

ABSTRACT
Here we show that cancer stem cells amount in human lung adenocarcinoma cell line A549 depends on E-cadherin expression. In fact, downregulation of E-cadherin expression enhanced expression of pluripotent genes (c-MYC, NESTIN, OCT3/4 and SOX2) and enriched cell population with the cells possessing the properties of so-called ‘cancer stem cells’ via activation of Wnt/β-catenin signaling. Repression of E-cadherin also stimulated cell proliferation and migration in vitro, decreased cell amount essential for xenografts formation in nude mice, increased tumors vascularization and growth. On the other hand, E-cadherin upregulation caused opposite effects i.e. diminished the number of cancer stem cells, decreased xenograft vascularization and decelerated tumor growth. Therefore, agents restoring E-cadherin expression may be useful in anticancer therapy.
KEYWORDS: B-catenin, cancer stem cells (CSCs), E-cadherin, tumor progression, wnt-signaling
Introduction
E-cadherin is a transmembrane calcium-dependent adhesion molecule expressed in almost all epithelial cells. It plays essential roles in embryogenesis and maintenance of adult epithelia, while alterations of its expression/function contributes to tumor development.1,2 Aberrant adhesion junctions, E-cadherin downregulation or complete shutdown of its expression (through E-cadherin gene mutation, epigenetic or transcriptional silencing, etc.) were found in many human carcinomas. These facts allowed considering it as a tumor suppressor.3 Inactivating germ-line mutations of CDH1 gene coding E-cadherin increase the risk of development of familial gastric carcinomas,4,5 while loss of E-cadherin during tumor progression may result in epithelium to mesenchyme transition (EMT).6-8 Noteworthy, a correlation between E-cadherin expression and survival of cancer patients was shown.9-11
The cells passing through EMT lose polarity, acquire spindle-like shape and enhanced migratory and invasive capacities.12-14 Nonetheless, some studies showed a crucial necessity in a reverse process – mesenchyme to epithelium transition (MET) when re-expression of E-cadherin was crucial for efficient metastasis formation.15-17
During the last few years a connection between EMT and acquisition of so-called [cancer stem cells] (CSCs) phenotype was described.18-22 According to the CSCs model, heterogenic cancer cells are arranged in a hierarchical manner with a small cell population on the top harboring tumorigenic capacity. These tumorigenic cells are called CSCs or tumor initiating cells (TICs). It is proposed that they play a leading role in tumor progression and metastasis while an impact of other cancer cells seems to be less prominent. CSCs could originate from the long-term or transient amplifying normal stem cells23,24 and possess certain properties, allowing their isolation as a definite minor population in the bulk of tumor cells. They can be both quiescent and capable of self-renewing; they can also contribute to tumor growth by giving rise to cells with high proliferating potential.25,26 However recent studies revealed a complicated mechanism maintaining a certain ratio of CSCs in the tumor cells population, particularly as a result of conversion of other cancer cells.27-29
The ways cancer cell turn into CSCs are poorly studied. It has been shown in many experiments that up- or downregulation of several genes (for example, TAZ, WNT, SHH, DKK-1, PTEN, BMI-1) could influence the proportion of CSCs in cancer cell populations.30-33 Taking into consideration that disappearance of E-cadherin intercellular adhesions is a major manifestation of EMT,13,14 that is often accompanied by acquisition of cell features ascribed to CSCs18-22 we decided to determine whether changes in E-cadherin expression could represent one of the driving forces in acquisition of CSCs phenotype. Previous experimental data concerning this idea were quite controversial. Some results supported this assumption. In fact, it has been shown for breast cancer HMLER and colon cancer SW480 cells that CDH1 gene knockdown led to enrichment of CD24−/CD44+ cells – a CSCs phenotype for these tumors.34,35 Moreover, CDH1 gene downregulation increased tumorigenic potential of HMLER cells34 – a basic feature of CSCs. On the other hand, there were reports showing that only E-cadherin-positive prostate cancer cells exhibited a CSCs phenotype, and that it's overexpression led to an increase in the proportion of CSCs.36 Further indirect evidence that E-cadherin can affect CSCs based on our previous results.37 Inactivation of human endothelial growth factor VEGF-C decreased population of CSCs in colon carcinoma HCT116 and lung carcinoma A549 cells and increased E-cadherin expression was observed.
To assess in more detail the effects of changes in E-cadherin expression on manifestations of CSCs phenotype we constructed lentiviral vectors able to inhibit or increase CDH1 gene expression and as result created human lung cancer A549 sublines with down- and upregulated E-cadherin.
Materials and methods
Cells
Human lung carcinoma A549 cell line (ATCC #CCL-185) expressing CDH1 or siRNA specific for CDH1. For E-cadherin expression cDNA of human CDH1 (GenBank: Z13009.1; cloned and verified into pBlEc plasmid38 and kindly provided by Prof. Sergey M. Troyanovsky) was inserted into the lentiviral pLenti6 vector (Invitrogen). For expression of siRNAs specific for CDH1, the hairpin structures containing 21-bp sequences corresponding to 5 different parts of CDH1 mRNA (Fig. 1A) were synthesized and AgeI/EcoRI cloned into the lentiviral pLKO.1-puro vector (Addgene, plasmid #8453). pLKO.1-shGFP-puro targeting eGFP (GenBank Accession No. pEGFP U55761) was used as a control. Oligonucleotides synthesis and DNA sequencing was performed by Evrogen (www.evrogen.com).
Figure 1.
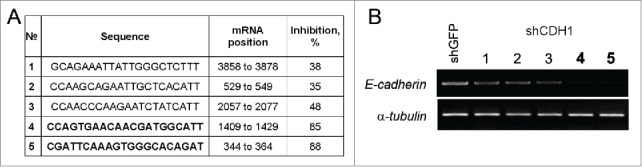
Obtaining constructs expressing shRNA specific for CDH1. (A) The sequences of 21 nucleotide regions corresponding to the human CDH1 mRNA (Sequence ID: ref NM_004360.3), validated as siRNA #4 and #5 (bold) – most effective targets. (B) Effect of transduction of lentiviral constructs pLKO.1-shCDH1 on expression of E-cadherin in A549. RT-PCR analysis of CDH1 expression with corresponding shRNAs (#1–5). α-tubulin mRNA was analyzed as loading control.
The pLenti6 and pLKO lentiviral DNA constructs together with the pΔR8.2 and pVSV-G packaging plasmids were transfected into 293FT cells using TurboFect Transfection Reagent (R0531, Thermo Scientific). Virus-containing supernatants collected 24 to 48 h after transfection and were used to infect recipient A549 cells in the presence of polybrene (8 μg/mL). As hairpin structures shCDH1#4 and shCDH1#5 were the most effective and had similar biological effects, supernatants containing these lentiviruses were combined and applied together. Infected cell cultures were selected for 5–6 d in medium containing 4 mg/ml blasticidin (R210-01, Invitrogen) for pLenti6 constructs or 1 μg/ml puromycin (P8833, Sigma) for pLKO constructs. Cells were cultured in DMEM medium supplemented with 10% fetal bovine serum and penicillin/streptomycin.
Reverse transcription-PCR assay
Total mRNA was isolated with SV Total RNA Isolation System (Promega) according to the manufacturer's protocols. To detect the corresponding gene expression using endpoint PCR, the following primers and conditions were used:
E-cadherin: F 5′-GTCTGTAGGAAGGCACAGCC-3′,
R 5′–TGCAACGTCGTTACGAGTCA–3′, 32 amplification cycles;
c-myc: F 5′–GGCTTCTCAGAGGCTTGGCGGGAAA–3′,
R 5′–CCTGGGGGATCAAGCGGGAGG–3′, 40 amplification cycles;
Nestin F 5′–GCGGCTGCGGGCTACTGAAA–3′,
R 5′–ATCCAAGACGCCGGCCCTCT–3′, 40 amplification cycles;
Sox2: F 5′–TGGACAGTTACGCGCACAT–3′,
R 5′–CGAGTAGGACATGCTGTAGGT–3′, 40 amplification cycles;
Oct3/4: F 5′–GTGTTCAGCCAAAAGACCATCT–3′,
R 5′–GGCCTGCATGAGGGTTTCT–3′, 40 amplification cycles;
α-tubulin: F 5′–GTTGGTCTGGAATTCTGTCAG–3′,
R 5′–AAGAAGTCCAAGCTGGAGTTC–3′, 30 amplification cycles;
initial denaturation 95°C 60 s; amplification: denaturation 95°C 30 s, annealing 60°C 30 s (for all primers), extension 72°C 30 s; final extension 5 min at 72°C.
The detection of PCR bands was performed using Chemi-Smart 3000 Imaging System (Vilber Lourmat).
For Real-time qPCR analysis IQ5 Biorad PCR Detection System (Biorad) was used. Real-time PCR was performed using 2.5x SYBR Green real-time PCR mix (Syntol, Russia) according to manufacturer protocol with addition of 0.4 µM of primers (forward, reverse) and 50 ng of cDNA and DEPC-treated water to the final volume of 25 µL. PCR conditions were as follows: 94°C for 5 min, followed by 40 cycles of amplification: 94°C for 10 s, 60°C for 10 s, and 72°C for 20s. For all genes, except Nestin (F 5′–CTCCAGAAACTCAAGCACC–3′, R 5′–TCCTGATTCTCCTCTTCCA-3′) and Sox2 (F 5′–TTCACATGTCCCAGCACTACCAGA–3′, R 5′–TCACATGTGTGAGAGGGGCAGTGTGC-3′), the same primers in qPCR were used. The PCR products were checked for specificity with agarose gel electrophoresis and melt-curve analysis. Data was analyzed with CFX Manager Software (BioRad). Alpha-tubulin gene was used for data normalization. Samples were collected from 3 independent cultures. Data was analyzed based on 2−ΔΔCt method.
Western blot analysis, total and nuclear protein extract preparation
was performed as previously described.39 Primary antibodies specific to E-cadherin (M126, Takara), vimentin (M0725, Dako), β-catenin (M3539, Dako), α-tubulin (sc-23948, Santa Cruz Biotechnology), histone H3 (9715, Cell Signaling) and Alexa488-conjugated secondary antibodies were used, band detection was performed using variable mode imager Typhoon9410 (GE Healthcare). The quantitation of protein bands was performed using TotalLab v.2.01 software.
Immunofluorescent microscopy
Cells on cover slips were fixed on the forth day after plating with 1% PFA for 15 min and treated with methanol for or 5 min at −20°C. Cells were incubated with primary (see 2.3) and secondary AlexaFluor594- or Alexa488-conjugated (Invitrogen) antibodies. DAPI (Life technologies) was applied for nuclear staining. Images were acquired using fluorescent Axioplan 2 microscope and AxioVision (Carl Zeiss Imaging Systems) software.
Luciferase reporter assay
was performed using commercially available Lenti TCF/LEF reporter (Cignal Lenti TCF/LEF Reporter (luc) Kit: CLS-018L, Qiagen) and Steady-Glo Luciferase Assay System (E2510, Promega) according to the manufacturer's protocols; the values were normalized in relation to protein concentration.
Boyden chamber cell migration assay
was performed using transwell Matrigel-coated chambers with 8-μm pore-size membranes (BD Biosciences) according to manufacturer instructions with 5 × 104 A549 cells. The migration activity was quantified by blind counting of the migrated cells of at least 10 fields per chamber.
Cell cultures growth rate
Cells (104) were plated on the 6-well culture plates and grown without passaging. Cell counts were performed each 48 hours using a hemocytometer (3 wells per point). The measurement proceeded until monolayer formation.
FACScan analysis
Cells were collected from subconfluent culture and the final concentration of 106 cells/ml in pre-warmed Hank's balanced salt solution containing 2% fetal bovine serum was used. The cells were incubated with 1 mg/ml Rhodamine123 dye (R8004, Sigma) for 30 min in dark at 37°C with intermittent mixing. At the end of the incubation, cells were spun down and washed in cold Hank's balanced salt solution containing 2% fetal bovine serum at 4°C and were labeled with 10mg/ml propidium iodide (P4170, Sigma) to distinguish live from dead cells before analysis. Rhodamine123 staining was detected using a BD FACSCanto II flow cytometer (BD Biosciences), exciting at 488 nm and detecting Rhodamine123 with a 530/30 broad pass (BP) filter and propidium iodide with a 630/22 BP filter. 107 events within the live gate were tracked for further analysis using BD FACSDiva Software (BD Biosciences) and WinMDI (Joseph Trotter) software.
Clonogenic assay
200 cells were added to 2,6% methylcellulose (Fluka), mixed with 2× DMEM supplemented with 20% fetal bovine serum, plated on non-adherent Petri dishes and cultured at 37°C for 14 d. The colonies were stained with crystal violet and incubated at 4°C overnight. Colonies were counted with TotalLab v.2.01 software, module Colony Counter (Nonlinear Dynamics).
Nude mice assay
D2 x J nude mice (10 animals for each experimental group) were inoculated subcutaneously with dosages of cells suspended in 100 ml of PBS: 106; 0,5 × 106; 0,25 × 106; 0,125 × 106 and 0,005 × 106. Tumor sizes were measured every 3 d and their volumes were calculated as (width2) × (length) × 0.5. After 25 d of observation, explanted tumors were isolated and analyzed immunohistochemically. These experiments were repeated 3 times to confirm the results.
The animal experimental protocols were approved by the Committee for Ethics of Animal Experimentation and the experiments were conducted in accordance with the Guidelines for Animal Experiments in Blokhin Memorial Russian Cancer Research Center.
Immunohistochemistry
Antigen retrieval was achieved by heating (95°C) of the sections in target retrieval solution pH 6.0 (S1699, Dako) for 40 min. The sections were incubated with primary antibodies to CD34 (553731, BD Biosciences PharMingen) at room temperature for 1 hour, followed by incubations with biotin-conjugated (559286 BD Biosciences PharMingen) secondary antibodies for 30 min at room temperature. To identify the target Streptividin-HRP (Dako) was added for 15 min at room temperature. The reactivity was visualized with DAB+ (diaminobenzidine) (K3468, Dako) according to the manufacturer's instructions. Sections were counterstained with Mayer's hematoxylin and mounted. Vessels and capillaries, identified by positive staining for CD34 and appropriate morphology, were counted. Angiogenic properties were scored as average number of relatively large microvessels (the size of lumens P>100 µm in length). On average, immunohistochemistry quantification was performed by taking pictures from maximum possible fields per tumor, at least 15 fields of vision per tumor type, and imaging at least 6 tumors per cell subline type using AxioVision software (Carl Zeiss Imaging Systems).
Statistical analysis
All experiments were performed in triplicate and data are expressed as mean ± SD, as indicated in figure legends. Statistical analysis was performed using unpaired Student's t test. P values ≤ 0.05 were considered to be significant. The GraphPad Prizm statistical software package ver. 5.01 was used.
Results
To investigate the influence of E-cadherin expression on properties of lung adenocarcinoma cells and possible role in cancer stem cell phenotype formation we obtained A549 derivatives with overexpressed or silenced E-cadherin. For E-cadherin overexpression ORF of human CDH1 gene was cloned into modified lentiviral vector pLenti6. The vector has the CMV promoter for driving constitutive expression of the target gene and the blasticidin selection marker for stable selection in mammalian cells. Based on MISSION® shRNA Library (Sigma-Aldrich) 5 validated siRNA targets of human CDH1 mRNA were tested on A549 cells and 2 most effective targets (#4,5 Fig. 1) were cloned into pLKO.1 lentiviral vector for siRNA expression for stable E-Cadherin repression (Fig. 1).
A549 cells with silenced E-cadherin expression acquired a more mesenchymal phenotype while cells with overexpressed E-cadherin looked more epithelial and had enhanced tight junctions. Inactivation of E-cadherin was accompanied with a slight increase of total vimentin levels (Fig. 2A). To prove the fact, since changes of vimentin protein amounts were not prominent upon E-cadherin up- and downregulation, A549 cells were infected 3 times independently and independent western blots were accomplished. Average densitometry results confirmed slight increasing of vimentin amounts (up to1,5–2times) caused by CDH1 repression (Fig. 2B). Despite this fact, significant vimentin filaments formation in cells with down-regulated E-cadherin expression was observed (Fig. 2C). E-cadherin downregulation caused disappearance of E-cadherin junctions, decrease of total β-catenin amount with its' translocation and slight accumulation in the nuclei (Fig. 2D). As nuclear β-catenin accumulation was not prominent and was detected with strong fluorescent enhancement (Fig. 2E) we analyzed β-catenin content in the nuclear extracts. Nuclear β-catenin was detected only in cells with repressed CDH1 gene (Fig. 2F). Histone H3 as a nuclear marker was used. Nuclear translocation of β-catenin is usually accompanied by its transcriptional function induction, so we tested the Wnt/β-catenin signaling pathway activation upon changes of E-cadherin expression. Using luciferase TCF/Lef-dependent reporter (Qiagen) we have shown that E-cadherin repression led to activation of TCF/Lef-dependent transcription (Fig. 2G). As the conserved Wnt/β-catenin pathway regulates stem cell pluripotency and cell fate decisions during development,36,40-42 we analyzed mRNA levels of some of the pluripotency genes.
Figure 2.
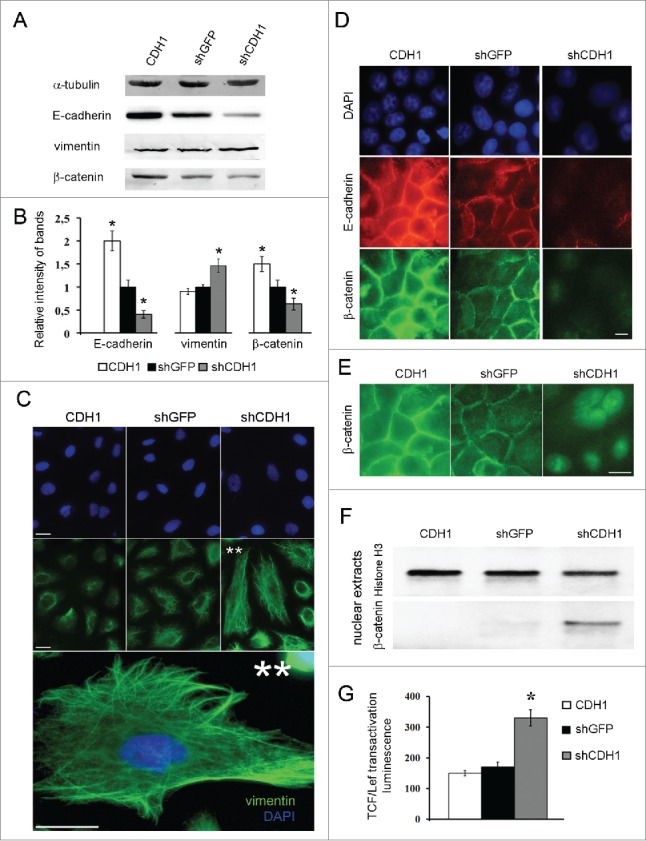
Effects of E-cadherin expression alterations on morphology features and activation of Wnt/β -catenin signaling pathway in A549 cells. (A) A representative Western blot analysis of E-cadherin, vimentin and β-catenin expression in A549 cell sublines. α-tubulin was analyzed as loading control. (B) Average results of E-cadherin, vimentin and β-catenin bands densitometry analysis normalized by tubulin expression of 3 independent experiments (Mean ± SD, * - p ≤ 0,05). (C) Immunofluorescent staining of A549 cell sublines with antibodies to vimentin. Bars 5 μm. ** - Enlarged A549shCDH1 cells with well-detected vimentin filaments. (D) Immunofluorescent staining of A549 cell sublines stained with E-cadherin and β-catenin antibodies. Bars 5 μm. (E) Enlarged and strongly enhanced immunofluorescent images of β-catenin staining of A549 cell sublines for nuclear β-catenin detection. Bars 5 μm. (F) Nuclear extracts Western blot analysis of β-catenin in A549 cell sublines. Histone H3 was used as loading control. (G) The reporter construct expressing luciferase gene under control of TCF/Lef was introduced into A549 cell sublines, luciferase activity was measured in 3 experiments; average data are presented (Mean ± SD, * p ≤ 0,05).
Increased transcription of c-MYC, NESTIN, OCT3/4 and SOX2 genes was detected in A549 cells with silenced CDH1 using end-point PCR (Fig. 3A) and confirmed by Real-time qPCR (Fig. 3B). As increased expression of these genes is connected not only with “normal” stem cells but is also observed in “cancer” stem cells,42-44 we also studied the influence of E-cadherin expression alterations on the proportion of cells with the CSCs traits in A549 cell sublines. For this purpose generally accepted approaches were used. Firstly we investigated the activity of ABC-transporters analyzing Rhodamine123 content in A549 cells with modified E-cadherin expression using FACS flow cytometry. Amount of side-population cells with enhanced ABC-transporter activity, one of the features of CSCs,21,44 was increased in the A549 subline with inactivated E-cadherin (shCDH ) and decreased in the subline with overexpressed E-cadherin (CDH1) (Fig. 4A). Then we tested cell abilities to form colonies in semisolid medium under non-adherent conditions. Subline with repressed E-cadherin formed up to 4 times more colonies than control A549 cells and more than 10 times compared with overexpressed E-cadherin subline (Fig. 4B). Also A549-shCDH1-formed colonies had noticeably larger size. In addition, more mesenchymal cells with downregulated E-cadherin demonstrated increased migration and invasive activities in vitro, when Boyden chamber assay with matrigel-coated filters were used. Upregulation of E-cadherin diminished these properties (Fig. 4C).
Figure 3.
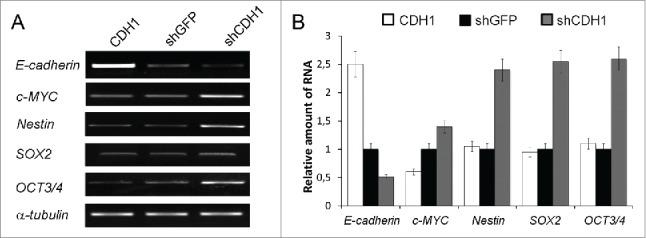
Effect of E-cadherin expression alterations on the pluripotency genes expression. (A) The result of one of the representative End-point PCR experiments. α-tubulin mRNA was analyzed as loading control. (B) Relative quantities of mRNAs estimated by Real-time qPCR. α-tubulin gene was used for data normalization (Mean ± SD).
Figure 4.

The effect of E-cadherin expression alterations on the percentage of cells with the CSCs features in A549 cell populations. (A) ABC-transporters activity by analysis of Rhodamine123 content in A549 sublines using flow cytometry. Left panel – results of quantitative estimation of side-population cells, (Mean ± SD, * p ≤ 0,05). Right panel – distribution of 107 cells according to their fluorescence; side populations of weakly fluorescent cells were counted in the left lower quadrants. (B) Clonogenic capacity of A549 cell sublines in non-adherent conditions. Results of quantitative estimation of the colony number (left panel) and colony size (right panel), (Mean ± SD, * p ≤ 0,05). (C) Invasion of A549 sublines in Boyden chambers through matrigel-coated membranes (Mean ± SD, * p ≤ 0,05).
The main trait of CSCs is tumorigenic ability in nude mice and injectable cell dose threshold is a very important characteristic.44 In fact, tumorigenic dosage for cells with overexpressed CDH1 was increased (Table 1), while shCDH1 A549 derivatives needed less cells for tumor formation. All these experiments indicate on the dependence of CSCs population on the levels of E-cadherin in A549 cells.
Table 1.
The effect of E-cadherin expression alterations on tumorigenic ability of A549 cells.
| Tumorigenicity (%)* |
|||||
|---|---|---|---|---|---|
| Sublines | 1×106 | 0,5×106 | 0,25×106 | 0,125×106 | 0,05 ×106 |
| CDH1 | 100% | 100% | 50% | 0% | 0% |
| shGFP | 100% | 100% | 75% | 15% | 0% |
| shCDH1 | 100% | 100% | 100% | 50% | 10% |
Number of mice with tumors/total number of mice with injected cells.
Besides enhancing CSCs properties, E-cadherin inactivation enhanced A549 cell proliferation in vitro (Fig. 5A) and stimulated vascularization and growth of tumor xenografts in nude mice (Fig. 5B,C). On the other hand, E-cadherin upregulation reduced the number of blood vessels and decelerated tumor growth (Fig. 5B,C).
Figure 5.
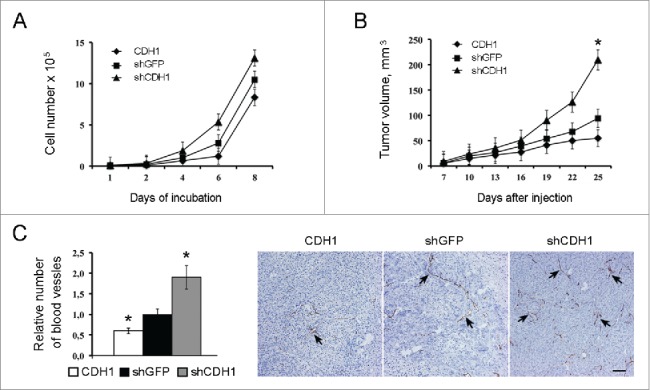
The effect of E-cadherin expression alterations on the growth kinetics of A549 cells in vitro, in vivo and on the xenographts vascularization. (A) Proliferation dynamics of cells. Typical result of one of 3 experiments is shown (Mean ± SD). (B) Dynamics of xenographts growth after subcutaneous injection (10 mice per group) of A549 cells. Typical result of one of 3 experiments is given (Mean ± SD, * p ≤ 0,05). (C) Immunohistochemical analysis for CD34 of A549 xenografts with modulated E-cadherin expression. Left panel - quantitative estimation of CD34-positive blood vessels larger 100 μm. 20–25 fields of view in each experimental group were analyzed (Mean ± SD, * p ≤ 0,05). Right panel - representative fields of view for each experimental group. Spikes – CD34-positive structures. Bars 100 μm.
It should be stressed, that A549 cells expressing shGFP was used as control in all experiments, and they did not differ from parental A549 culture in any analyzed characteristics or tests and showed similar results.
Discussion
To ascertain the effects of E-cadherin expression modulation on acquisition of cancer stem cell phenotype we generated the A549 human non-small-cell lung carcinoma (NSCLC) cells with different levels of E-cadherin expression: E-cadherinhigh and E-cadherinlow. We noticed that E-cadherinlow cells lost epithelial and acquired some mesenhymal features, such as slight increase of vimentin expression accompanied by twisted and strengthened vimentin filament network and enhanced migratory capacity. These findings confirmed that E-cadherinlow cell lines could acquire some features of epithelial-to-mesenhymal transition (EMT) whereas E-cadherinhigh cell lines and control cells retained epithelial phenotype.
As far as we noticed that cell lines with downregulated E-cadherin changed their morphology we examined how changes in E-cadherin expression would affect growth kinetics in vitro and in vivo. E-cadherin upregulation caused some deceleration of growth rate both in vitro and in vivo, while E-cadherin downregulation increased the growth rate, and the effect was particularly strong in vivo. In fact, xenografts with reduced CDH1 expression showed approximately 2-fold increases in final tumors volumes. As tumor vascularization is considered to be a major limiting factor of tumor development45,46 we examined the density of blood vessels in tumor xenografts. We demonstrated that the density of large vessels (larger than 100 µm) was significantly increased in E-cadherinlow tumors at least partially explaining their faster growth and larger sizes.
Conducting xenograft assay we noticed that the threshold cell dose for E-cadherinlow cell subline was significantly lower than that for control cell line while on the contrary the threshold dose for E-cadherinhigh cells was higher than for control cell lines. That is why we proposed that E-cadherin downregulation could lead to an increase of the proportion of CSCs as the ability to form new tumors is considered to be the main characteristic of CSCs. To prove this we performed a series of experiments that were believed to serve as a so-called “gold standard” for assessing the percentage of the CSCs: determined the cell dose threshold for tumor formation in nude mice; detected cells that effectively excluded Rhodamine123 and assessed colony formation in semisolid medium.44 As the result of all 3 tests we proved that E-cadherinlow A549 cells were enriched with CSCs, while in E-cadherinhigh cells the percentage of CSCs was reduced. This allows us to suggest that loss of E-cadherin expression could lead to the acquisition of CSCs phenotype.
To obtain more characteristics for cells with altered E-cadherin expression we analyzed expression of some stem cell markers, such as OCT3/4, SOX2, Nestin and the oncogene c-MYC. We observed elevated levels of mRNAs of all these genes in E-cadherinlow cells. So, downregulation of E-cadherin results in elevation of genes that are believed to maintain stem state of the cell. We suppose that E-cadherin downregulation might promote the increased expression of stem-maintaining transcriptional factors Oct3/4 and SOX2, which in turn could play a role in acquisition of cancer stem cell features. The influence of E-cadherin expression on CSCs phenotype is summarized on the scheme (Fig. 6). A549 cells were additionally cloned and after several passages different clones showed similar, between themselves and initial cell culture, characteristics in CSCs functional tests (data not shown). These results are in accordance with the hypothesis of dynamic CSCs status, dependent on different intra- and extra-cellular conditions.26-29
Figure 6.
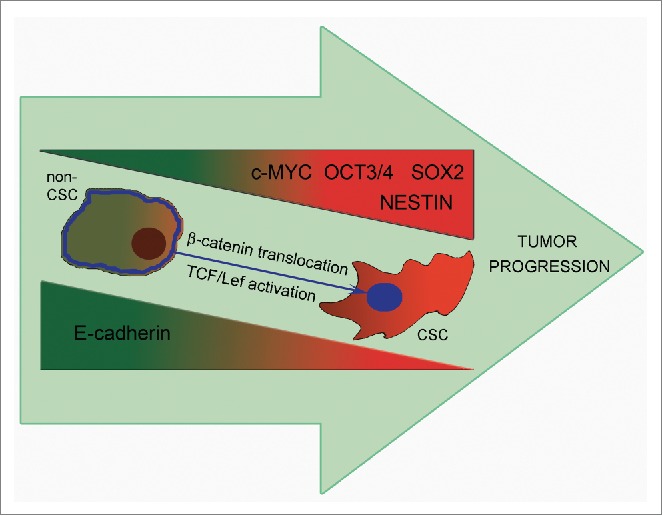
Upon tumor progression E-cadherin downregulation leads to an increase of the proportion of CSCs and acquisition of the ability to form secondary foci of tumor growth. E-cadherin repression results in elevation of genes maintaining stemness – Oct3/4, SOX2, Nestin and c-Myc playing a role in acquisition of CSCs features. A possible mechanism for the enhancement of CSCs properties may be the translocation of β-catenin to the nucleus and activation of the Wnt/β-catenin pathway (partly through c-MYC increased transcription).
Taken together the results of our work show that downregulation of CDH1 increased while its upregulation decreased various cell features ascribed to CSCs, including tumorigenic potential. This allowed considering changes in E-cadherin intercellular adhesions as one of the molecular determinants responsible for dynamic increase/decrease of manifestations of CSCs phenotype. Since CSCs are the driving force for tumor progression and metastasis, elimination of this tumor cell population seems to be extremely important in anticancer therapy. We believe that restoring E-cadherin function could be relatively safe in this respect.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
KPB is grateful to the Russian Science Foundation (RSCF), grant No. 14-15-00467 for support.
References
- [1].Stemmler MP. Cadherins in development and cancer. Mol Biosyst 2008; 4(8):835-50; PMID:18633485; http://dx.doi.org/ 10.1039/b719215k [DOI] [PubMed] [Google Scholar]
- [2].van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008; 65(23):3756-88; PMID:18726070; http://dx.doi.org/ 10.1007/s00018-008-8281-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rodriguez FJ, Lewis-Tuffin LJ, Anastasiadis PZ. E-cadherin's dark side: possible role in tumor progression. Biochim Biophys Acta 2012; 1826(1):23-31; PMID:22440943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chun N, Ford JM. Genetic testing by cancer site: stomach. Cancer J 2012; 18(4):355-63; PMID:22846738; http://dx.doi.org/ 10.1097/PPO.0b013e31826246dc [DOI] [PubMed] [Google Scholar]
- [5].Corso G, Marrelli D, Pascale V, Vindigni C, Roviello F. Frequency of CDH1 germline mutations in gastric carcinoma coming from high- and low-risk areas: metanalysis and systematic review of the literature. BMC Cancer 2012; 12:8; PMID:22225527; http://dx.doi.org/ 10.1186/1471-2407-12-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Spaderna S, Schmalhofer O, Hlubek F, Jung A, Kirchner T, Brabletz T. Epithelial-mesenchymal and mesenchymal-epithelial transitions during cancer progression. Verh Dtsch Ges Pathol 2007; 91:21-8; PMID:18314592 [PubMed] [Google Scholar]
- [7].Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev 2009; 28(1–2):151-66; PMID:19153669; http://dx.doi.org/ 10.1007/s10555-008-9179-y [DOI] [PubMed] [Google Scholar]
- [8].Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res 2010; 8(5):629-42; PMID:20460404; http://dx.doi.org/ 10.1158/1541-7786.MCR-10-0139 [DOI] [PubMed] [Google Scholar]
- [9].Kim A, Bae YK, Gu MJ, Kim JY, Jang KY, Bae HI, Lee HJ, Hong SM. Epithelial-mesenchymal transition phenotype is associated with patient survival in small intestinal adenocarcinoma. Pathology 2013; 45(6):567-73; PMID:24018801; http://dx.doi.org/ 10.1097/PAT.0b013e3283650bab [DOI] [PubMed] [Google Scholar]
- [10].Bae YK, Choi JE, Kang SH, Lee SJ. Epithelial-mesenchymal transition phenotype is associated with clinicopathological factors that indicate aggressive biological behavior and poor clinical outcomes in invasive breast cancer. J Breast Cancer 2015; 18(3):256-63; PMID:26472976; http://dx.doi.org/ 10.4048/jbc.2015.18.3.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kobayashi H, Sugimoto H, Onishi S, Nakano K. Novel biomarker candidates for the diagnosis of ovarian clear cell carcinoma. Oncol Lett 2015; 10(2):612-18; PMID:26622542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 2006; 172(7):973-81; PMID:16567498; http://dx.doi.org/ 10.1083/jcb.200601018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139(5):871-90; PMID:19945376; http://dx.doi.org/ 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- [14].Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119(6):1420-8; PMID:19487818; http://dx.doi.org/ 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al.. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013; 339(6119):580-4; PMID:23372014; http://dx.doi.org/ 10.1126/science.1228522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Thiery JP, Lim CT. Tumor dissemination: an EMT affair. Cancer Cell 2013; 23(3):272-3; PMID:23518345; http://dx.doi.org/ 10.1016/j.ccr.2013.03.004 [DOI] [PubMed] [Google Scholar]
- [17].Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al.. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports 2013; 2(1):78-91; PMID:24511467; http://dx.doi.org/ 10.1016/j.stemcr.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al.. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133(4):704-15; PMID:18485877; http://dx.doi.org/ 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Abell AN, Johnson GL. Implications of mesenchymal cells in cancer stem cell populations: relevance to EMT. Curr Pathobiol Rep 2014; 2(1):21-26; PMID:25530923; http://dx.doi.org/ 10.1007/s40139-013-0034-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009; 9(4):265-73; PMID:19262571; http://dx.doi.org/ 10.1038/nrc2620 [DOI] [PubMed] [Google Scholar]
- [21].Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29(34):4741-51; PMID:20531305; http://dx.doi.org/ 10.1038/onc.2010.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu X, Fan D. The epithelial-mesenchymal transition and cancer stem cells: functional and mechanistic links. Curr Pharm Des 2015; 21(10):1279-91; PMID:25506898; http://dx.doi.org/ 10.2174/1381612821666141211115611 [DOI] [PubMed] [Google Scholar]
- [23].Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414(6859):105-11; PMID:11689955; http://dx.doi.org/ 10.1038/35102167 [DOI] [PubMed] [Google Scholar]
- [24].Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 2012; 22(5–6):396-403; PMID:22554795; http://dx.doi.org/ 10.1016/j.semcancer.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8(10):755-68; PMID:18784658; http://dx.doi.org/ 10.1038/nrc2499 [DOI] [PubMed] [Google Scholar]
- [26].Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med 2009; 15(9):1010-2; PMID:19734877; http://dx.doi.org/ 10.1038/nm0909-1010 [DOI] [PubMed] [Google Scholar]
- [27].Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011; 146(4):633-44; PMID:21854987; http://dx.doi.org/ 10.1016/j.cell.2011.07.026 [DOI] [PubMed] [Google Scholar]
- [28].Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al.. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011; 108(19):7950-5; PMID:21498687; http://dx.doi.org/ 10.1073/pnas.1102454108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem 2013; 59(1):168-79; PMID:23220226; http://dx.doi.org/ 10.1373/clinchem.2012.184655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, et al.. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011; 147(4):759-72; PMID:22078877; http://dx.doi.org/ 10.1016/j.cell.2011.09.048 [DOI] [PubMed] [Google Scholar]
- [31].Cai C, Zhu X. The Wnt/β-catenin pathway regulates self-renewal of cancer stem-like cells in human gastric cancer. Mol Med Rep 2012; 5(5):1191-6; PMID:22367735 [DOI] [PubMed] [Google Scholar]
- [32].Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006; 66(12):6063-71; PMID:16778178; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-0054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol 2009; 7(6):e1000121; PMID:19492080; http://dx.doi.org/ 10.1371/journal.pbio.1000121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009; 138(4):645-59; PMID:19682730; http://dx.doi.org/ 10.1016/j.cell.2009.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ye J, Wu D, Shen J, Wu P, Ni C, Chen J, Zhao J, Zhang T, Wang X, Huang J. Enrichment of colorectal cancer stem cells through epithelial-mesenchymal transition via CDH1 knockdown. Mol Med Rep 2012; 6(3):507-12; PMID:22684815 [DOI] [PubMed] [Google Scholar]
- [36].Bae KM, Su Z, Frye C, McClellan S, Allan RW, Andrejewski JT, Kelley V, Jorgensen M, Steindler DA, Vieweg J, et al.. Expression of pluripotent stem cell reprogramming factors by prostate tumor initiating cells. J Urol 2010; 183(5):2045-53; PMID:20303530; http://dx.doi.org/ 10.1016/j.juro.2009.12.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Khromova N, Kopnin P, Rybko V, Kopnin BP. Downregulation of VEGF-C expression in lung and colon cancer cells decelerates tumor growth and inhibits metastasis via multiple mechanisms. Oncogene 2012; 31(11):1389-97; PMID:21804602; http://dx.doi.org/ 10.1038/onc.2011.330 [DOI] [PubMed] [Google Scholar]
- [38].Chitaev NA, Troyanovsky SM. Adhesive but not lateral E-cadherin complexes require calcium and catenins for their formation. J Cell Biol. 1998; 142(3):837-46; PMID:9700170; http://dx.doi.org/ 10.1083/jcb.142.3.837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zamkova M, Khromova N, Kopnin BP, Kopnin P. Ras-induced ROS upregulation affecting cell proliferation is connected with cell type-specific alterations of HSF1/SESN3/p21Cip1/WAF1 pathways. Cell Cycle 2013; 12(5):826-36; PMID:23388456; http://dx.doi.org/ 10.4161/cc.23723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ying L, Mills JA, French DL, Gadue P. OCT4 coordinates with WNT signaling to pre-pattern chromatin at the SOX17 locus during human ES cell differentiation into definitive endoderm. Stem Cell Reports 2015; 5(4):490-8; PMID:26411902; http://dx.doi.org/ 10.1016/j.stemcr.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang P, Chang WH, Fong B, Gao F, Liu C, Al Alam D, Bellusci S, Lu W. Regulation of induced pluripotent stem (iPS) cell induction by Wnt/β-catenin signaling. J Biol Chem 2014; 289(13):9221-32; PMID:24482235; http://dx.doi.org/ 10.1074/jbc.M113.542845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yi XJ, Zhao YH, Qiao LX, Jin CL, Tian J, Li QS. Aberrant Wnt/β-catenin signaling and elevated expression of stem cell proteins are associated with osteosarcoma side population cells of high tumorigenicity. Mol Med Rep 2015; 12(4):5042-8; PMID:26134785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dahlrot RH, Hermansen SK, Hansen S, Kristensen BW. What is the clinical value of cancer stem cell markers in gliomas? Int J Clin Exp Pathol 2013; 6(3):334-48; PMID:23412423 [PMC free article] [PubMed] [Google Scholar]
- [44].Richard V, Nair MG, Santhosh Kumar TR, Pillai MR. Side population cells as prototype of chemoresistant, tumor-initiating cells. Biomed Res Int 2013; 2013:517237; PMID:24294611; http://dx.doi.org/ 10.1155/2013/517237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 2007; 8(6):464-78; PMID:17522591; http://dx.doi.org/ 10.1038/nrm2183 [DOI] [PubMed] [Google Scholar]
- [46].Khromova NV, Kopnin PB, Stepanova EV, Agapova LS, Kopnin BP. p53 hot-spot mutants increase tumor vascularization via ROS-mediated activation of the HIF1/VEGF-A pathway. Cancer Lett 2009; 276(2):143-51; PMID:19091459; http://dx.doi.org/ 10.1016/j.canlet.2008.10.049 [DOI] [PubMed] [Google Scholar]