PMID:23824538; http://dx.doi.org/ 10.1101/gad.219642.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 2014; 59:505-17; PMID:23913306; http://dx.doi.org/ 10.1002/hep.26659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu XD, Yao J, Tripathi DN, Ding Z, Xu Y, Sun M, Zhang J, Bai S, German P, Hoang A, et al.. Autophagy mediates HIF2alpha degradation and suppresses renal tumorigenesis. Oncogene 2015; 34:2450-60; PMID:24998849; http://dx.doi.org/ 10.1038/onc.2014.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen HY, White E. Role of autophagy in cancer prevention. Cancer Prevention Res 2011; 4:973-83; PMID:21733821; http://dx.doi.org/ 10.1158/1940-6207.CAPR-10-0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, De Zio D, Nazio F, Antonioli M, D'Orazio M, et al.. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol 2015; 17:20-30; PMID:25438055; http://dx.doi.org/ 10.1038/ncb3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al.. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/ 10.1016/j.ccr.2006.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kang R, Tang D, Lotze MT, Zeh HJ 3rd. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 2012; 8:989-91; PMID:22722139; http://dx.doi.org/ 10.4161/auto.20258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Strohecker AM, White E. Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy 2014; 10:384-5; PMID:24362353; http://dx.doi.org/ 10.4161/auto.27320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011; 25:1510-27; PMID:21764854; http://dx.doi.org/ 10.1101/gad.2051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differentiation 2015; 22:377-88; PMID:25257172; http://dx.doi.org/ 10.1038/cdd.2014.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Thibault B, Castells M, Delord JP, Couderc B. Ovarian cancer microenvironment: implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev 2014; 33:17-39; PMID:24357056; http://dx.doi.org/ 10.1007/s10555-013-9456-2 [DOI] [PubMed] [Google Scholar]
- [21].Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res 2010; 316:2713-22; PMID:20451516; http://dx.doi.org/ 10.1016/j.yexcr.2010.04.032 [DOI] [PubMed] [Google Scholar]
- [22].Yang G, Rosen DG, Zhang Z, Bast RC Jr, Mills GB, Colacino JA, Mercado-Uribe I, Liu J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A 2006; 103:16472-7; PMID:17060621; http://dx.doi.org/ 10.1073/pnas.0605752103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729-40; PMID:17667954; http://dx.doi.org/ 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- [24].Hans F, Dimitrov S. Histone H3 phosphorylation and cell division. Oncogene 2001; 20:3021-7; PMID:11420717; http://dx.doi.org/ 10.1038/sj.onc.1204326 [DOI] [PubMed] [Google Scholar]
- [25].Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB life 2000; 50:279-89; PMID:11327322; http://dx.doi.org/ 10.1080/15216540051081010 [DOI] [PubMed] [Google Scholar]
- [26].Agarwal ML, Agarwal A, Taylor WR, Chernova O, Sharma Y, Stark GR. A p53-dependent S-phase checkpoint helps to protect cells from DNA damage in response to starvation for pyrimidine nucleotides. Proc Natl Acad Sci U S A 1998; 95:14775-80; PMID:9843965; http://dx.doi.org/ 10.1073/pnas.95.25.14775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res 2005; 65:3980-5; PMID:15899785; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-3995 [DOI] [PubMed] [Google Scholar]
- [28].Yang ST, Huang AC, Tang NY, Liu HC, Liao CL, Ji BC, Chou YC, Yang MD, Lu HF, Chung JG. Bisdemethoxycurcumin-induced S phase arrest through the inhibition of cyclin A and E and induction of apoptosis via endoplasmic reticulum stress and mitochondria-dependent pathways in human lung cancer NCI H460 cells. Environmental toxicol 2015. Sept 15 [Epub ahead of print]; http://dx.doi.org/ 10.1002/tox.22191 [DOI] [PubMed] [Google Scholar]
- [29].Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36:30-8; PMID:20728362; http://dx.doi.org/ 10.1016/j.tibs.2010.07.007 [DOI] [PubMed] [Google Scholar]
- [30].Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest 2013; 123:3685-92; PMID:23999443; http://dx.doi.org/ 10.1172/JCI69741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Halestrap AP, Wilson MC. The monocarboxylate transporter family–role and regulation. IUBMB Life 2012; 64:109-19; PMID:22162139; http://dx.doi.org/ 10.1002/iub.572 [DOI] [PubMed] [Google Scholar]
- [32].Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res 2011; 71:6921-5; PMID:22084445; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1457 [DOI] [PubMed] [Google Scholar]
- [33].Pervaiz S, Clement MV. Superoxide anion: oncogenic reactive oxygen species? Int J Biochem Cell Biol 2007; 39:1297-304; PMID:17531522; http://dx.doi.org/ 10.1016/j.biocel.2007.04.007 [DOI] [PubMed] [Google Scholar]
- [34].Burhans WC, Heintz NH. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radical Biol Med 2009; 47:1282-93; PMID:19486941; http://dx.doi.org/ 10.1016/j.freeradbiomed.2009.05.026 [DOI] [PubMed] [Google Scholar]
- [35].Chen QM, Liu J, Merrett JB. Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem J 2000; 347:543-51; PMID:10749685; http://dx.doi.org/ 10.1042/bj3470543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Martinez-Outschoorn UE, Balliet RM, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Hereditary ovarian cancer and two-compartment tumor metabolism: epithelial loss of BRCA1 induces hydrogen peroxide production, driving oxidative stress and NFkappaB activation in the tumor stroma. Cell Cycle 2012; 11:4152-66; PMID:23047606; http://dx.doi.org/ 10.4161/cc.22226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, et al.. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256-76; PMID:20814239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, Birbe RC, Howell A, Pavlides S, Gandara R, et al.. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011; 10:1772-83; PMID:21558814; http://dx.doi.org/ 10.4161/cc.10.11.15659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sattler UG, Meyer SS, Quennet V, Hoerner C, Knoerzer H, Fabian C, Yaromina A, Zips D, Walenta S, Baumann M, et al.. Glycolytic metabolism and tumour response to fractionated irradiation. Radiotherapy Oncol 2010; 94:102-9; PMID:20036432; http://dx.doi.org/ 10.1016/j.radonc.2009.11.007 [DOI] [PubMed] [Google Scholar]
- [40].Sattler UG, Mueller-Klieser W. The anti-oxidant capacity of tumour glycolysis. Int J Radiation Biol 2009; 85:963-71; PMID:19895273; http://dx.doi.org/ 10.3109/09553000903258889 [DOI] [PubMed] [Google Scholar]
- [41].Wenzel U, Schoberl K, Lohner K, Daniel H. Activation of mitochondrial lactate uptake by flavone induces apoptosis in human colon cancer cells. J Cell Physiol 2005; 202:379-90; PMID:15452831; http://dx.doi.org/ 10.1002/jcp.20129 [DOI] [PubMed] [Google Scholar]
- [42].Connor KM, Hempel N, Nelson KK, Dabiri G, Gamarra A, Belarmino J, Van De Water L, Mian BM, Melendez JA. Manganese superoxide dismutase enhances the invasive and migratory activity of tumor cells. Cancer Res 2007; 67:10260-7; PMID:17974967; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1204 [DOI] [PubMed] [Google Scholar]
- [43].Hempel N, Carrico PM, Melendez JA. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anti-Cancer Agents Medicinal Chem 2011; 11:191-201; PMID:21434856; http://dx.doi.org/ 10.2174/187152011795255911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev 2010; 29:351-78; PMID:20386957; http://dx.doi.org/ 10.1007/s10555-010-9225-4 [DOI] [PubMed] [Google Scholar]
- [45].Chakraborty P, Sk UH, Bhattacharya S. Chemoprotection and enhancement of cancer chemotherapeutic efficacy of cyclophosphamide in mice bearing Ehrlich ascites carcinoma by diphenylmethyl selenocyanate. Cancer Chemotherapy Pharmacol 2009; 64:971-80; PMID:19221751; http://dx.doi.org/ 10.1007/s00280-009-0950-8 [DOI] [PubMed] [Google Scholar]
- [46].Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med 2009; 13:3644-54; PMID:20196784; http://dx.doi.org/ 10.1111/j.1582-4934.2009.00663.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Meshkini A, Yazdanparast R. Involvement of oxidative stress in taxol-induced apoptosis in chronic myelogenous leukemia K562 cells. Exp Toxicologic Pathol 2012; 64:357-65; PMID:21074392; http://dx.doi.org/ 10.1016/j.etp.2010.09.010 [DOI] [PubMed] [Google Scholar]
- [48].Yang L, Lai D. Ovarian cancer stem cells enrichment. Methods Mol Biol 2013; 1049:337-45; PMID:23913228; http://dx.doi.org/ 10.1007/978-1-62703-547-7_25 [DOI] [PubMed] [Google Scholar]
- [49].Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al.. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025-40; PMID:15657430; http://dx.doi.org/ 10.1128/MCB.25.3.1025-1040.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Khaled AR, Kim K, Hofmeister R, Muegge K, Durum SK. Withdrawal of IL-7 induces Bax translocation from cytosol to mitochondria through a rise in intracellular pH. Proc Natl Acad Sci U S A 1999; 96:14476-81; PMID:10588730; http://dx.doi.org/ 10.1073/pnas.96.25.14476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu Y, Hitomi H, Diah S, Deguchi K, Mori H, Masaki T, Nakano D, Kobori H, Nishiyama A. Roles of Na(+)/H(+) exchanger type 1 and intracellular pH in angiotensin II-induced reactive oxygen species generation and podocyte apoptosis. J Pharmacological Sci 2013; 122:176-83; PMID:23800993; http://dx.doi.org/ 10.1254/jphs.12291FP [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.