

ABSTRACT
Natural killer (NK) cells are now recognized to exhibit characteristics akin to cells of the adaptive immune system. The generation of adaptive memory is linked to epigenetic reprogramming including alterations in DNA methylation. The study herein found reproducible genome wide DNA methylation changes associated with human NK cell activation. Activation led predominately to CpG hypomethylation (81% of significant loci). Bioinformatics analysis confirmed that non-coding and gene-associated differentially methylated sites (DMS) are enriched for immune related functions (i.e., immune cell activation). Known DNA methylation-regulated immune loci were also identified in activated NK cells (e.g., TNFA, LTA, IL13, CSF2). Twenty-one loci were designated high priority and further investigated as potential markers of NK activation. BHLHE40 was identified as a viable candidate for which a droplet digital PCR assay for demethylation was developed. The assay revealed high demethylation in activated NK cells and low demethylation in naïve NK, T- and B-cells. We conclude the NK cell methylome is plastic with potential for remodeling. The differentially methylated region signature of activated NKs revealed similarities with T cell activation, but also provided unique biomarker candidates of NK activation, which could be useful in epigenome-wide association studies to interrogate the role of NK subtypes in global methylation changes associated with exposures and/or disease states.
KEYWORDS: Differentially methylated regions, digital PCR, DNA methylation, innate immunity, NK cells, NKp46
Abbreviations
- NK
natural killer cell
- ddPCR
droplet digital polymerase chain reaction
- DMR
differentially methylated region
- DMS
differentially methylated site
- EWAS
epigenome wide association study
- TCR
T cell receptor
- CMV
cytomegalovirus
- LCMV
lymphocytic choriomeningitis virus
- CpG
cytosine guanine dinucleotide
- T1D
type I diabetes
- NKT
natural killer T cell
- NCR
natural killer cell receptor
- MS
multiple sclerosis
- MS-PCR
methylation specific polymerase chain reaction
- GWAS
genome-wide association study
- PMA
phorbol 12-myristate 13-acetate
- FACS
fluorescence-activated cell sorting
- IL2
interleukin 2
- CSF2
granulocyte-macrophage colony-stimulating factor
- MALT
mucosa-associated lymphoid tissue.
Introduction
Epigenetic modifications, including DNA methylation, dictate programmed lineage differentiation within the immune system.1 Remodeling the epigenome during development leads to progressively restricted immune subtypes. DNA methylation is thought to provide a chemically stable mark that serves to initiate and reinforce these cell fate decisions.2 Large-scale methylation studies of specific lineage pathways have been performed on differentiating erythroid,3,4 T lymphocyte,5,6 B lymphocyte,7 and myeloid4 cells. These important developmental studies reveal insights into the regulation of cell fate decisions but also serve as a potential source of biomarkers that help characterize different immune compartments during the immune response. The best known example of a single gene epigenetic marker of immune cell lineage is demethylation within the FOXP3 gene that provides a marker of CD4 T regulatory cells (Treg).8 FOXP3 expression drives the transcriptional program of Tregs.5 Another example of lineage specific methylation, which marks human natural killer cells (NK), is represented by demethylated CpG sites within the NK cell activation receptor NCR1/NKp46 gene locus that predicts the numbers and proportions of NK cells in blood and tissue.9
Less understood is the extent to which stable and heritable changes in DNA methylation might be produced during immune activation within cells of a common lineage identity. Although transcriptomic studies implicate genome wide alterations in gene expression during pathogen- and cytokine-induced immune activation in different cell types,10-14 the exact role of DNA methylation in the genesis of these effects is not known. However, a few individual loci have been studied intensively. For example, the production of interferon γ (IFNG) in response to IL2 treatments or T cell receptor (TCR) activation leads to the demethylation of the IFNG locus in CD4 Th1 cells.15,16 Studies of CD8 T cell activation and memory generation point to a profound plasticity in DNA methylation within the CD8A TCR co-receptor locus,17 which correlates with different cytokine exposures and conditions of Th1/2 polarization. Studies using broader CpG scans showed that CD4 helper T cells undergo dynamic and flexible methylation changes both in a genome-wide and a targeted manner during T cell activation and differentiation.18 In addition, ex vivo differentiation of human CD14 blood monocytes to immature and mature dendritic cells led to significant remodeling of the monocyte/macrophage methylome.19
Although the NK cell methylome has not been extensively studied during cytokine or NCR receptor activation, some work has been done with cytomegalovirus (CMV) infected mice and humans. A clonal expansion of NKG2C receptor positive NK cells has been shown to occur, and in some HCMV infected individuals, subsets of memory-like NK cells deficient in B-cell and myeloid signaling proteins, including the tyrosine kinase SYK, have been identified. Furthermore, the downregulation of SYK occurred in the context of DNA hypermethylation within a restricted segment of the SYK gene promoter.20 HCMV-associated NK cells also displayed deficiencies and hypermethylation in signaling adaptors such as EAT-2 and FCER1G as well as the ZBTB16/PLZF transcription factor.21
We predicted that remodeling of the NK methylome following NKp46 engagement would be a feature of NK activation based on the prior demonstration that NK cells display a type of immunologic memory that shares many features with CD8 T effector and memory cells following antigen activation.22 The work herein studied the DNA methylation profiles of resting and activated NK cells; NK activation was achieved by ex vivo antibody-ligation of the CD2 and NKp46 receptors and cell culture in the presence of rIL2. We note similarities in the induced methylation alterations with those occurring in other antigen and cytokine stimulated cell lineages.
A specific aim of the study was to identify promising single gene candidates that might provide biomarkers of NK cell activation. We developed a single gene qPCR assay of demethylation within the BHLHE40 transcription factor using the highly sensitive droplet digital PCR (ddPCR) technique23 and consider its potential as a biomarker of NK activation within peripheral whole blood.
Results
Characterization of naïve and activated NK cells
Untouched human NK cells were isolated from a total of 12 individuals over 2 phases of this study (Suppl Fig. 1). Isolated naïve and activated NK cell populations were confirmed by flow cytometric analyses (Suppl Fig. 2). On average, 96.1% (±2.7%) of NK cells were activated at day 9 (n = 12; data not shown) using CD54 as a measure of activation, compared with no activation at T = 0. The killing phenotype of activated cells was confirmed using a target cell lysis assay and the ability of activated cells to demonstrate antibody-dependent cell cytotoxicity measured in the presence and absence of the monoclonal antibody therapeutic agent Rituximab (Fig. 1).
Figure 1.
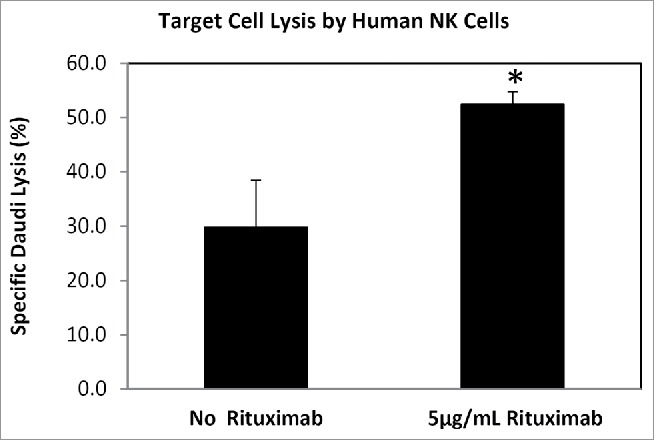
Phenotype confirmation of IL2/CD2/CD335 in vitro activated human NK cells. Activated NK cells were challenged with target cells (the human B-lymphoblast Daudi cell line), at a ratio of 5:1. These activated NK cells killed 29.8 % (±8.6%) of target cells in an antibody-independent manner, and had significantly enhanced killing [52.4% (±2.4%); P-value = 0.01] in the presence of Rituximab. This increased Daudi cell lysis is likely through an ADCC-mediated mechanism as the medium lacks functional complement.
The experimental design of the study was based on a discovery phase followed by a replication phase (Suppl Fig. 1). During the discovery phase, isolated human NK cells were harvested at 3 time points: just after MACS isolation (T = 0), 16 h after initiation of activation (T = 16 h), and 9 d post-activation (T = 9 d). Cell viability, purity, and activation status were verified using flow cytometric techniques (Suppl Fig. 2 and Suppl Table 1). Hierarchical clustering analysis of the discovery phase array data indicated there was no significant difference between the methylation patterns at T = 0 and 16 h (data not shown). Somewhat surprisingly, this included no indication of any altered DNA methylation status for genes known to be involved in cell cycle regulation. Hence, the replication studies focused on T=0 and 9 d post-activation.
NK cells were isolated from 6 additional donors for the replication phase of the study. As with the discovery set, these were analyzed by hierarchical clustering, which was able to determine differential methylation patterns between naïve and activated NK cells (Fig. 2). Additionally, the genomic location of the top 1000 loci was conserved across the discovery and replication data sets (data not shown). The discovery and replication datasets were compared in order to generate a list of the top significant loci in common between these 2 sets (Suppl Table 3). This analysis returned 385 loci containing 259 identified genes, microRNA, and coding regions. Fig. 3 shows the genomic distribution characteristics and locations of the significant probes within gene regions.
Figure 2.

Hierarchical clustering of the most significant differentially methylated loci in in vitro activated human NK cells. [A] A heatmap of β-values illustrating the most significant loci common to the discovery and replication phases were identified, yielding a data set of 385 overlapping loci. In an unsupervised analysis, naïve and activated NK populations were easily differentiated. The naïve group (T = 0) also contains 2 commercially prepared/purchased NK samples (T = 0*) which clustered among the (experimental) isolated naïve NK cells. [B] The gene-associated loci from the 385 overlapping loci all had highly significant q-values (<10−3), so loci were listed by priority category (Suppl Fig. 1B) and the top 27 genes (65 loci) of the list were selected for illustration. This includes all 21 gene-associated loci from Class I and all 44 loci from Class II. Loci and gene names can be found in Suppl Table 3.
Figure 3.
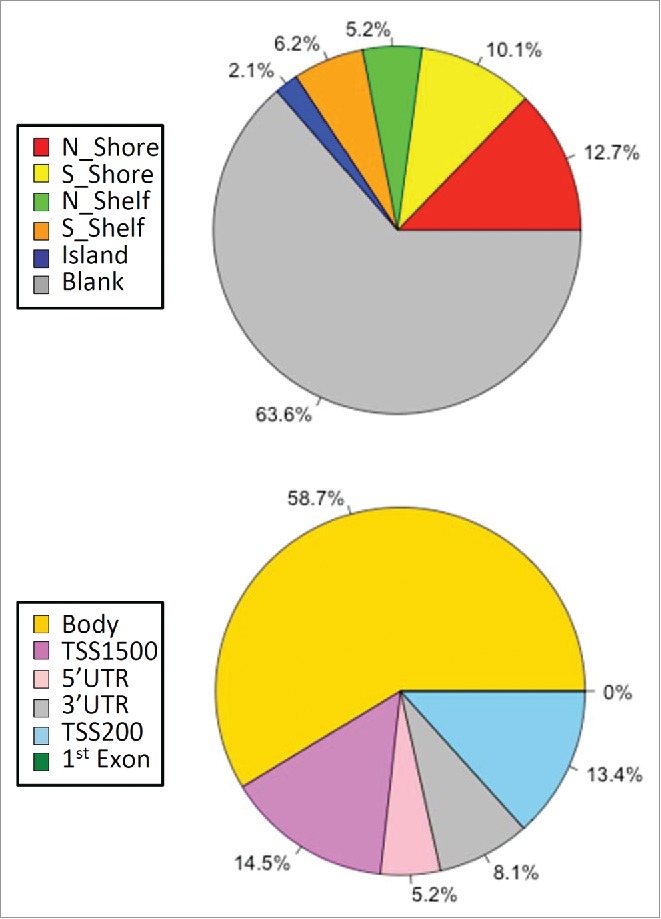
Distribution of significant DMSs by genomic probes and locations. Genomic distribution of Illumina 450K-annotated CpG probe classes (top pie chart) and distinct gene group locations (bottom chart) from the top 385 significant DMSs common to both study phases are illustrated. CpG probe classes were obtained from the Illumina annotation file. Designations reflect the probe location relative to the CpG island (based on UCSC criteria), where north and south shores (N_Shore and S_Shore) reflect 2 kb up- and down-stream of an island, respectively, and “shelf” refers to 2 kb up- and down-stream of the shores (N_Shelf and S_Shelf). “Ocean” has been used to denote probes not assigned to any of the CpG categories by Illumina. The six gene group locations categorized are: TSS1500 (within 1500 bps of a transcription start site “TSS”), TSS200 (within 200 bps of a TSS), body (within a gene), 1st exon, 5′UTR (5′ untranslated region) and 3′UTR.
In an effort to more completely investigate the nature of the altered loci observed in NK activation, we interrogated the lineage of origin of the top 500 replicated loci by applying the algorithm of Houseman24 to a well-known library of the DNA methylation profiles of the common cell types in peripheral blood.25 This algorithm has been validated as a tool to discern lineage of unknown peripheral blood mixtures, using known profiles from isolated cells of a single lineage.26 This analysis revealed that there were 7 DMSs that were found in activated NK cells that were not differentially methylated in other cell types, including cg11783497 (IL1RN), cg07996532 (ECE1), cg17566874 (CSF2), cg05550420 (DCHS1), cg06169746, cg26174398, cg13157980 (EIF2C2). This implies some level of specificity of DNA methylation in activated NK cells (although these genes may be expressed in other immune cell types).
Pathway-based analyses
To identify the most phenotypically important loci, a series of prioritizing criteria were developed (Suppl Fig. 1B) that weighted effect size and regional coherence (multiple significant probes). This identified several genes with considerable differential methylation between naïve and activated NKs; these genes were further analyzed for altered gene expression. These included genes that: i) ranked highest on the priority list (including BHLHE40, MYO1G, NCF4); ii) were reported by others to have altered expression in NKs treated with CMV (such as BHLHE40, KSR1, MAML2, TNFAIP3, ZBTB32);11 iii) appear on our list of top 27 genes (e.g. BHLHE40, IL13, MYO1G and NCF4); and/or iv) genes known to play a role in activation of other immune cells (BHLHE40, KSR1, NCF4). All of the candidates tested displayed significant hypomethylation and significantly increased mRNA expression was observed for 6 of the tested genes while a small but significant decrease in expression was found for one. The remaining 3 genes exhibited no change in expression (Suppl Fig. 4).
Bioinformatics analyses of the 285 most significant coding sequence-associated loci in common between the discovery and replication data sets were assessed for functional annotation enrichment and for pathway associations. Of these 285 coding sequence-associated loci, 278 were successfully mapped by DAVID and generated 15 significant, functionally similar clusters (enrichment score > 1), with the highest enrichment for regulation of leukocyte/lymphocyte/cell activation (enrichment score of 4.03; Table 1A). The functional annotation chart module returned 164 significant (P < 0.05) overrepresented terms (Table 1B). Approximately 50% of these are directly immune function related (e.g., cytokine/cytokine receptor interaction, regulation of leukocyte activation, immune response) while many of the remaining are clearly basic cellular functions, but likely require enhanced activity during cell activation (e.g., regulation of phosphorylation, protein transport and protein secretion, etc.) Additionally, Ingenuity Pathway Analysis using the “canonical pathway” module revealed significant enrichment of immune-related signaling pathways (e.g., NF-kappa β, TNFR2 and IL-10 signaling pathways and cytokine production pathways), differentiation pathways (e.g., T-helper and dendritic cell maturation), as well as disease-related pathways with key immune cell components (e.g., asthma and multiple sclerosis). Significant (Benjamini-Hochberg P-value < 0.05) immune related pathways were observed as the top hits regardless of whether the analysis was constrained by immune cell filters or not (data not shown).
Table 1A.
DAVID functional annotation and enrichment analysis results. DAVID clustering analysis identified 15 significant functional groups ranging in enrichment from 4.03 to 1.65, using the “highest” stringency setting. Several of the highest ranking clusters are associated with cell activation, the expected physiological phenotype.
| Cluster | Description | Enrich-ment score | # genes | P-value range | Genes |
|---|---|---|---|---|---|
| Cluster 1 | positive regulation of leukocyte/lymphocyte/cell activation | 4.03 | 9 | 5.38E-5 – 1.4E-4 | CD38, IL2RA, CD80, CD3E, IL4R, IL13, RARA, ITPKB, TNFRSF4 |
| Cluster 2 | regulation of leukocyte/lymphocyte/cell activation | 3.4 | 10 | 1.97E-4 -6.77E-4 | CD38, IL2RA, CD80, ADORA2A, CD3E, IL4R, IL13, RARA, ITPKB, TNFRSF4 |
| Cluster 3 | regulation of leukocyte/lymphocyte/mononuclear cell proliferation | 2.63 | 6 | 9.84E-4 – 0.006 | CD38, IL2RA, CD80, CD3E, IL13, TNFRSF4 |
| Cluster 4 | tumor necrosis factor | 2.56 | 4 | 0.001 – 0.003 | TNF, TNFSF4, LTA, TNFSF8 |
| Cluster 5 | regulation of lymphocyte/Tcell differentiation | 2.55 | 5 | 0.001 – 0.011 | IL2RA, CD80, IL4R, RARA, ITPKB |
| Cluster 6 | leukocyte/lymphocyte/mononuclear cell proliferation | 2.55 | 5 | 0.002– 0.003 | SHB, ZBTB32, TNFSF4, CCND3, TNFRSF4 |
| Cluster 7 | phosphorylation-associated | 2.42 | 7 | 0.001 – 0.043 | CSF2, CARD14, TNF, CCND3, CD80, CD3E, CD81 |
| Cluster 8 | protein transport and localization | 2.15 | 7 | 0.005 – 0.012 | TNF, TNFSF4, NFKBIE, ADORA2A, IL13, TNFRSF4, LTA |
| Cluster 9 | T-helper/CD4 α β T cell differentiation | 2.12 | 3 | 0.003 – 0.017 | CD80, IL4R, RARA |
| Cluster 10 | C2 membrane targeting protein | 2.11 | 7 | 0.002 – 0.018 | PLCL2, HECW2, RGS3, SYT16, TC2N, RASA3, PRKCB |
| Cluster 11 | regulation of immunoglobulin/cytokine/protein secretion | 2.01 | 3 | 0.006 – 0.020 | TNF, TNFSF4, TNFRSF4 |
| Cluster 12 | regulation of phosphorylation/phosphorous metabolic process | 1.84 | 14 | 0.012 – 0.016 | CSF2, TNF, ADORA2A, CD3E, CCDC88C, NPRL2, TNFRSF4, CISH, CARD14, CD80, CCND3, RGS3, CD81, ZFYVE28 |
| Cluster 13 | cell death/apoptosis | 1.8 | 13 | 0.015 – 0.016 | CD38, PRUNE2, TNFRSF9, AHRR, TNF, IL2RA, ADORA2A, CD3E, MMP9, LGALS12, ARHGEF12, PDCD6, LTA, TNFSF8 |
| Cluster 14 | SANT family (DNA binding) | 1.78 | 4 | 0.005–0.038 | CRAMP1L, TRERF1, RERE, NCOR2 |
| Cluster 15 | calcium/metal ion homeostasis | 1.65 | 8 | 0.013 – 0.046 | S1PR3, CD38, CD55, CCR5, CCR1, TPT1, LOC389787, LOC285741, CCL5, PRKCB |
Table 1B.
DAVID functional annotation and enrichment analysis results. The top 10 most significant (by p-value) out of 164 significant (P-value < 0.05) enriched/overrepresented terms returned by the functional annotation chart module.
| Category | Term | Count | % | P-Value | Benjamini | Genes | Fold Enrich-ment |
|---|---|---|---|---|---|---|---|
| GOTERM_BP_FAT | GO:0051251˜positive regulation of lymphocyte activation | 9 | 0.398 | 5.38E-05 | 0.077 | CD38, IL2RA, CD80, CD3E, IL4R, IL13, RARA, ITPKB, TNFRSF4 | 6.748 |
| SP_PIR_KEYWORDS | phosphoprotein | 120 | 5.300 | 7.28E-05 | 0.023 | KCNC4, LTBP2, ITPKB, TBC1D16, SHB, VPS13D, TPT1, LOC389787, RARA, GNL1, SAMD4A, KIF13B, CDH23, PTPRJ, PDXK, STMN3, CSNK1G1, CD3E, NCF4, TC2N, TRERF1, CECR6, RPTOR, CCDC6, TNS1, CCR5, LOC100128265, TNFAIP3, SH3GL1, C9ORF25, TM4SF19, NFKBIE, CRAMP1L, CAPZB, TAGLN3, PRUNE2, ECE1, CNR2, BHLHE40, RAP1GAP2, IFT140, SREBF1, ASPSCR1, DNMT3A, TRIP4, CPA6, OSBPL8, LPP, PMF1, GAS7, ARMC2, NOP14, CD55, SYNE2, RGS3, KIAA0922, CD59, GRK6, HIVEP3, SPTBN2, BEGAIN, SLC5A10, NCOR2, RERE, E2F1, E2F2, HECW2, FOXK1, LOC729799, PDIA6, C12ORF32, MBP, SLC1A2, TRIM3, IL4R, CASP7, ANKRD11, TNIP2, FAM83G, NSF, SLC43A2, CTBP2, CCDC88C, ARHGEF12, LDLRAP1, RAD50, TTF2, SIGIRR, PRKCB, TNFRSF9, CARD14, CCND3, ZFYVE28, SRGAP3, SCN11A, KSR1, PPP1R15A, WDR46, SEMA4A, TNF, CCR1, CREM, ZBTB12, SEC14L1, PLCL2, RGS12, PLEKHG6, KRT7, PIK3R6, LOC285741, RASA3, NFATC1, BGLAP, IL2RA, NFASC, DPYSL3, DPYSL2, TAB2, DUSP4, PDE2A, RPS6KA1, RASSF1, NFIC, SETD3, ARAP1 | 1.329 |
| KEGG_PATHWAY | hsa04060:Cytokine-cytokine receptor interaction | 15 | 0.663 | 9.22E-05 | 0.009 | CSF2, TNFSF4, IL2RA, TNF, CCR1, IL21R, IL13, CCL5, TNFRSF4, CCL17, TNFSF8, TNFRSF9, CCR5, IL4R, LTA | 3.385 |
| GOTERM_BP_FAT | GO:0002696˜positive regulation of leukocyte activation | 9 | 0.398 | 1.01E-04 | 0.073 | CD38, IL2RA, CD80, CD3E, IL4R, IL13, RARA, ITPKB, TNFRSF4 | 6.175 |
| GOTERM_BP_FAT | GO:0046634˜regulation of α-β T cell activation | 6 | 0.265 | 1.21E-04 | 0.058 | CD80, ADORA2A, CD3E, IL4R, RARA, ITPKB | 12.12 |
| GOTERM_BP_FAT | GO:0050867˜positive regulation of cell activation | 9 | 0.398 | 1.40E-04 | 0.051 | CD38, IL2RA, CD80, CD3E, IL4R, IL13, RARA, ITPKB, TNFRSF4 | 5.897 |
| GOTERM_BP_FAT | GO:0006954˜inflammatory response | 15 | 0.663 | 1.52E-04 | 0.044 | TNFSF4, IL2RA, TNF, ADORA2A, CCR1, IL1RN, IL13, CCL5, TNFRSF4, SIGIRR, CCL17, S1PR3, CD55, CCR5, CNR2 | 3.357 |
| GOTERM_BP_FAT | GO:0001775˜cell activation | 14 | 0.618 | 1.61E-04 | 0.039 | CSF2, ZBTB32, TNFSF4, TNF, ADORA2A, CD3E, IL21R, ITPKB, TNFRSF4, SHB, CD80, CCND3, LTA, KIF13B | 3.548 |
| GOTERM_BP_FAT | GO:0006955˜immune response | 23 | 1.016 | 1.88E-04 | 0.039 | CSF2, GALNT2, TNF, TNFSF4, IL2RA, NCF4, CCR1, IL1RN, IL13, CCL5, TNFRSF4, SIGIRR, CCL17, MBP, TNFSF8, CD96, CD55, CCR5, IL4R, CNR2, TGFBR3, GNL1, LTA | 2.424 |
| GOTERM_BP_FAT | GO:0051249˜regulation of lymphocyte activation | 10 | 0.442 | 1.97E-04 | 0.036 | CD38, IL2RA, CD80, ADORA2A, CD3E, IL4R, IL13, RARA, ITPKB, TNFRSF4 | 4.914 |
All but one of these is significant using the conservative Benjamini-Hochberg multiple comparison correction for false discovery rate. Most terms are directly immune function related, primarily associated with activation. Both this and the analysis shown in Table 1A confirm the importance of the gene-associated DMSs identified in our analysis as playing an important role in NK cell activation.
The frequency of differential methylation within the transcription factor binding sites for 161 transcription factors was assessed using bioinformatics techniques to mine the ENCODE database (Suppl Table 6). We hypothesized there would be enrichment for a limited number of transcription factors that might suggest their importance in mediating the activation phenotype. Separate exploration of significant hypo- and hypermethylated DMS revealed a surprisingly similar list of transcription factor binding sites perturbed by changes in methylation. Significant associations involved EP300, CEBPB, FOS, MYC, GATA2, STAT3, RUNX3 and others. EP300, STAT3, and RUNX3 play well-known roles in NK differentiation; however, they are also expressed in other immune cell lineages. A novel observation in our data was the common occurrence of GATA2 among the TFBSs; GATA2 has recently been shown essential in the terminal maturation of CD56 dim NK cells that are most enabled for cytotoxicity (Suppl Table 6). Two TFBS perturbations were unique to the hypermethylated CpG: GRp20 (glucocorticoid receptor) and ZKSCAN1. There were 16 TFBS that had hypomethylated DMS that were not altered by hypermethylation.
In other functional analyses, the 100 non-coding loci were evaluated using the GREAT software application (Suppl Table 7). Results of these analyses align with alterations in immune activity, including iNKT development, T cell effector and memory function and STAT3 signaling. This analysis also revealed enrichment of targets of miR-155, including SOCS1. Recent studies show that miR-155 is upregulated in NK cells by activating receptor ligation and cytokines (IL-12, IL-15, IL-18) and regulates INFG production. Interestingly miR-155 is involved in the expansion of virus-specific NK cells, the latter being mediated through specific inhibition of SOCS1.27 Taken together, these annotation analyses confirm the importance of the differentially methylated loci identified in our study as playing an important role in NK cell activation.
Further analysis was made by comparison of the most significant 385 differentially methylated loci associated from this study to a known activated NK model cell line, the genetically altered NK-92 cells (Conkwest). Unlike the parental NK-92 strain, the CD16.176V.NK-92 line has been transfected to express the high affinity variant of the CD16 Fc receptor, making them optimized for ADCC. A hierarchical clustering analysis of freshly isolated naïve NK, experimentally activated NK, and genetically modified NK-92 strain showed, as expected, the NK-92 to cluster more closely with the chemically activated NKs than naïve cells (Fig. 4). To better understand the similarity between these cells optimized for ADCC and the in vitro activated NK cells, a comparison of significant differentially methylated sites from both groups was performed. The analysis revealed an overlap of 169 loci, 111 of which were associated with gene/coding sequence and 78 loci (representing 58 coding sequences) unique to NK-92 (Suppl Table 4). Based on the DAVID functional enrichment analysis of this overlapping dataset, the most significant enriched functional annotations were biological processes of immune response, inflammatory response, positive regulation of lymphocyte activation and regulation of lymphocyte activation (P-value range: 9.07 × 10−7 to 3.65 × 10−5; data not shown). The functional annotation clustering identified the most important members were associated with positive regulation of lymphocyte, leukocyte and cell activation (3 separate “GOTERM_BP_FAT” terms), with an enrichment score of 4.34 (data not shown). Enrichment analysis of the 58 sequence-associated DMSs for NK-92 revealed a significant enrichment of genes for cell death/apoptosis (enrichment score 1.75) and transcription regulation (enrichment score 1.22; data not shown).
Figure 4.

Hierarchical clustering of DMSs from fresh isolated naïve human NK, in vitro activated isolated human NK cells and genetically enhanced NK-92 cells. The 50 most significant differentially methylated loci were determined for the NK-92 cell line relative to naïve human NK cells (from the replication dataset). As expected, the NK-92 cells, which are genetically primed for killing by expressing the high affinity variant of the CD16 Fc receptor, clustered more closely with the activated isolated NK cells than with their naïve counterparts. (The list of loci can be obtained in Supplemental Table 5.)
Development of a biomarker for active NK cells
A number of potential markers of activated NK cells were tested for suitability for a digital PCR assay. In an effort to rank order the changes in DNA methylation, the Illumina HumanMethylation450 BeadChip DMS candidates were prioritized by statistical significance (P- and q-values), extent of coverage (multiple positive probes), effect size, known biological role, and novelty (data not shown). Of the trial candidates, BHLHE40 showed the greatest potential for assay design. A region of exon 5 that included 3 CpGs interrogated by the array and 25 other CpGs, was bisulfite sequenced (Fig. 5). A combination of differential methylation and sequence amenable to assay design resulted in the use of 2 particular CpGs as the target region to assess the activation phenotype.
Figure 5.

Bisulfite sequence for exon 5 of BHLHE40 and validation of the ddPCR methylation assay. Over nineteen separate clones were isolated and bisulfite sequenced for naïve and activated NK cells for 3 individuals. A schematic diagram of the location of sequencing and the corresponding dot plot of results for the first 28 CpGs of BHLHE40 exon 5, are shown in panel A. For reference, the CpGs interrogated by the Illumina HumanMethylation450 beadchip have boxes around the CpG position (CpGs 9, 15, and 24) and are shown as gray bars in panel B. [B] Summary bar graph of the differential methylation at each of the 28 CpGs sequenced, revealing a pattern of lower methylation in activated NK cells compared to naïve cells. Data revealed an area within exon 5 to be an excellent target region for assessment of the NK activation phenotype; 2 loci (highlighted in dark gray, panel A) were incorporated into the final ddPCR assay, which demonstrated a close linear correspondence with DNA sequenced reference DNA [C].
The newly developed BHLHE40 demethylation ddPCR assay was applied to the naïve and activated NK cells from 3 donors (of the 12 used in the NK study; Fig. 6). Additionally, 8 different isolated cell types were assessed to determine the specificity of BHLHE40 demethylation as a marker for activated NK cells (not subjects from this study; Fig. 6). The in vitro activated NK cells consistently measured ∼60% demethylation as compared with ∼15–35% demethylation from naïve NKs. Some demethylation was also detectable in other isolated cell types, primarily from T- and B-cells. The robustness of the assay was also assessed by measuring BHLHE40 demethylation in whole blood samples from 6 individual donors (not subjects from this study; Fig. 6 inset). The assay demonstrated linear kinetics in a 3 log dilution series (Rsquare > 0.98; data not shown).
Figure 6.
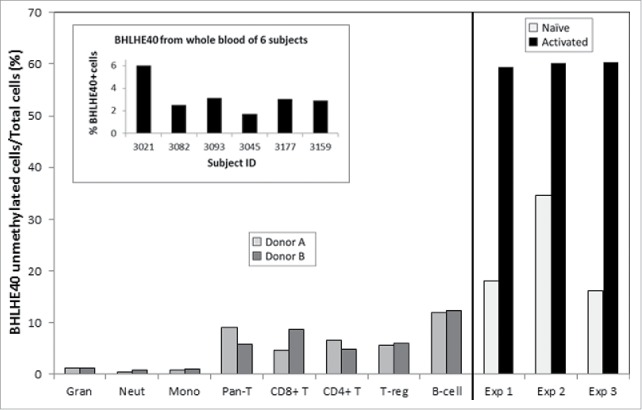
BHLHE40 ddPCR assay in isolated naïve and activated NK pairs, various leukocyte subtypes and in whole blood. The newly developed ddPCR assay for a BHLHE40 demethylation marker was tested on: a subset of the experimental naïve and activated NK cell pairs (far right graph); on isolated cell types (2 different donors per cell type); on whole blood samples from 6 different individuals (inset). Activated NK cells show significant demethylation relative to naïve NKs, as well as in comparison to other immune cell types. The ddPCR assay robustness was demonstrated by the ability to measure BHLHE40 hypomethylation in the complex mixture of whole blood DNA.
Discussion
The Illumina HumanMethylation450 BeadChip arrays were applied to paired donor primary naïve and in vitro activated NK cells in an effort to characterize the profile of DNA alterations associated with NK activation. This same methodology can be utilized to investigate and develop additional biomarkers of NK activation.
Long-term but not short-term activation leads to methylome remodeling
A significant observation from these experiments is that NK cells display no detectable changes in DNA methylation 16 h following activation with antibody mediated receptor ligation and IL2 stimulation. In addition, no DNA methylation changes in genes associated with cell cycle regulation were observed. The short-term kinetics of DNA methylation changes in stimulated immune cells has been controversial. While rapid changes in gene expression have been described in naïve T cells differentiating to effector and memory cells, little consensus exists on the methylation alterations that accompany these early events.10,13 Our observations at 16 h are consistent with previously published data studying CD4 T cells undergoing in vitro activation with CD3 and CD28 antibodies.6 Further studies are necessary to determine if the lack of early kinetic change in methylation is due to steps involving active TET-mediated hydroxymethylation or a passive dilution and lack of methylation maintenance. In contrast to the lack of short-term changes, NK cells at day 9 post-activation (this study) showed great numbers of highly significant changes in DNA methylation, reflecting a broad remodeling of the methylome.
A discovery and replication experimental design was employed, applying strict false discovery rate thresholds, that identified significant differential methylation (predominately hypomethylation) localizing to intragenic regions. We identified several hypomethylated loci that are among those previously shown to be differentially methylated in the immune system, whereas, many other loci discovered have never been associated with differential methylation in activated immune cells, including NKs. The functional annotation analyses confirmed the biological relevance of the differentially methylated loci to NK cell activation. The 111 gene/coding sequence-associated loci found in common between the in vitro activated human NK cells and the genetically modified NK-92 cell line enhanced for ADCC may represent candidate genes (and critical loci) that will be useful in pharmacologic manipulation to enhance NK activation.
Established gene regions regulated by differential DNA methylation
A prominent focus of demethylation in NK cell activation involved the chromosome 6p21.3 region that contains the lymphotoxin α (LTA), tumor necrosis factor α (TNFA) and lymphotoxin β (LTB) genes. This 10 kb region has been intensively studied for epigenetic regulation of pivotal inflammatory mediators.28 Both LTA and TNFA were demethylated in our activated NK cells. Previous work showed that the DNA methylation status of the proximal promoter of the TNFA gene is highly correlated with gene transcription.28 Three sites within exon 1 of LTA in NK cells overlapped with loci demethylated within naïve CD4 T cells in Sjogren's syndrome.29
This study found additional members of the TNF family outside the 6p21 locus were also differentially methylated in activated NK cells. These include members of the TNF (ligand) superfamily, members 4, 8, and 10, and TNF receptor superfamily members 4 and 9. All of these exhibited significantly hypomethylated loci in activated NK cells except TNFSF8, which showed a significant hypermethylated DMS upon activation. Changes in methylation status for TNFRSF4, 9 and TNFA were observed during Treg differentiation, also exhibiting demethylation.30 Table 2 illustrates more examples of differentially methylated genes from other activated immune cells that appear to be common with NK cell activation.30-39
Table 2.
Examples of stimulatory or activating molecules common to NK and other immune cells.
| Gene | Role | Ref |
|---|---|---|
| CD80 | Primary co-stimulatory molecule in T-cell cytotoxicity | Tatari-Calderone, Semnani et al., 200231 |
| CD81 and CD82 | Tetraspanin proteins involved in cytoskeletal rearrangement associated with TCR activation | Levy S, 201433 |
| CD96 | Late phase T and NK cell immune response involving adhesive interactions with CD155/poliovirus receptor | Chan, Martinet et al., 201434 Martinet and Smyth, 201535 |
| IL21R | Binding of ligand (IL21) promotes growth and differentiation in T, B and NK cells; ligand binding induces various signaling pathways including JAK/STAT | Kratholm, Iverson, et al., 201336 |
| KSR1 | Scaffolding protein with role in MAPK signaling pathway (T-cell activation); hypomethylation observed in Treg activation | Zhang, Maksimovic et al., 201330 |
| RPTOR | mTORC1 component; Coordinate regulation of immunological activity and metabolic transition that accompanies cell activation, effector differentiation and proliferation; hypomethylation observed in Treg activation | Man and Kallies, 201537 ; Zhang, Maksimovic et al., 201330 |
| SEMA4A | Among other functions, immunoregulatory; plays a role in DC and T cell activation, cell-cell interaction, migration and differentiation (Th1) | Vadasz and Toubi, 201432 |
| TNFA, TNFSF4, TNFRSF4, TNFRSF9 | Survival and activation signaling; hypomethylation of TNF/TNFR superfamily members with Treg activation | Hanna, Bechtel et al., 200438; Zhang, Maksimovic et al., 201330 |
| ZBED2 (antisense CD96) | Function largely unknown, but expression array meta-analysis indicates high enrichment in Th1 and 2 cells | Benita, Cao et al., 201039 |
This study identified differential methylation in a number of genes with known stimulatory or activation functions in other immune cells. The table provides several examples of such genes and their roles. The literature consistently reveals striking commonality between NK and T cells.
The Th2 cytokine locus has been intensively studied as a model of chromatin dynamics directing gene expression in the immune system. The genes encoding the Th2 cytokines IL4, IL5 and IL13 are clustered in a 160 kb region of chromosome 5q31 in humans. Significant methylation changes were observed in loci associated with IL5 and IL13 (Suppl Fig. 3). One of the strongest and most consistent differentially methylated gene loci in our experiments was the demethylation of CpG sites within 1 kb of the proximal promoter of the IL13 gene in activated NK cells. Eight consecutive probes displayed highly consistent demethylation in activated compared to naïve NK cells, which was associated with marked upregulation of mRNA synthesis (Suppl Fig. 4). Studies by others showed demethylation in this region was characteristic of human Th2 polarized CD4/CD28 activated T cells leading to increased IL13 synthesis.40
Our studies extend the association of immune activation with demethylation of the proximal IL13 promoter to NK cells. This is of interest as NK cells are typically thought of in relationship to Th1 inflammatory cytokines, such as IFNG. Upregulation of IFNG was confirmed in our NK cells by flow cytometry, however, NK cells also produce IL13 and other Th2 cytokines,41 a phenotype implicated in allergic disease.42 Additionally, there is evidence of the existence and regulation of 2 distinct resting peripheral NK cell subsets producing either Th1 or Th2 cytokines exclusively.43 Our studies are unable to discern whether DNA methylation states of the IFNG and IL13 genes are coordinated within individual NK cells.
Also located at 5q31 is CSF2, the gene coding for the cytokine that controls production, differentiation, and function of granulocytes and macrophages. We found 3 CpGs sites reproducibly demethylated at day 9 including a site (cg02325250) previously identified as being rapidly demethylated in PMA/I stimulated mouse CD4 cells and associated with enhanced CSF2 expression.18 However, in our experiments at 16 h post-stimulation, none of the probes in CSF2 were significantly different from those of naïve NK cells, suggesting different kinetics in NK and T cells for this gene locus. By 9 d, NK cells displayed consistently lower methylation. We note that BHLHE40 activity is required for CSF2 production, and both genes are targets of demethylation in activated NK cells.44
Common co-stimulatory and activation loci in NK and T cells
Demethylation of numerous genes encoding established (for other immune cell types) immune stimulatory and activation loci in NK cells underscores the common epigenetic programs that are shared between the innate and adaptive immune systems. This is particularly true with respect to NK and T cells,11 and is further supported by findings in this study. Table 2 provides several examples of stimulatory and activation-related genes in T cells that also exhibit significant differential methylation during NK cell activation.
Discrete demethylation within the Interferon γ (IFNG) locus in activated NK cells
The IFNG gene was broadly demethylated in resting NK cells with no statistically significant genome-wide differential methylation following activation, although a small increased IFNG expression was observed by FACs analysis. Our DNA methylation result is consistent with previous studies showing that IFNG production in peripheral NK cells does not require DNA methylation remodeling. This aspect of the circulating blood NK cell methylome before and after activation is in contrast to the significant IFNG demethylation required by naïve CD4 cells differentiating into IFNG competent Th1 polarized cells.45-47 The IFNG locus of naïve NK cells has thus been claimed to be constitutively demethylated.48 However, a recent detailed bisulfite sequencing study of NK cell IFNG loci indicated that a highly restricted region of IFNG becomes demethylated during cell activation and that this alteration confers a higher IFNG competence to fully differentiated NK cells.49 Our array experiments contained 2 CpG probes (cg00848007, cg26227465) located within this influential region in exon 1 and the immediate 5′ sequence. Targeted analysis of these specific probes confirmed significant demethylation at these loci following NK activation (data not shown), although the changes would not meet genome-wide significance. These results speak to the fact that subtle and highly localized epigenetic remodeling can be obscured when using only genome wide discovery.
Transcription factors: BHLHE40, ZBTB32/FAZF, NFATC1, BACH2
Reprograming of DNA methylation within regulatory regions of transcription factor genes has been observed during immune activation. In murine CD8 cells following infection with the lymphocytic choriomeningitis virus (LCMV),50 methylation changes were evident at day 8 post-infection in cells demonstrating a fully differentiated effector CD8 cell phenotype. Our NK cells at day 9 post-activation share important methylation features with LCMV activated CD8 T cells including the demethylation of BHLHE40 (also known as Dec1, Stra13, Sharp2, or Bhlhb2). BHLHE40 belongs to a family of basic helix-loop-helix transcriptional regulators that is an attractive candidate for activation as it is induced in NK cells in response to cytokines (IL-2, IL-12, IL-15, INFG, TNFα) that enhance cytotoxic function.51 Additional roles of BHLHE40 and other differentially methylated transcription factors can be found in Table 3.13,52-57
Table 3.
Prominent transcription factors hypomethylated in activated NK cells. Large changes in methylation status of transcription factors were observed, with 4 prominent examples shown here, with their putative functional role in activation.
| Transcription Factor | Immune-Related Roles/Functions | Relevant Studies |
|---|---|---|
| BHLHE40 | Key regulator of a pan-differentiation transcriptional module, involved in several haematopoietic lineages | |
| Regulates several physiological processes, including the cell cycle, apoptosis, and circadian rhythms | ||
| Important roles in CD8 T cell memory development and function | Hu and Chen et al., 201313 | |
| Involved in the development of natural killer T (NKT) cells and the differentiation of murine dendritic cells and CD4 Tregs | Miller, Brown et al., 201253; Miyazaki, Miyazaki et al., 201054 | |
| ZBTB32/FAZF | Essential cell-intrinsic and lineage-specific role in promoting NK cell expansion after viral infection | Beaulieu, Zawislak et al., 201455 |
| NFATC1 (or NFAT2) | Regulates T-cell cytokine gene expression by binding to the promoter/enhancer regions of antigen-responsive genes, usually in cooperation with heterologous DNA-binding partners | Macian, 200552 |
| BACH2 | “Master transcription factor” associated with super-enhancer in B-, T- and CD56 cells; important in determining and maintaining cell identity; disregulation implicated in susceptibility to several autoimmune diseases | Roychoudhuri, Hirahara, et al., 201357, Hnisz, Abraham et al., 2013 |
Based on its role in T and NKT development and apparent significance in NK cell activation, several ddPCR assays for quantitating BHLHE40 demethylation were designed and evaluated. Cloning and bisulfite sequencing revealed broad and heterogeneous demethylation within the exon 5 region. Based on results, 2 CpG sites upstream of the array CpG probes were targeted for assay design. This upstream region contains a higher CpG density making it more favorable for MS-PCR design. This ddPCR assay accurately predicted the levels of unmethylated CpG sites in the exon 5 region. Consistent with its role in several lymphocyte lineages but not in myeloid cells, we observed low levels of BHLHE40 methylation in T, B and NK cells but high levels in granulocytes and monocytes. Among non-activated cells the highest prevalence of unmethylated BHLHE40 was found in naïve NK cells.
Among transcription factors, significant hypomethylation of the proximal ZBTB32 gene (also known as the Fanconi anemia zinc finger, or FAZF) and many fold induction of its expression stand out as notable features of NK cell activation in our studies. ZBTB32 is a member of the BTB-ZF transcription factor family, which can recruit chromatin-remodeling co-factors to regulate gene expression,58 and plays an essential cell-intrinsic and lineage-specific role in promoting NK cell expansion after viral infection.55 Recent studies focused on BTB-ZF family members as targets of epigenetic reprograming in CMV infection showed unmethylated CpGs in ZBTB32/FAPZ in HCMV memory NK cells but its expression was not elevated compared with naïve NK cells21 in contrast to our observed 600-fold increase in mRNA expression.
Among other functions, the NFAT family of transcription factors regulates cytokine gene expression by binding to the promoter/enhancer regions of antigen-responsive genes, usually in cooperation with heterologous DNA-binding partners. NFATC1 serves as regulator for many immune cell types such as T cells and natural killer cells.56 This study identified a locus in intron 9 that displayed significant, large demethylation (Δβ = −0.4; P =1.3 × 10−12; q = 5.6 × 10−8). The demethylation of intron 9 in activated NK deserves further study to understand what, if any, regulatory role it may have.
Activated NK cells contained a DMS in BACH2. BACH2 is a transcriptional repressor expressed by mature lymphoid cells that imposes regulation on humoral and cellular immunity. BACH2 helps maintain the balance between immune-stimulating and immune-regulating cells, or self-tolerance. Variants of BACH2 have been implicated in the susceptibility to several autoimmune diseases and allergy.59,60 Although a role for BACH2 in NK cells has not been determined, it plays an important role in lineage stabilization of Tregs.57 Given the significant commonalities between the cell types, BACH2 may function similarly in NK cells.
Novel methylation sites in signaling pathways: MAML2, TAGLN3, CISH, RPS6KA1/RSK1, PI3KR6
The CpG loci with the largest difference in DNA methylation upon NK activation occurred within the Mastermind-like 2 (MAML2) gene. MAML2 is a transcriptional co-activator of NOTCH,61 which is a highly conserved cell-cell communication pathway involved in numerous processes during embryogenesis, development, and hematopoiesis.62 Notch signaling enhances NK cell maturation and increases cytolytic effector capacity and cytokine secretion.63 Little information exists on the epigenetic features of MAML2. One study found the MAML2 promoter became hypermethylated in senescent mesenchymal stem cells after long term culture.64 We found another Notch related gene, the actin-binding protein involved in cytoskeletal organization,65 TAGLN3, demethylated in activated NK cells.
Significant hypomethylation of loci within the CISH gene was found in this study. CISH encodes a protein belonging to the cytokine-induced STAT inhibitor (CIS), also known as suppressor of cytokine signaling (SOCS) protein family. CIS family members are known to be cytokine-inducible negative regulators of cytokine signaling. Previous studies observed that DNA methylation levels within the second exon DMS of CISH was 100% in naïve CD4+ T cells, 52% in effector cells, and 13% in memory cells; an additional 17% of DMRs were hypermethylated in naïve cells, intermediately methylated in effector cells, and hypomethylated in memory cells.66
Since hypermethylation was so uncommonly observed in these activated NK cells, it is of interest to note RPS6KA1, ribosomal protein S6 kinase, polypeptide 1. Also known as p90RSK, this is a member of the ribosomal S6 protein serine/threonine kinases that regulate diverse cellular processes such as cell growth and differentiation. Putative methylation-based regulation of RPS6KA1 may result in modulation of the mTOR pathway, which has recently been recognized as important in the coordinate functioning of immune and metabolic activities in T cells.37
Novel NK activation loci that co-localize with GWAS hits in immune related disease: CLEC16A, NCF4 and TNFAIP3
It is interesting to note that genomic regions previously identified in GWAS studies of immune related diseases co-localize with novel CpG loci differentially methylated in activated NK cells in the current study. The convergence of GWAS with EWAS of NK activation is an indirect but suggestive clue to the potential importance of DMRs in immune regulation.
A robust association of CLEC16A demethylation with NK activation points to another novel pathway previously unappreciated in NK biology. CpG site cg03995533 was significantly unmethylated in activated NK cells. This site at 16p13.13 lies in intron 18 of CLEC16A, a region identified in GWAS as a susceptibility locus for several autoimmune diseases including T1D and multiple sclerosis.67 CLEC16A function is largely unexplored but it is thought to play a role in regulating mitophagy, a selective form of autophagy necessary for mitochondrial quality control.68 CLEC16A is expressed in NK cells67 but is also abundant in CNS white matter and blood monocyte-derived dendritic cells from MS patients, in which it strongly co-localized with human leukocyte antigen class II. Alleles of the intron 18 GWAS SNP rs12708716, are associated with CLEC16A expression in pancreatic islet cells68 and thymic tissue. A very recent GWAS of common variable immunodeficiency disorder identified rs17806056, also in intron 18, as a significant susceptibility factor for that disease.69 This SNP lays 1,995 bp upstream of the activated NK DMS. The convergence of replicated GWAS signals with regions affected by differential DNA methylation during NK activation supports the plausible regulatory importance of these epigenetic changes.
Multiple CpGs within intron 1 and the proximal promoter regions of the NCF4 gene were hypomethylated in activated NK cells. The NCF4 protein is a cytosolic regulatory component of the superoxide-producing NADPH-oxidase whose function is most strongly associated with the antibactericidal action of reactive oxygen species produced by myeloid linage cells. In other immune cells, NADPH oxidase plays an important role at several levels in autophagy by regulating signaling, antigen processing and phagosomal pH.70,71 NCF4 deficiency leads to aberrant degradation and accumulation of the pro-inflammatory cytokine IL1B, which contributes to granulomatous colitis in inflammatory bowel disease.72 It is of interest that GWAS SNPs implicated in Crohn's disease include NCF4 with the most consistent associations (rs4821544) localizing to intron 1 of the gene.73 Our strong hypomethylated signal at cg02532700 of intron 1 is 849 bps downstream of rs4821544. Other unmethylated loci occurred nearby and in the proximal promoter region of the gene.
Tumor necrosis family α induced protein 3 (TNFAIP3) is a well-known negative regulator of the NF-kB activation pathway. TNFAIP3 can attenuate the NF-kB activity triggered by signaling from TNF and Toll-like receptors,74 and is implicated in several autoimmune diseases. In activated NK cells, we found the CpG 08919597 site consistently hypomethylated; this site appears to be highly conserved, located within a TF binding cluster and a very strong DNaseI site/enhancer in ENCODE experiments. It also lays approximately 1200 bp from GWAS SNP candidates for autoimmune disease. TNFAIP3 is considered a tumor suppressor inactivated in cancer through deletion, mutation or alternatively by DNA hypermethylation. TNFAIP3 hypermethylation occurs in MALT lymphoma but not in chronic lymphocytic leukemia nor in multiple myeloma.75 The tumor related DNA methylation occurs within 18 CpGs sites of the gene promoter in contrast to the intronic location that we identified.
Contrasts between the in vitro activated NK methylome and HCMV memory-like NK cells: FCERG1, SYK, EAT2 and ZBTB16
Dramatic and stable epigenetic alterations within innate immune cells after infection by human pathogens have been documented. Long-lived “memory-like” NK cells have been identified that have distinct cell surface phenotype and functional properties. Recent studies implicate DNA methylation as an important mechanism leading to the cardinal features of these cells. Specifically, NK cells from HCMV-infected individuals include distinct subsets of memory-like NK cells that are deficient in multiple transcription factors (ZBTB16/PAZF) and signaling proteins, the latter including the tyrosine kinase SYK20 and the signaling adaptors FCERG1 and EAT2.21 These insights led us to ask if these CMV activation features were also present in our NKp46/CD2/IL2 activated NK cells. The analysis included 2 of the SYK CpG sites that were reported to be hypermethylated in memory NK cells following CMV infection.20 Β-values for these 2 probes were both only sparsely methylated; thus do not indicate significant differences in SYK methylation. This suggests hypermethylation at these sites may be unique to NK memory. Similarly FCERG1 and EAT2 loci implicated in HCMV were broadly hypomethylated and unaffected by NK activation. ZBTB16 was also hypomethylated and, in contrast, the ZBTB12 member was hypermethylated. These contrasting methylation features may help to distinguish NK cells activated through disparate exposures.
Implications for population-based research and therapeutics
Our data also have important implications for the proper interpretation of epigenome-wide association studies carried out in whole blood. As demonstrated convincingly by Bauer et al. (2015),76 associations of small changes in DNA methylation of specific loci associated with environmental exposures can be explained by exposure-associated changes in the leukocyte composition. The authors show that smoking-associated alterations of a CD3 lymphocyte subtype explain the reports by multiple authors of tobacco-associated DNA methylation alterations at the GPR15 locus. Because it is possible that tobacco smoking activates NK cells, we examined whether the long list of loci reported to have altered methylation associated with smoking77 might also be found as DMSs indicative of NK activation. We observed that change in methylation of the AHRR locus is both a marker of NK activation and smoking78 and that several other DMSs associated with NK activation that we found here have been reported by others to be associated with smoking, including: MYO1G (cg07826859), NCF4 (cg02532700), and GPR68 (cg05875421). This adds evidence to support the need for cautious interpretation of EWAS data, suggesting that the underlying leukocyte biology is responsive to exposure and may represent the signal captured in the EWAS data.
Detailed understanding of the methylome of activated NK cells has implications for emerging therapeutics. NK cells are seen as promising agents for cell-based cancer therapies.79 However, there is a great need to develop more effective protocols to activate and expand high numbers of NK cells ex vivo to be able to infuse sufficient numbers of functional NK cells to cancer patients. The methylation profile established here provides an epigenetic template to evaluate such protocols. In addition, new classes of epigenetic drugs that target important transcriptional mechanisms are being developed that have high specificity and biological activity.80 For example, EP300, which was identified in differential methylated loci in both hyper- and hypo-methylated NK loci, is a bromodomain containing transactivator that has been successfully targeted with small molecule inhibitors.81 The methylation profile of activated NK cells described here would predict that bromodomain inhibitors targeting EP300 could modulate NK activation, which may be beneficial or undesirable depending on the specific application.
Limitations of the study
The Illumina 450K panel is extensive but only interrogates a fraction of existing CpG sites in the genome. The activation protocol we used is an artificial ex vivo approach that may not represent physiologically relevant in vivo immune reactions. Different activation protocols may lead to different modifications of the methylome, thus our profile may be one of many possible signatures. Also, the output of arrays may reflect the average of subsets within the cell population. Thus, the differential methylation signature of activated NK cells described here may be a composite reflecting the sum of several subsets. Our aim here was to search for potential markers that would faithfully reflect NK activation status (not predict protein levels); additional functional study of DMSs identified here will therefore be necessary to reveal the relationship between differential methylation and protein level and function.
Conclusions
Long-term culture and stimulation but not shorter-term activation times were associated with robust and reproducible changes in DNA methylation within human NK cells. At day 9 post-activation, most methylation changes consisted of hypomethylation that occurred predominately outside of CpG islands and instead featured gene bodies or CpG sparse regions. Genes previously identified as induced in activated lymphocytes, especially T cells, and that are regulated in part through DNA methylation, were prominently represented among the most significant differentially methylated sites in this study. Well-known immune activation loci included TNFA family members, CD96, Th2 cytokines (IL13) and co-stimulatory loci. We observed other novel loci including transcription factors previously identified in lymphocyte activation but not implicated as targets of DNA methylation (BHLHE40, ZBTB32, NFATC1). Genes implicated in specific haematopoietic motility (e.g., MYO1G) and Notch signaling (MAML2, TGLN3) were also identified for the first time. Also noteworthy were less well characterized genes implicated through genetic studies in autoimmune disease (CLEC16A, NCF4, BACH2) and for whom the relevant genetic susceptibility loci co-localized with the regions affected by DNA methylation changes in NK cells. Our ability to target BHLHE40 demethylation using ddPCR provides a potential new tool for NK studies. The detection of BHLHE40 demethylation in the whole blood of (healthy) individuals suggests this marker could be investigated in clinical populations. Potential applications include assessing immunomodulatory therapies or monitoring the levels of ex vivo activated NK cells in cell mediated therapies. Finally, the sum of many DMSs identified here could be used in population EWAS studies to identify and adjust for the contribution of activated NK cells as a strategy to avoid confounding by the effects of immune activation on DNA methylation profiles.
Methods
Overview of activation and DMR discovery pipeline
A discovery-replication experimental design was applied to guide our identification of valid CpG loci associated with NK cell activation (Suppl Fig. 1A). A strict false discovery threshold (q < 0.003) was applied to the 450K methylation data after filtering out those CpG loci that may have been affected by gender (X,Y chromosomes) or autosomal polymorphisms (SNP >0.05). The discovery set consisted of 6 different blood donors whose NK cells were studied at time 0 and day 9 post-activation, 4 of whom had additional samples taken at time 16 h. The replication phase consisted of a second set of 6 subjects processed and arrayed independently at a later time. Only loci that met the threshold criteria (q < 0.003) in both phases were included in the final data set.
NK cell isolation and culture
Within 3–5 h of blood draw, freshly generated leukoreduction filters were used to isolate NK cells; the filters were supported on a clamp/ring-stand and back-flushed with ∼250 ml of room temperature, sterile, PBS (without calcium or magnesium), pH 7.2, 2 mM EDTA (Miltenyi Rinsing Solution, cat. no. 130-091-222). Flushing was carried out with ˜50 ml at a rate of approximately 2 min per syringe of PBS into a single use sterile Erlenmeyer flask, while gently massaging the filter to enhance cell release. Flushed cell suspensions were subjected to Ficoll Paque gradients (GE cat. no. 17-1440-02, density 1.077 g/ml) in 50 ml conical tubes. The resulting buffy coat layers were collected and washed several times to remove platelets prior to the NK cell enrichment procedure.
Untouched NK cells were obtained using the Miltenyi NK Cell Isolation Kit (cat. no. 130-092-657) per manufacturer's instructions. Immediately following isolation, sub-samples were removed for both flow cytometry sample preparation and nucleic acids extraction. The remaining cells were activated using the Miltenyi NK Cell Activation & Expansion Kit (cat. no. 130-094-483), with beads prepared as directed by the manufacturer. This activation is a combination of CD2 and CD335 (NKp46) coated microbeads and exposure to 500 IU/ml IL2 (Miltenyi 130-097-744). The NK cells were cultured at a density of 1–1.5 × 106 cells/ml for 9 d under standard conditions (37°C, 5%CO2) in CellGenix SCGM (cat. no. 20802-0500) supplemented with 10% heat inactivated FBS (Life Technologies cat. no. 16000-044) and 1x L-glutamine/Pen/Strep mix (2 mM L-glut, 100 U/ml penicillin, 100 µg/ml streptomycin; Life Technologies cat. no. 10378016).
Flow cytometry (FACS)
Fresh, magnetic-activated cell sorting (MACS) isolated and 9-day cultured NK cells were characterized by flow cytometric techniques. Samples were analyzed for viability, NK purity and activation status using a 5-stain panel (Suppl Fig. 2 and Suppl Table 1). Compensation controls were prepared using anti-mouse compensation beads (Life technologies ABC Anti-mouse Bead kit, cat. no. A10344). Heat killed cells (65°C, 1 min followed by 1 min on ice) were used as compensation controls for viability dye. All cells were washed twice and suspended in protein-free PBS. When IFN-γ was measured, an intracellular stain was employed. An aliquot of cells for IFN-γ staining were first incubated with a Golgi blocker, brefeldin A (eBioscience cat. no. 00-4506), for 4 h. Cells were stained for surface markers per manufacturer's instructions, and fixed in IC Fixation Buffer (eBioscience cat. no. 00-8222). Intracellular staining was carried out after permeabilizing cells (Permeabilization Buffer, eBioscience cat. no. 00-8333), as directed by the manufacturer. All fixed flow cytometry samples were stored at 4°C in the dark and analyzed within ∼36 h on a FACS Aria (UCSF Laboratory for Cell Analysis) or FACS AriaUII (Brown University Flow Cytometry and Sorting Facility).
Killing assay
A killing assay was developed at Janssen Pharmaceutical and employed at Brown University to assess the killing phenotype of activated NK cells, using target Daudi cells. Killing was determined by measuring free GAPDH in the media/supernatant. Daudi cells (ATCC CCL-213) were cultured in RPMI 1640 supplemented with L-glutamine, 1% Pen-Strep and 10% heat inactivated FBS, and incubated at 37°C in a humidified incubator with 5% CO2. Cultures were maintained at a density between 3 × 105–3 × 106 cells/ml. Killing assay wells of U-bottom tissue culture plates were set up as follows: medium only (background control), Daudi cells only for target cell spontaneous lysis, NK cells only for effector cell spontaneous lysis, Daudi cells only for maximum target cell lysis via chemical disruption, effector:target (5:1), and effector:target (5:1) in the presence of 5 µg/ml Rituximab. Wells that included Rituximab were incubated with 40,000 viable Daudi cells in a final volume of 75 µL medium at room temperature for 15 min for opsonization. Plates were centrifuged (300 x g, 5 min) and medium removed. Washed, viable, day 9 activated NK cells (2 × 105) in complete medium were added to wells as described above. Cells were gently pelleted (300 x g, 1 min) to promote cell contact, and incubated at 37°C for 4 h for NK killing of Daudi cells. Following incubation, 10 µL of lysis buffer (Component 6, part #3035 from aCellaTox kit, Cell Technologies) was added to the subset of Daudi only wells designated for the maximum lysis control and incubated 15 min at room temperature. All wells were brought to a final volume of 200 µL with medium and centrifuge 750 x g, 1 min. Detection of GAPDH in the medium/ supernatant was carried out using reagents from the eBioscience GAPDH InstantOne ELISA kit (cat. no. 85-86131-11) with some procedural modification. Fifty microliters of experimental supernatant was added to 50 µL of 1:1 InstantOne detection and capture antibodies in the wells of the provided InstantOne ELISA assay plate. Plates were sealed and mixed (140–300 RPM) for 1 h at room temperature. Following incubation, the cell supernatant/antibody mixture was removed and plate wells washed 3 times with 200 µL /well wash buffer. One hundred microliters of detection reagent was added per well and incubated at room temperature for 10–15 min on an orbital shaker at 50 RPM. Reactions were stopped by the addition of 100 µL /well of kit stop reagent. Absorbance at 450 nm was read on a Molecular Devices Spectromax M2.
Specific NK killing percentages were calculated as follows:
Where experimental lysis is the signal from NK + Daudi cells, spontaneous lysis is the sum of signal from NK-only and Daudi-only wells, and maximum lysis is Daudi cells in the presence of chemical lysis solution.
Nucleic acid extraction, bisulfite conversion and methylation array
NK cells were harvested at T = 0 (inactivated/naïve), T = 16 h (activated; discovery phase only), and T = 9 d (activated), and DNA extracted using the Norgen RNA/DNA Purification kit (cat. no. 48600) for discovery phase samples, and Zymo Universal Quick DNA Miniprep kit (cat. no. D4068) for replication phase samples. DNA was bisulfite treated and converted using the Zymo EZ DNA Methylation kit (cat. no. D5001) per manufacturer's instructions, including the alternative incubation conditions recommended for the Illumina InfiniumMethylation450 BeadChip array samples. Bisulfite converted DNA was sent to the UCSF Center for Human Genetics, Genomics Core facility for processing using Illumina chemistry, instruments and protocols.
Gene expression
Total RNA was purified from NK cell pellets using ALLPrep DNA/RNA Mini Kits (Qiagen cat. no. 80204) and cDNA created with random primers and SuperScript II Reverse Transcriptase (Invitrogen cat. no. 18604-022) based on vendor's instructions. Gene expression was assessed by comparative CT method (ΔΔCT) using IPO8, B2M, and GAPDH as the endogenous references genes. All expression assays were inventory Taqman Gene Expression Assays (LifeTechnologies; Suppl Table 2). Assays were carried out per manufacturer's instructions, using 2 µl of cDNA per 20 µl PCR. Reactions were run in triplicate on an Applied Biosystems 7300. ΔΔCT was calculated using 9 d activated samples as the “test” and T = 0 (freshly isolated NK cells) as the “calibrator.”
Cloning and bisulfite sequencing
Genomic DNA for bisulfite sequencing was extracted from NK cells using ALLPrep DNA/RNA Mini Kits (Qiagen cat. no. 80204) and bisulfite treated with EZ DNA Methylation Kit (Zymo cat. no. D5001).
Bisulfite converted DNA was amplified with bisulfite-specific and methylation insensitive primers. PCR products were purified using either a Gel Extraction Kit (Qiagen cat. no. 28604) or PCR Purification Kit (Qiagen cat no 28104), and PCR products ligated into plasmid vectors using the TOPO TA Cloning system (Invitrogen cat. no. K4500-0). Plasmid-transformed Escherichia coli were cultured overnight on Luria-Bertani agar plates and the positive colonies were cultured overnight in Luria-Bertani media. Plasmid DNA containing PCR products were isolated with QuickLyse MiniPrep Kits (Qiagen cat. no. 27405) and 20 colonies from each experiment group were sequenced in both directions using an ABI 373 automated sequencer with dye primer chemistry and standard M13 primers.
Droplet digital PCR (ddPCR)
As described in Suppl Fig. 1B, the most significant loci from the analysis were further prioritized for PCR assay development and ranked using 3 additional features: number of statistically significant probes within a gene, effect size and CpG density. Gene loci were scored based on the presence of multiple probe hits within a gene and the effect size (>0.3 β-value difference between activated and non-activated conditions), as well as the density of CpGs surrounding the top hit probe. Based on these feasibility criteria, several candidate genes were selected and tested for potential quantitative methylation assay development (data not shown). BHLHE40 was chosen for further exploration as a putative biomarker of NK activation.
Four methylation-specific PCR (MSP) designs were selected for BHLHE40 and tested using the QX100 Droplet Digital PCR system (Bio-Rad)(data not shown). The following assay was selected: forward primer 5′- TGGAGGTGGGTGATAAGTTG-3′, reverse primer 5′- ACTCAAATTTCCAAACATATACCC-3′, and probe 6FAM-5′- AAACACAAAAACACTCAAAACCTAA-3′-NFQ. Master mixes for design testing included: Droplet PCR Supermix (Bio-Rad, cat. no. 186-3024), primers and probe (Applied Biosystems), and EB buffer (Qiagen cat. no. 19086). The so-called “C-Less” reaction (forward primer 5′-TTGTATGTATGTGAGTGTGGGAGAGA-3′, reverse primer 5′- TTTCTTCCACCCCTTCTCTTCC-3′ and probe 5′- 6FAM-CTCCCCCTCTAACTCTAT-NFQ-3)’ was used to quantify the total input of bisulfite converted DNA into each sample and BHLHE40 copy number expressed as a ratio to the total number of copies.82
BHLHE40 and C-Less total input master mixes were aliquoted into 96-well plates and PCR reactions performed in monoplex using 80 ng of bisulfite converted DNA for all reactions. Samples were transferred one column at a time to an 8 channel Droplet Generator Cartridge (Bio-Rad, cat no 186-3008). QX100 Droplet Generator Oil (Bio-Rad, cat no 186-3005) was added, and droplets generated using the QX100 Droplet Generator (Bio-Rad). Typically 10,000 to 14,000 droplets were generated for each sample and transferred to an Eppendorf 96-well PCR plate (cat no 951020362), foil heat sealed, and placed into a S1000 thermal cycler (Bio-Rad). The PCR cycling conditions for gradient temperature testing were as follows: 95°C for 10 min, 40 cycles of 94°C for 30 sec and 63°C-56°C for 1 min, and ending with 98°C for 10 min and 4°C hold. Following PCR, the plate was transferred to the QX100 Droplet Reader (Bio-Rad) for droplet analysis/determination of either positive or negative for fluorescence/amplification. Data was analyzed using the QuantaSoft software from BioRad.
BHLHE40 ddPCR assay performance was examined using activated NK cells as a positive standard of demethylated exon 5, which was cloned and bisulfite sequenced. Samples of activated NK DNA were diluted with hypermethylated DNA from granulocytes to simulate the detection of BHLHE40 within a background of excess hypermethylated DNA. Dilution of activated NK DNA with the background copies observed in completely hypermethylated DNA was used to estimate the assay limit of quantification and dynamic range (data not shown).
Gene enrichment and pathway analyses
Gene-associated hits from the overlapping loci (discovery and replication data sets) were analyzed for functional enrichment as well as pathway association. Functional enrichment was evaluated using the National Institute of Allergy and Infectious Disease DAVID Bioinformatics Database v6.7 http://david.abcc.ncifcrf.gov/.83,84 P-value (or EASE score) was set at 0.05 (please refer to https://david.ncifcrf.gov/helps/functional_annotation.html#E3 for more detailed explanation). The lower the P-value (or EASE score), the more enriched the gene terms in the dataset compared with the representation of that gene-term in the human genome. Reference data was limited to using only human genes found in the selected (“Official Gene Symbol” setting in DAVID) annotation databases. Pathway analyses (“canonical pathway” module) were run on Ingenuity Pathway Analysis software (Qiagen). Analyses were run using a stringent filter for “human,” which returned results most relevant to humans. Additionally, analyses were run using all tissue and cell types, then repeated constraining the input/reference to immune cell information only. Non-coding associated loci were evaluated in a similar manner using the open access program GREAT (version 3.0.0; Suppl table 7). This program was designed to interrogate numerous annotation/ontology databases to calculate enrichment for proximal and distal, non-coding genomic regions. Analysis was carried out using the program default association rule parameters.
The significant differentially methylated loci (385) were also assessed in the context of transcription factor binding site. The TFBS for 161 transcription factors from the ENCODE database was interrogated using the “Txn Factor ChIP” track and wgEncodeRegTfbsClusteredV3 table through the UCSC Genome Browser (hg19 assembly) site.
Statistical analysis
Methylation signals from Illumina 450K methylation arrays were preprocessed using the functional normalization method “preprocessFunnorm” in the minfi package85 86 to control for unwanted variation among plates and samples. To avoid interference from polymorphisms affecting probe hybridization, all probes containing single nucleotide polymorphisms (SNPs) were removed as were probes contained on the X- and Y-chromosomes. Additionally, cross-reactive probes were removed using criteria from Chen, et al. 2013.87 Models for time 0 vs. 16 h activation, and 0 vs. 9 d activation found F statistics and P-values using an empirical Bayes method. These calculations were made using the function “eBayes” in the “limma” package.88 False discovery rate (FDR) was corrected for using the Benjamini-Hochberg adjustment for multiple testing.89 CpG loci were ordered by FDR-adjusted q-value, and the top 1000 loci selected for further consideration. Subsequent analyses focused on gene-associated CpG loci.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge the helpful suggestions and discussions of Dr. Shichun Zheng.
Funding
JKW was supported by the Robert Magnin Newman endowment in Neuro-oncology. This work was supported by Janssen Pharmaceuticals.
References
- 1.Calvanese V, Fernandez AF, Urdinguio RG, Suarez-Alvarez B, Mangas C, Perez-Garcia V, Bueno C, Montes R, Ramos-Mejia V, Martinez-Camblor P, et al.. A promoter DNA demethylation landscape of human hematopoietic differentiation. Nucleic Acids Res 2012; 40:116-31; PMID:21911366; http://dx.doi.org/ 10.1093/nar/gkr685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6-21; PMID:11782440; http://dx.doi.org/ 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- 3.Yu Y, Mo Y, Ebenezer D, Bhattacharyya S, Liu H, Sundaravel S, Giricz O, Wontakal S, Cartier J, Caces B, et al.. High resolution methylome analysis reveals widespread functional hypomethylation during adult human erythropoiesis. J Biol Chem 2013; 288:8805-14; PMID:23306203; http://dx.doi.org/ 10.1074/jbc.M112.423756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karpurapu M, Ranjan R, Deng J, Chung S, Lee YG, Xiao L, Nirujogi TS, Jacobson JR, Park GY, Christman JW. Kruppel like factor 4 promoter undergoes active demethylation during monocyte/macrophage differentiation. PloS one 2014; 9:e93362; PMID:24695324; http://dx.doi.org/ 10.1371/journal.pone.0093362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitagawa Y, Ohkura N, Sakaguchi S. Molecular determinants of regulatory T cell development: the essential roles of epigenetic changes. Front Immunol 2013; 4:106; PMID:23675373; http://dx.doi.org/ 10.3389/fimmu.2013.00106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komori HK, Hart T, LaMere SA, Chew PV, Salomon DR. Defining CD4 T cell memory by the epigenetic landscape of CpG DNA methylation. J Immunol 2015; 194:1565-79; PMID:25576597; http://dx.doi.org/ 10.4049/jimmunol.1401162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee ST, Xiao Y, Muench MO, Xiao J, Fomin ME, Wiencke JK, Zheng S, Dou X, de Smith A, Chokkalingam A, et al.. A global DNA methylation and gene expression analysis of early human B-cell development reveals a demethylation signature and transcription factor network. Nucleic Acids Res 2012; 40:11339-51; PMID:23074194; http://dx.doi.org/ 10.1093/nar/gks957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wieczorek G, Asemissen A, Model F, Turbachova I, Floess S, Liebenberg V, Baron U, Stauch D, Kotsch K, Pratschke J, et al.. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res 2009; 69:599-608; PMID:19147574; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2361 [DOI] [PubMed] [Google Scholar]
- 9.Accomando WP, Wiencke JK, Houseman EA, Butler RA, Zheng S, Nelson HH, Kelsey KT. Decreased NK cells in patients with head and neck cancer determined in archival DNA. Clin Cancer Res 2012; 18:6147-54; PMID:23014525; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW, Immunological Genome Project C . Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat Immunol 2013; 14:404-12; PMID:23396170; http://dx.doi.org/ 10.1038/ni.2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bezman NA, Kim CC, Sun JC, Min-Oo G, Hendricks DW, Kamimura Y, Best JA, Goldrath AW, Lanier LL, Immunological Genome Project C . Molecular definition of the identity and activation of natural killer cells. Nat Immunol 2012; 13:1000-9; PMID:22902830; http://dx.doi.org/ 10.1038/ni.2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dybkaer K, Iqbal J, Zhou G, Geng H, Xiao L, Schmitz A, d'Amore F, Chan WC. Genome wide transcriptional analysis of resting and IL2 activated human natural killer cells: gene expression signatures indicative of novel molecular signaling pathways. BMC Genomics 2007; 8:230; PMID:17623099; http://dx.doi.org/ 10.1186/1471-2164-8-230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu G, Chen J. A genome-wide regulatory network identifies key transcription factors for memory CD8(+) T-cell development. Nat Commun 2013; 4:2830; PMID:24335726; http://doi:10.1038/ncomms3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma D, Cao W, Kapur A, Felder M, Scarlett CO, Patankar MS, Li L. Differential expression of proteins in naive and IL-2 stimulated primary human NK cells identified by global proteomic analysis. J Proteomics 2013; 91:151-63; PMID:23806757; http://dx.doi.org/ 10.1016/j.jprot.2013.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melvin AJ, McGurn ME, Bort SJ, Gibson C, Lewis DB. Hypomethylation of the interferon-gamma gene correlates with its expression by primary T-lineage cells. Eur J Immunol 1995; 25:426-30; PMID:7875204; http://dx.doi.org/ 10.1002/eji.1830250218 [DOI] [PubMed] [Google Scholar]
- 16.Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol 2003; 4:235-40; PMID:12548284; http://dx.doi.org/ 10.1038/ni887 [DOI] [PubMed] [Google Scholar]
- 17.Harland KL, Day EB, Apte SH, Russ BE, Doherty PC, Turner SJ, Kelso A. Epigenetic plasticity of Cd8a locus during CD8(+) T-cell development and effector differentiation and reprogramming. Nat Commun 2014; 5:3547; PMID:24675400; http://dx.doi.org/ 10.1038/ncomms4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Chen G, Ma L, Ohms SJ, Sun C, Shannon MF, Fan JY. Plasticity of DNA methylation in mouse T cell activation and differentiation. BMC Mol Biol 2012; 13:16; PMID:22642378; http://dx.doi.org/ 10.1186/1471-2199-13-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Ulm A, Somineni HK, Oh S, Weirauch MT, Zhang HX, Chen X, Lehn MA, Janssen EM, Ji H. DNA methylation dynamics during ex vivo differentiation and maturation of human dendritic cells. Epigenetics Chromatin 2014; 7:21; PMID:25161698; http://dx.doi.org/ 10.1186/1756-8935-7-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, Scott JM, Kamimura Y, Lanier LL, Kim S. Epigenetic Modification and Antibody-Dependent Expansion of Memory-like NK Cells in Human Cytomegalovirus-Infected Individuals. Immunity 2015; 42:431-42; PMID:25786175; http://dx.doi.org/ 10.1016/j.immuni.2015.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, Han H, Chiang SC, Foley B, Mattsson K, et al.. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015; 42:443-56; PMID:25786176; http://dx.doi.org/ 10.1016/j.immuni.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamimura Y, Lanier LL. Homeostatic control of memory cell progenitors in the natural killer cell lineage. Cell Rep 2015; 10:280-91; PMID:25578733; http://dx.doi.org/ 10.1016/j.celrep.2014.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiencke JK, Bracci PM, Hsuang G, Zheng S, Hansen H, Wrensch MR, Rice T, Eliot M, Kelsey KT. A comparison of DNA methylation specific droplet digital PCR (ddPCR) and real time qPCR with flow cytometry in characterizing human T cells in peripheral blood. Epigenetics 2014; 9:1360-5; PMID:25437051; http://dx.doi.org/ 10.4161/15592294.2014.967589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012; 13:86; PMID:22568884; http://dx.doi.org/ 10.1186/1471-2105-13-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Soderhall C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PloS one 2012; 7:e41361; PMID:22848472; http://dx.doi.org/ 10.1371/journal.pone.0041361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Accomando WP, Wiencke JK, Houseman EA, Nelson HH, Kelsey KT. Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol 2014; 15:R50; PMID:24598480; http://dx.doi.org/ 10.1186/gb-2014-15-3-r50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu LF, Gasteiger G, Yu IS, Chaudhry A, Hsin JP, Lu Y, Bos PD, Lin LL, Zawislak CL, Cho S, et al.. A Single miRNA-mRNA Interaction Affects the Immune Response in a Context- and Cell-Type-Specific Manner. Immunity 2015; 43:52-64; PMID:26163372; http://dx.doi.org/ 10.1016/j.immuni.2015.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falvo JV, Jasenosky LD, Kruidenier L, Goldfeld AE. Epigenetic control of cytokine gene expression: regulation of the TNF/LT locus and T helper cell differentiation. Adv Immunol 2013; 118:37-128; PMID:23683942; http://dx.doi.org/ 10.1016/B978-0-12-407708-9.00002-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altorok N, Coit P, Hughes T, Koelsch KA, Stone DU, Rasmussen A, Radfar L, Scofield RH, Sivils KL, Farris AD, et al.. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjogren's syndrome. Arthritis Rheumatol 2014; 66:731-9; PMID:24574234; http://dx.doi.org/ 10.1002/art.38264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Maksimovic J, Naselli G, Qian J, Chopin M, Blewitt ME, Oshlack A, Harrison LC. Genome-wide DNA methylation analysis identifies hypomethylated genes regulated by FOXP3 in human regulatory T cells. Blood 2013; 122:2823-36; PMID:23974203; http://dx.doi.org/ 10.1182/blood-2013-02-481788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tatari-Calderone Z, Semnani RT, Nutman TB, Schlom J, Sabzevari H. Acquisition of CD80 by human T cells at early stages of activation: functional involvement of CD80 acquisition in T cell to T cell interaction. J Immunol 2002; 169:6162-9; PMID:12444120; http://dx.doi.org/ 10.4049/jimmunol.169.11.6162 [DOI] [PubMed] [Google Scholar]
- 32.Vadasz Z, Toubi E. Semaphorins: their dual role in regulating immune-mediated diseases. Clin Rev Allergy Immunol 2014; 47:17-25; PMID:23397481; http://dx.doi.org/ 10.1007/s12016-013-8360-4 [DOI] [PubMed] [Google Scholar]
- 33.Levy S. Function of the tetraspanin molecule CD81 in B and T cells. Immunol Res 2014; 58:179-85; PMID:24522698; http://dx.doi.org/ 10.1007/s12026-014-8490-7 [DOI] [PubMed] [Google Scholar]
- 34.Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol 2014; 15:431-8; PMID:24658051; http://dx.doi.org/ 10.1038/ni.2850 [DOI] [PubMed] [Google Scholar]
- 35.Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol 2015; 15:243-54; PMID:25743219; http://dx.doi.org/ 10.1038/nri3799 [DOI] [PubMed] [Google Scholar]
- 36.Kratholm SK, Iversen MB, Reinert L, Jensen SK, Hokland M, Andersen T, Rankin A, Young D, Frische S, Paludan SR, et al.. Interleukin-21 receptor signalling is important for innate immune protection against HSV-2 infections. PloS one 2013; 8:e81790; PMID:24358128; http://dx.doi.org/ 10.1371/journal.pone.0081790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Man K, Kallies A. Synchronizing transcriptional control of T cell metabolism and function. Nat Rev Immunol 2015; 15:574-84; PMID:26272293; http://dx.doi.org/ 10.1038/nri3874 [DOI] [PubMed] [Google Scholar]
- 38.Hanna J, Bechtel P, Zhai Y, Youssef F, McLachlan K, Mandelboim O. Novel insights on human NK cells' immunological modalities revealed by gene expression profiling. J Immunol 2004; 173:6547-63; PMID:15557145; http://dx.doi.org/ 10.4049/jimmunol.173.11.6547 [DOI] [PubMed] [Google Scholar]
- 39.Benita Y, Cao Z, Giallourakis C, Li C, Gardet A, Xavier RJ. Gene enrichment profiles reveal T-cell development, differentiation, and lineage-specific transcription factors including ZBTB25 as a novel NF-AT repressor. Blood 2010; 115:5376-84; PMID:20410506; http://dx.doi.org/ 10.1182/blood-2010-01-263855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santangelo S, Cousins DJ, Winkelmann NE, Staynov DZ. DNA methylation changes at human Th2 cytokine genes coincide with DNase I hypersensitive site formation during CD4(+) T cell differentiation. J Immunol 2002; 169:1893-903; PMID:12165514; http://dx.doi.org/ 10.4049/jimmunol.169.4.1893 [DOI] [PubMed] [Google Scholar]
- 41.Hoshino T, Winkler-Pickett RT, Mason AT, Ortaldo JR, Young HA. IL-13 production by NK cells: IL-13-producing NK and T cells are present in vivo in the absence of IFN-gamma. J Immunol 1999; 162:51-9; PMID:9886369 [PubMed] [Google Scholar]
- 42.Culley FJ. Natural killer cells in infection and inflammation of the lung. Immunology 2009; 128:151-63; PMID:19740372; http://dx.doi.org/ 10.1111/j.1365-2567.2009.03167.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loza MJ, Peters SP, Zangrilli JG, Perussia B. Distinction between IL-13+ and IFN-gamma+ natural killer cells and regulation of their pool size by IL-4. Eur J Immunol 2002; 32:413-23; PMID:11813160; http://dx.doi.org/ 10.1002/1521-4141(200202)32:2%3c413::AID-IMMU413%3e3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- 44.Brettingham-Moore KH, Sprod OR, Chen X, Oakford P, Shannon MF, Holloway AF. Determinants of a transcriptionally competent environment at the GM-CSF promoter. Nucleic Acids Res 2008; 36:2639-53; PMID:18344520; http://dx.doi.org/ 10.1093/nar/gkn117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yano S, Ghosh P, Kusaba H, Buchholz M, Longo DL. Effect of promoter methylation on the regulation of IFN-gamma gene during in vitro differentiation of human peripheral blood T cells into a Th2 population. J Immunol 2003; 171:2510-6; PMID:12928400; http://dx.doi.org/ 10.4049/jimmunol.171.5.2510 [DOI] [PubMed] [Google Scholar]
- 46.Winders BR, Schwartz RH, Bruniquel D. A distinct region of the murine IFN-gamma promoter is hypomethylated from early T cell development through mature naive and Th1 cell differentiation, but is hypermethylated in Th2 cells. J Immunol 2004; 173:7377-84; PMID:15585862; http://dx.doi.org/ 10.4049/jimmunol.173.12.7377 [DOI] [PubMed] [Google Scholar]
- 47.Jones B, Chen J. Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J 2006; 25:2443-52; PMID:16724115; http://dx.doi.org/ 10.1038/sj.emboj.7601148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tato CM, Martins GA, High FA, DiCioccio CB, Reiner SL, Hunter CA. Cutting Edge: Innate production of IFN-gamma by NK cells is independent of epigenetic modification of the IFN-gamma promoter. J Immunol 2004; 173:1514-7; PMID:15265878; http://dx.doi.org/ 10.4049/jimmunol.173.3.1514 [DOI] [PubMed] [Google Scholar]
- 49.Luetke-Eversloh M, Cicek BB, Siracusa F, Thom JT, Hamann A, Frischbutter S, Baumgrass R, Chang HD, Thiel A, Dong J, et al.. NK cells gain higher IFN-gamma competence during terminal differentiation. Eur J Immunol 2014; 44:2074-84; PMID:24752800; http://dx.doi.org/ 10.1002/eji.201344072 [DOI] [PubMed] [Google Scholar]
- 50.Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol 2013; 191:3419-29; PMID:23956425; http://dx.doi.org/ 10.4049/jimmunol.1301395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kato Y, Kawamoto T, Fujimoto K, Noshiro M. DEC1/STRA13/SHARP2 and DEC2/SHARP1 coordinate physiological processes, including circadian rhythms in response to environmental stimuli. Curr Top Dev Biol 2014; 110:339-72; PMID:25248482; http://dx.doi.org/ 10.1016/B978-0-12-405943-6.00010-5 [DOI] [PubMed] [Google Scholar]
- 52.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 2005; 5:472-84; PMID:15928679; http://dx.doi.org/ 10.1038/nri1632 [DOI] [PubMed] [Google Scholar]
- 53.Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al.. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol 2012; 13:888-99; PMID:22797772; http://dx.doi.org/ 10.1038/ni.2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyazaki K, Miyazaki M, Guo Y, Yamasaki N, Kanno M, Honda Z, Oda H, Kawamoto H, Honda H. The role of the basic helix-loop-helix transcription factor Dec1 in the regulatory T cells. J Immunol 2010; 185:7330-9; PMID:21057086; http://dx.doi.org/ 10.4049/jimmunol.1001381 [DOI] [PubMed] [Google Scholar]
- 55.Beaulieu AM, Zawislak CL, Nakayama T, Sun JC. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat Immunol 2014; 15:546-53; PMID:24747678; http://dx.doi.org/ 10.1038/ni.2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller MR, Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol 2010; 10:645-56; PMID:20725108; http://dx.doi.org/ 10.1038/nri2818 [DOI] [PubMed] [Google Scholar]
- 57.Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, Sciume G, Zare H, Vahedi G, Dema B, et al.. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature 2013; 498:506-10; PMID:23728300; http://dx.doi.org/ 10.1038/nature12199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee SU, Maeda T. POK/ZBTB proteins: an emerging family of proteins that regulate lymphoid development and function. Immunological reviews 2012; 247:107-19; PMID:22500835; http://dx.doi.org/ 10.1111/j.1600-065X.2012.01116.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grant SF, Hakonarson H. Genome-wide association studies in type 1 diabetes. Current diabetes reports 2009; 9:157-63; PMID:19323961; http://dx.doi.org/ 10.1007/s11892-009-0026-5 [DOI] [PubMed] [Google Scholar]
- 60.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al.. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nature genetics 2010; 42:1118-25; PMID:21102463; http://dx.doi.org/ 10.1038/ng.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin SE, Oyama T, Nagase T, Harigaya K, Kitagawa M. Identification of new human mastermind proteins defines a family that consists of positive regulators for notch signaling. J Biol Chem 2002; 277:50612-20; PMID:12386158; http://dx.doi.org/ 10.1074/jbc.M209529200 [DOI] [PubMed] [Google Scholar]
- 62.Kojika S, Griffin JD. Notch receptors and hematopoiesis. Exp Hematol 2001; 29:1041-52; PMID:11532344; http://dx.doi.org/ 10.1016/S0301-472X(01)00676-2 [DOI] [PubMed] [Google Scholar]
- 63.Felices M, Ankarlo DE, Lenvik TR, Nelson HH, Blazar BR, Verneris MR, Miller JS. Notch signaling at later stages of NK cell development enhances KIR expression and functional maturation. J Immunol 2014; 193:3344-54; PMID:25172483; http://dx.doi.org/ 10.4049/jimmunol.1400534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choi MR, In YH, Park J, Park T, Jung KH, Chai JC, Chung MK, Lee YS, Chai YG. Genome-scale DNA methylation pattern profiling of human bone marrow mesenchymal stem cells in long-term culture. Exp Mol Med 2012; 44:503-12; PMID:22684242; http://dx.doi.org/ 10.3858/emm.2012.44.8.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ratie L, Ware M, Jagline H, David V, Dupe V. Dynamic expression of Notch-dependent neurogenic markers in the chick embryonic nervous system. Front Neuroanat 2014; 8:158; PMID:25565981; http://doi:10.3389/fnana.2014.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hashimoto S, Ogoshi K, Sasaki A, Abe J, Qu W, Nakatani Y, Ahsan B, Oshima K, Shand FH, Ametani A, et al.. Coordinated changes in DNA methylation in antigen-specific memory CD4 T cells. J Immunol 2013; 190:4076-91; PMID:23509353; http://dx.doi.org/ 10.4049/jimmunol.1202267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hakonarson H, Grant SF, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Casalunovo T, Taback SP, Frackelton EC, et al.. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 2007; 448:591-4; PMID:17632545; http://dx.doi.org/ 10.1038/nature06010 [DOI] [PubMed] [Google Scholar]
- 68.Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, Spruce LA, Kushner JA, Groop L, Seeholzer SH, et al.. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 2014; 157:1577-90; PMID:24949970; http://dx.doi.org/ 10.1016/j.cell.2014.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li J, Jorgensen SF, Maggadottir SM, Bakay M, Warnatz K, Glessner J, Pandey R, Salzer U, Schmidt RE, Perez E, et al.. Association of CLEC16A with human common variable immunodeficiency disorder and role in murine B cells. Nat Commun 2015; 6:6804; PMID:25891430; http://dx.doi.org/ 10.1038/ncomms7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang J, Brumell JH. NADPH oxidases contribute to autophagy regulation. Autophagy 2009; 5:887-9; PMID:19550142; http://dx.doi.org/ 10.4161/auto.9125 [DOI] [PubMed] [Google Scholar]
- 71.Lam GY, Huang J, Brumell JH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol 2010; 32:415-30; PMID:20803017; http://dx.doi.org/ 10.1007/s00281-010-0221-0 [DOI] [PubMed] [Google Scholar]
- 72.de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, Begun J, Plantinga TS, Joosten LA, van der Meer JW, et al.. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A 2014; 111:3526-31; PMID:24550444; http://dx.doi.org/ 10.1073/pnas.1322831111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn's disease in a population-based cohort. Genes Immun 2008; 9:561-5; PMID:18580884; http://dx.doi.org/ 10.1038/gene.2008.49 [DOI] [PubMed] [Google Scholar]
- 74.Wertz I, Dixit V. A20–a bipartite ubiquitin editing enzyme with immunoregulatory potential. Adv Exp Med Biol 2014; 809:1-12; PMID:25302362; http://dx.doi.org/ 10.1007/978-1-4939-0398-6_1 [DOI] [PubMed] [Google Scholar]
- 75.Chanudet E, Huang Y, Ichimura K, Dong G, Hamoudi RA, Radford J, Wotherspoon AC, Isaacson PG, Ferry J, Du MQ. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia 2010; 24:483-7; PMID:19907439; http://dx.doi.org/ 10.1038/leu.2009.234 [DOI] [PubMed] [Google Scholar]
- 76.Bauer M, Linsel G, Fink B, Offenberg K, Hahn AM, Sack U, Knaack H, Eszlinger M, Herberth G. A varying T cell subtype explains apparent tobacco smoking induced single CpG hypomethylation in whole blood. Clin Epigenetics 2015; 7:81; PMID:26246861; http://dx.doi.org/ 10.1186/s13148-015-0113-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harlid S, Xu Z, Panduri V, Sandler DP, Taylor JA. CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the Sister Study. Environ Health Perspect 2014; 122:673-8; PMID:24704585; http://doi:10.1289/ehp.1307480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Monick MM, Beach SR, Plume J, Sears R, Gerrard M, Brody GH, Philibert RA. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am J Med Genet B Neuropsychiatr Genet 2012; 159B:141-51; PMID:22232023; http://dx.doi.org/ 10.1002/ajmg.b.32021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dahlberg CI, Sarhan D, Chrobok M, Duru AD, Alici E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front Immunol 2015; 6:605; PMID:26648934; http://dx.doi.org/ 10.3389/fimmu.2015.00605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gelato KA, Shaikhibrahim Z, Ocker M, Haendler B. Targeting epigenetic regulators for cancer therapy: modulation of bromodomain proteins, methyltransferases, demethylases, and microRNAs. Expert opinion on therapeutic targets 2016; 1-17; PMID:26799480; http://dx.doi.org/ 10.1517/14728222.2016.1134490 [DOI] [PubMed] [Google Scholar]
- 81.Hammitzsch A, Tallant C, Fedorov O, O'Mahony A, Brennan PE, Hay DA, Martinez FO, Al-Mossawi MH, de Wit J, Vecellio M, et al.. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci U S A 2015; 112:10768-73; PMID:26261308; http://dx.doi.org/ 10.1073/pnas.1501956112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campan M, Weisenberger DJ, Trinh B, Laird PW. MethyLight. Methods Mol Biol 2009; 507:325-37; PMID:18987824; http://dx.doi.org/ 10.1007/978-1-59745-522-0_23 [DOI] [PubMed] [Google Scholar]
- 83.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 84.Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1-13; PMID:19033363; http://dx.doi.org/ 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014; 30:1363-9; PMID:24478339; http://dx.doi.org/ 10.1093/bioinformatics/btu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, Hansen KD. Functional normalization of 450 k methylation array data improves replication in large cancer studies. Genome Biol 2014; 15:503; PMID:25599564; http://dx.doi.org/ 10.1186/s13059-014-0503-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013; 8:203-9; PMID:23314698; http://dx.doi.org/ 10.4161/epi.23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43(7):e47; PMID:25605792; http://doi.org/10.1093/nar/gkv007 2218183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med 1990; 9:811-8; PMID:2218183; http://dx.doi.org/ 10.1002/sim.4780090710 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.