

The vast majority of deaths from cancer are due to metastatic disease.1 Cancer cells loose cell-to-cell adhesion and apicobasal polarity and gain invasive abilities.2 These changes are partly due to the Epithelial-Mesenchymal Transition (EMT) which includes decreased expression of epithelial genes coupled with the activation of mesenchymal genes, among other factors.3 The transition is reversible and may be required for establishment of distant metastases. One integral part of the EMT program includes variations in chromatin configuration and may be related to altered expression of histone variants. Previously, one histone variant, macroH2A, has been reported to suppress the progression of melanoma through direct transcriptional upregulation of CDK8.4
We observed that decreased histone H2A.X expression led to morphological changes in HCT116 cells as well as increased invasiveness. H2A.X plays key roles in DNA double-strand break repair and genome stability, and is classified as a tumor suppressor.5 Because little is known about possible H2A.X involvement in cancer progression, we examine if dysregulation of H2A.X expression could impact the evolution of cancer. This endeavor led to our recently published study, “The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition”.6 We set out to determine the molecular pathways underlying these changes. Using genome-wide differential gene expression analysis comparing H2A.X-deficient and control HCT116 cells, we found a significant correlation between H2A.X loss and the activation of a set of mesenchymal genes (SNAI2 (Slug), ZEB1, VIM, THBS1, VCAN, TGFB2, ITGB4 among others) coupled with repression of key epithelial genes (CDH1, RAB25, SERPINB5, MAGI1), all transitions characteristic of the EMT (Fig. 1). Co-silencing the key EMT-related transcription factors Slug and ZEB1 led to EMT reversal, indicating that Slug and ZEB1 mediate the EMT driven by H2A.X loss. Similar observations were made using H2A.X knockout HCT116 cells. H2A.X removal might be expected to alter chromatin, but would it be specific? When, macroH2A.1, involved in metastasis, and H2A.Z, involved in cancer progression, were each individually depleted in HCT116 cells, there were no obvious changes in relation to EMT. Importantly, ectopic H2A.X re-expression led to a partial EMT reversal as did Slug and ZEB1 silencing, further implicating a role for H2A.X in EMT. Interestingly, we correlated the expression levels of H2A.X with those of Slug and ZEB1 in the panel of 233 human adenocarcinoma samples available in the Cancer Genome Atlas (TCGA). The transcript levels of H2A.X, Slug and ZEB1 were found significantly correlated, supporting a potential role of H2A.X in the regulation of EMT-related transcription factors in vivo.
Figure 1.
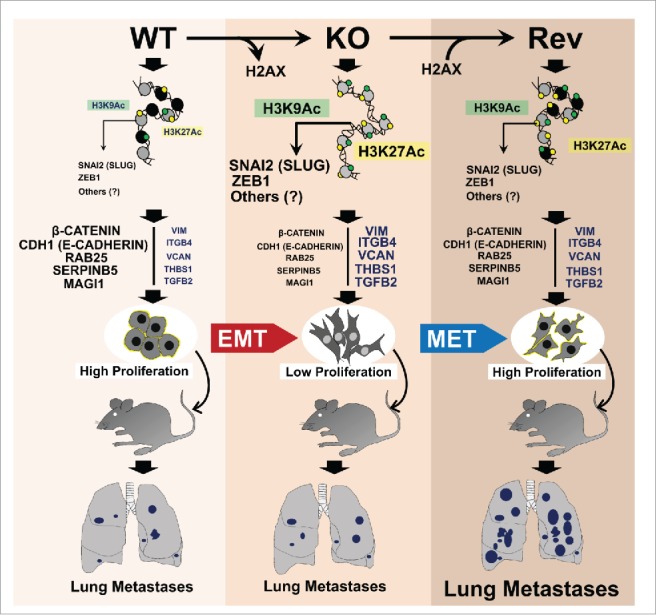
Role of H2A.X in EMT. H2A.X loss enhances Slug and ZEB1 promoter activities (high H3K9ac / H3K27ac). Consequently, increased expression of Slug and ZEB1 leads to activation and repression of mesenchymal and epithelial genes respectively, thus promoting EMT. H2A.X reintroduction in H2A.X null cells enables partial MET, increased cell proliferation and resistance to stresses, thus promoting metastatic colonization.
We next studied the mechanism underlying H2A.X loss-induced EMT. H2A.X loss was found to enhance Slug and ZEB1 promoter activities, as well as leading to an accumulation of the active transcription marks H3K9ac and H3K27ac. We surmised that H2A.X removal from the Slug and ZEB1 promoters enhanced their transcriptional activation through chromatin relaxation, leading to EMT activation (Fig. 1).
EMT is one of the hallmarks of metastasis, but there is increasing evidence for a requirement for the reverse transition (MET) in metastatic colonization.7 Utilizing immuno-compromised mice, we investigated the effects of H2A.X removal and its reintroduction on metastasis in vivo. Cells re-expressing H2A.X exhibited a significant increase in the numbers of metastatic lung nodules compared to both the H2A.X knockout and parental cells. These observations suggested some factors we had not considered. Why had the invasive H2A.X-deficient cells failed to establish high metastasis in vivo? We speculated that slow growth and a deficient DNA damage response in H2A.X-null cells made them prone to environmental stresses and defective in resuming proliferation in vivo.
Several findings support this hypothesis. First, H2A.X re-expression is correlated with the rate of cell proliferation. Second the revertant cell's ability for DNA repair and survival phenocopied parental cells, with enhanced capacity to form colonies after genotoxic stress. Third, revertants with a non-phosphorylatable H2A.X protein failed to reverse EMT and remained defective in DNA repair, indicating that EMT and DNA repair are coupled. Fourth, H2A.X revertant cells partially maintained their invasive properties and many mesenchymal markers, which may have enabled them to resume proliferation and colonize distant sites (Fig. 1). Inducible silencing of H2A.X followed by its re-expression in vivo may provide more conclusive evidence on the role of H2A.X in metastasis.
One may wonder how H2A.X loss and its re-expression could occur in vivo in a specific cell type. First, gene profiles from Gene Expression Omnibus, GEO (GSE17708), indicate that the H2A.X gene is down-regulated in the A549 lung adenocarcinoma cell line in a model of TGF-β-induced EMT. Second, (http://www.targetscan.org/ and http://mirdb.org/miRDB/) revealed that miR-24 is a putative regulator of H2A.X transcript. Perhaps variations in the regulation of the above factors could be key in H2A.X-driven EMT. Also of interest is investigating the roles of several EMT inducers, including inflammatory cytokines, oncogenic/metabolic stressors and hypoxia in H2A.X gene regulation. Finally, our findings raise the question whether H2A.X-driven EMT occurs in epithelial cells of different origins, and whether the EMT program utilizes the same molecular mechanism. Above all, our findings open a new avenue of research toward the role of histone variants in the regulation of EMT and cancer progression.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases, Radiation/Nuclear Countermeasures Program and the Intramural Research Program of the National Cancer Institute, Center for Cancer Research, National Institutes of Health.
References
- [1].Coghlin C, Murray GI. Current and emerging concepts in tumour metastasis. J Pathol 2010; 222:1-15; PMID:20681009; http://dx.doi.org/ 10.1002/path.2727 [DOI] [PubMed] [Google Scholar]
- [2].Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139:871-90; PMID:19945376; http://dx.doi.org/ 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- [3].Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15:178-96; PMID:24556840; http://dx.doi.org/ 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kapoor A,Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, et al.. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010; 468:1105-9; PMID:21179167; http://dx.doi.org/ 10.1038/nature09590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer 2008; 8:957-67; PMID:19005492; http://dx.doi.org/ 10.1038/nrc2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Weyemi U, Redon CE, Choudhuri R, Aziz T, Maeda D, Boufraqech M, Parekh PR, Sethi TK, Kasoji M, Abrams N, et al.. The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition. Nat Commun 2016; 7:10711; PMID:26876487; http://dx.doi.org/ 10.1038/ncomms10711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22:725-36; PMID:23201165; http://dx.doi.org/ 10.1016/j.ccr.2012.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]