

ABSTRACT
The development and progression of invasive breast cancer is characterized by alterations to the genome and epigenome. However, the relationship between breast tumor characteristics, disease subtypes, and patient outcomes with the cumulative burden of these molecular alterations are not well characterized. We determined the average departure of tumor DNA methylation from adjacent normal breast DNA methylation using Illumina 450K methylation data from 700 invasive breast tumors and 90 adjacent normal breast tissues in The Cancer Genome Atlas. From this we generated a novel summary measure of altered DNA methylation, the DNA methylation dysregulation index (MDI), and examined the relation of MDI with tumor characteristics and summary measures that quantify cumulative burden of genetic mutation and copy number alterations. Our analysis revealed that MDI was significantly associated with tumor stage (P = 0.017). Across invasive breast tumor subtypes we observed significant differences in genome-wide DNA MDIs (P = 4.9E–09) and in a fraction of the genome with copy number alterations (FGA) (P = 4.6E–03). Results from a linear regression adjusted for subject age, tumor stage, and estimated tumor purity indicated a positive significant association of MDI with both MCB and FGA (P = 0.036 and P < 2.2E–16). A recursively partitioned mixture model of all 3 somatic alteration burden measures resulted in classes of tumors whose epigenetic and genetic burden profile were associated with the PAM50 subtype and mutations in TP53, PIK3CA, and CDH1. Together, our work presents a novel framework for characterizing the epigenetic burden and adds to the understanding of the aggregate impact of epigenetic and genetic alterations in breast cancer.
KEYWORDS: Breast cancer, DNA methylation, deregulation, dysregulation, epigenome, TCGA
Introduction
Invasive breast carcinoma is the most common non-keratinocyte cancer in the US with an estimated 230,000 new cases and 40,000 deaths expected in 2015.1 Both genetic and epigenetic alterations are recognized contributors to breast carcinogenesis and invasive tumors accumulate additional genetic and epigenetic aberrations as the disease progresses.2 Recently, studies have demonstrated that summary measures that quantify burden of genetic alterations, including the fraction of the genome affected by copy number alterations (FGA) and somatic mutation count burden (MCB), may be useful in predicting patient outcomes.3-5 However, there is a corresponding lack of available resources that summarize the departure of DNA methylation in tumors from its component normal.
Epigenetic alterations, such as changes in DNA methylation, are well-established early events in breast cancer that serve to disrupt normal gene expression and increase chromosomal instability.6,7 Indeed, previous studies have identified that widespread differences in DNA methylation profiles exist between pre-invasive breast cancer (ductal carcinoma in situ) and normal breast tissue.8,9 As indicated by these earlier studies, the DNA methylation levels in cancer diverge in a direction dependent upon genomic location and are non-random. For example, concentrated regions of CpG dinucleotides known as CpG islands, which are typically unmethylated in non-neoplastic cells, demonstrate elevated levels of methylation in cancer that have the potential for gene silencing.10 In contrast, CpG dinucleotides in less concentrated contexts (i.e., distal from gene promoters or associated with repeat elements) are typically methylated in non-neoplastic cells to support chromosomal stability and undergo hypomethylation in cancer.10 Furthermore, regions that border CpG islands (i.e., CpG island shores) exhibit significantly altered DNA methylation in tumor compared with normal tissue.11 As such, a summary measure that quantifies the total departure of DNA methylation in tumor cells from their component normal requires a consideration of different potential directions of DNA methylation alterations in tumor compared with normal tissue depending on genomic context.
Breast tumors demonstrate heterogeneity in patterns of genetic and epigenetic alterations, both across breast cancer patients and within breast cancer molecular subtypes. The patterns of DNA methylation in relation to breast tumor characteristics, disease subtypes, and patient demographics have been well characterized. Previous research has highlighted that DNA methylation patterns vary based on age,12 molecular subtypes,13 hormone receptor status,14 and with the presence of specific mutations.15 However, the relation of a global measure of epigenetic dysregulation with tumor characteristics, such as disease subtype and patient prognosis, remains unclear. In addition, an integrative assessment of combined epigenetic and genetic burden measures represents an opportunity to better understand tumor biology and to identify potential prognostic factors.
In this study, we utilized DNA methylation data measured on the Illumina HumanMethylation450 array available from The Cancer Genome Atlas (TCGA) database and investigated the departure of DNA methylation in breast tumors from its component normal. We developed and applied a novel approach that summarizes the CpG site-specific dysregulation of tumor DNA methylation compared with referent normal tissue in a breast cancer data set. We then examined the relation of this summary measure with both mutation burden and the fraction of genome with copy number alterations. Additionally, an analysis of methylation dysregulation stratified by genomic context demonstrated that regions with differential epigenetic dysregulation are related with tumor subtypes and patient characteristics. Finally, we used an integrative approach to identify classes of tumors via their combined profile of epigenetic and genetic aberrations to characterize tumors beyond gene expression-based subtypes. Overall, this work demonstrates that a summary measure of DNA methylation dysregulation is associated with tumor stage and may have utility as a marker of cancer progression within specific tumor subtypes.
Results
Genome-wide and stratified CpG island region DNA methylation alterations
The clinical and pathological characteristics of the 700 breast cancer patients in this study are summarized in Table 1. To investigate genome-wide DNA methylation alterations in breast tumors (n = 700) and normal breast tissues (n = 90) we first calculated the mean DNA methylation β-values for each subject stratified by genomic context region as defined by relation to CpG islands (CGI; Open Sea, North Shelf, North Shore, CGI, South Shore, and South Shelf), as DNA methylation levels are known to be dependent upon genomic context.10 We then compared the mean methylation status of tumors with mean methylation status of normal samples as a function of genomic context. As anticipated, mean methylation of CpG loci in both CGI and CGI shores (regions up to 2 kb distant from CGI) were significantly higher in tumor samples compared with normal samples (CGI; Wilcoxon, P = 1.5E–33, North Shore; Wilcoxon P = 4.3 E–06, South Shore; Wilcoxon P = 6.9E–05; Fig. 1). Extending to CGI shelves (2–4 kb from CGI) and Open Sea loci (isolated CpGs), mean methylation levels were significantly lower in breast tumors compared with adjacent normal breast tissue (Open Sea; Wilcoxon P = 1.4E–07, North Shelf; Wilcoxon P = 8.9E–11, South Shelf; Wilcoxon 7.8E–11; Fig. 1). All P-values for tests of mean methylation differences between tumor and normal tissue by genomic context meet the Bonferroni corrected significance threshold of 8.3E–03.
Table 1.
Patient demographic and tumor characteristics.
| Covariates | All n = 700 (%) | CNA and Mutation n = 636 (%) | P-value | Unmatched n = 610 (%) | Matched n = 90 (%) | P-value |
|---|---|---|---|---|---|---|
| Age | ||||||
| Range | 26–90 | 26–90 | 26–90 | 28–90 | ||
| Median | 58 | 58 | 58 | 56 | ||
| Mean (sd) | 57.86 (13.1) | 58.06 (13.1) | 58.0 (12.8) | 57.18 (15.4) | ||
| Stage | ||||||
| I | 115 (16.4) | 107 (16.8) | 0.88 | 102 (16.7) | 13 (14.4) | 0.76 |
| II | 391 (55.9) | 357 (56.1) | 0.96 | 336 (55.1) | 55 (61.1) | 0.58 |
| III | 180 (25.7) | 161 (25.3) | 0.9 | 160 (26.2) | 20 (22.2) | 0.61 |
| IV | 8 (1.1) | 6 (1.0) | 0.79 | 7 (1.1) | 1 (1.1) | 1 |
| Missing | 6 (0.9) | 5 (0.8) | 1 | 5 (0.8) | 1 (1.1) | 0.56 |
| Clinical Subtype | ||||||
| TNBC | 75 (10.7) | 67 (10.5) | 0.93 | 68 (11.1) | 7 (7.8) | 0.47 |
| HER2 Clinical | 79 (11.3) | 75 (11.8) | 0.8 | 66 (10.8) | 13 (14.4) | 0.38 |
| ER+ Other | 279 (39.9) | 256 (40.3) | 0.96 | 243 (39.8) | 36 (40.0) | 1 |
| Missing | 267 (38.1) | 238 (37.4) | 0.88 | 233 (38.2) | 34 (37.8) | 1 |
| PAM50 Subtype | ||||||
| Basal | 84 (12.0) | 79 (12.4) | 0.87 | 73 (12.0) | 11 (12.2) | 1 |
| HER2-enriched | 31 (4.4) | 30 (4.7) | 0.9 | 25 (4.1) | 6 (6.7) | 0.28 |
| Luminal A | 277 (39.6) | 267 (42.0) | 0.58 | 227 (37.2) | 50 (55.6) | 0.043* |
| Luminal B | 126 (18.0) | 120 (18.9) | 0.78 | 105 (17.2) | 21 (23.3) | 0.26 |
| Normal like | 17 (2.4) | 16 (2.5) | 1 | 16 (2.6) | 1 (1.1) | 0.71 |
| Missing | 165 (23.6) | 124 (19.5) | 0.15 | 164 (26.9) | 1 (1.1) | 5.60E–08 |
| TP53Mutation | ||||||
| No | 449 (70.6) | |||||
| Yes | 187 (29.4) | |||||
| PIK3CAMutation | ||||||
| No | 432 (67.9) | |||||
| Yes | 204 (32.1) | |||||
| CDH1 Mutation | ||||||
| No | 91 (14.3) | |||||
| Yes | 545 (85.7) |
Figure 1.
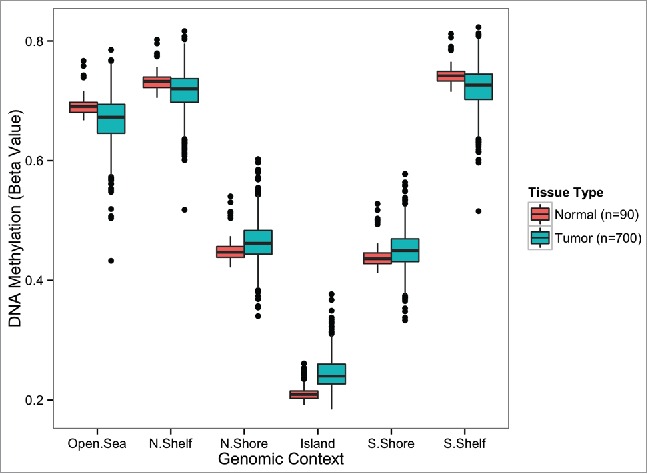
Genome-wide differences in average methylation levels between normal and tumor tissue stratified by genomic location. Average methylation levels at CGIs and CGI-shores are consistently higher in tumors compared with adjacent normal tissue (Wilcoxon rank sum test, P < 0.0005). Average methylation levels outside of CGIs (CGI-shelves and Open Sea) are consistently lower in tumors compared with adjacent normal tissue (Wilcoxon rank sum test, P < 0.0005). Significant differences are highlighted with a ‘*’ symbol. The number of autosomal CpG sites included in the calculation of average methylation for each genomic context: N.Shelf (16,455), N.Shore (49,626), Island (137,972), S.Shore (38,977), S.Shelf (21,758), Open Sea (127,120). N.Shelf, “North Shelf;“ N.Shore, “North Shore;” S.Shore, “South Shore;“ S.Shelf, “South Shelf.”
Genome-wide DNA methylation dysregulation indices
To evaluate dysregulation of tumor DNA methylation compared with normal tissue we developed a methylation dysregulation index (MDI). Briefly, our MDI measure represents the cumulative departure from normal DNA methylation in a CpG locus-specific manner calculated by summing the absolute difference in DNA methylation β-values at each CpG between each tumor sample (n = 700) and the median β-value for each CpG across all normal samples (n = 90), and then dividing by the total number of CpGs. The output from this genome-wide summary measure represents the average change in β-value for any given CpG in the tumor sample compared with normal. Therefore, a MDI value near 0 is taken to indicate a similar methylation profile to the companion normal samples while increasing levels of MDI indicate a greater extent of DNA methylation dysregulation. To evaluate the appropriateness of using a median methylation across normal breast tissues to calculate MDI in tumors we compared MDI calculated for tumors using available matched normal samples (n = 90), to the MDI in these same tumors calculated using median normal and observed high similarity of patient-matched normal and median-normal MDI values (Supplemental Fig. 1A). The distribution of MDI across tumors is shown in Supplemental Fig. 1B. In addition to calculating a genome-wide MDI, we calculated the MDI within each genomic context (i.e., Open Sea, CGI, CGI shores, and CGI shelves). The highest observed context-specific MDI levels were in the Open Sea region, and the lowest observed levels were in CGIs (Supplemental Fig. 1C).
Recently, Yang et al. developed a DNA methylation “instability” index to measure aberrant DNA methylation in cancer.16 In that study, the authors were motivated to determine whether distinct epigenetic pathways controlled hyper- and hypo-methylation in a pan-cancer analysis. Notably, the authors measured the hypermethylation deviation of cancerous tissues from normal via averaging Z-scores (HyperZ score) across probes in promoter CGIs while hypomethylation was determined by averaging Z-scores (HypoZ score) across Open Sea probes that were not located in promoter regions. With the autosomal probes available in our data set we generated HyperZ and HypoZ scores using the methods described in Yang et al. and compared those values to the CGI probe-specific and Open Sea probe-specific MDI values. Departure of tumor methylation from normal at CGIs and Open Sea probes were more highly correlated when MDI was used (Spearman; r = 0.65, P < 2.2E–16) in place of the Z-score approach (Spearman; r = 0.14, P = 2.0E–4, Supplemental Figs. 2A and 2B). As expected CpG Island MDI is strongly correlated with HyperZ score (Spearman; r = 0.85, P < 2.2E–16) and Open Sea MDI is strongly correlated with the HypoZ score (Spearman; r = 0.79, P < 2.2E–16, Supplemental Fig. 2C and 2D). Notably, a subset of samples had near zero HyperZ and HypoZ scores whereas the respective tumor MDI values for those samples were far greater than zero (Supplemental Fig. 2C and 2D).
Methylation Dysregulation Index (MDI) is associated with patient age and tumor characteristics
Subject age, tumor stage, and breast tumor subtype are each independently related with breast cancer prognosis. To understand whether methylation dysregulation increases with disease progression we examined the association between genome-wide MDI and patient and tumor characteristics related with prognosis. Results from a linear regression indicated significant positive associations of age (P = 1.3E–07, Supplemental Fig. 3A) and tumor stage (P = 0.013, Supplemental Fig. 3B) with MDI across all tumors adjusting for TCGA estimated tumor purity. While this association for age was consistent for each MDI value from specific genomic contexts, after applying the Bonferroni correction threshold, the positive association for tumor stage was only present in the CpG Island context (Supplemental Fig. 3B–D).
We next tested the relation of genome-wide MDI and genomic-context-specific MDI with clinical [ER+, HER2+, triple negative breast cancer (TNBC), available n = 433], and Prediction Analysis of Microarray 50-gene classifier (PAM50) tumor subtypes (Luminal A, Luminal B, Basal, HER2-enriched, and normal-like, available n = 535). Genome-wide MDI was significantly different among clinical subtypes (Kruskal P-value = 0.019, Supplemental Fig. 4A). Testing the relation of genomic-context-specific MDI with clinical subtype indicated a significant relation with CpG island MDI (Kruskal P = 4.7E–05, Supplemental Fig. 4B), though not other genomic regions suggesting that altered CpG island methylation is driving the relation of genome-wide MDI with clinical subtype. Further, we observed a significant difference in genome-wide MDI among PAM50 subtypes (P < 2.2E–16; Fig. 2A). Luminal B subtype tumors had significantly higher MDI than the other 4 subtypes (Fig. 2A) and this relationship was consistent for genomic-context-specific MDI (Supplemental Fig. 4C, all P-values < 2.2E–16).
Figure 2.
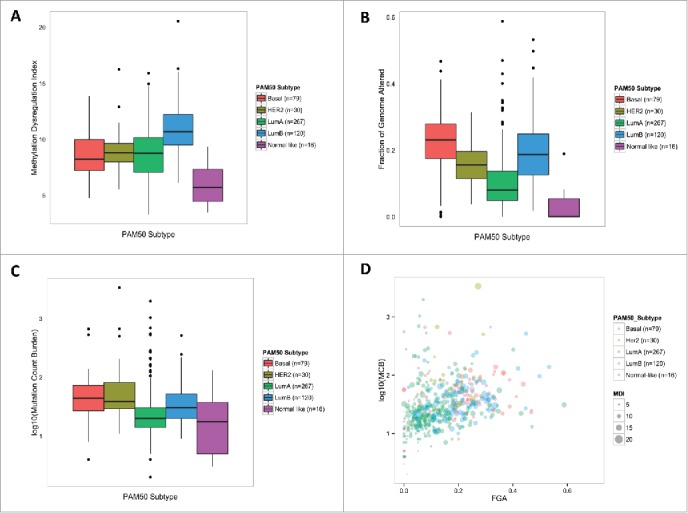
Differential molecular alteration burden among PAM50 subtypes. (A) Methylation dysregulation is significantly different among PAM50 subtypes (Kruskal, P = 1.2E–19). (B) Fraction of the genome affected by copy number alterations is significantly different among PAM50 subtypes (Kruskal, P = 5.3E–30). (C) Mutation Count Burden is significantly different among PAM50 subtypes (Kruskal, P = 3.3E–15) (D) Illustrates the relationship of the 3 molcular alteration burden measures among PAM50 subtypes. Log(MCB) is plotted versus FGA, while increasing bubble diameter corresponds with increasing MDI.
Genetic alteration burden measures are associated with MDI
To better understand the relationship between genetic alteration burden measures and MDI we used data on the fraction of the genome with copy number alterations (FGA), and mutation count burden (MCB) for these tumors. First testing the relation of genetic alteration summary measures with patient and tumor characteristics we observed that subject age was associated with a significant increase in MCB (P = 9.0E–03, Supplemental Fig. 5A), but age was not significantly associated with FGA (P = 0.84, Supplemental Fig. 5B). Interestingly, unlike the observed significant associations with MDI, increasing tumor stage was not associated with either MCB (P = 0.54) or FGA (P = 0.33). Further, both FGA and MCB were significantly different among clinical subtypes, with TNBC tumors exhibiting the highest levels in both genetic alteration burden measures (Kruskal, P = 5.7E–08 and P = 1.7E–07, respectively, Supplemental Fig. 6A and B). Similarly, FGA and MCB were significantly different among PAM50 subtypes (Kruskal, P = 5.3E–30 and P = 3.3E–15, respectively). Basal-like tumors exhibited the highest levels of FGA and MCB (high degree of overlap between TNBC and basal tumors), while Luminal A tumors had the lowest levels of the 2 genetic alteration measures (Fig. 2B and C).
Next, we sought to examine whether the genetic alteration burden summary measures of MCB and FGA were associated with MDI. Results from a linear regression that adjusted for subject age, tumor stage, and TCGA estimated tumor purity indicated a significant positive association of MDI with both MCB and FGA (P = 0.036 and P < 2.2E–16, respectively, Supplemental Fig. 7A–B). To determine whether a particular subtype was driving the observed association between MDI and genetic alterations, we performed clinical subtype and PAM50 subtype stratified linear regression models adjusting for subject age, tumor subtype, and estimated tumor purity. Overall, methylation dysregulation in all 3 clinical subtypes were positively associated with FGA, but only the HER2-positive subtype was positively associated with MCB (Table 2). Among the PAM50 subtypes, our analyses revealed disparate associations between MDI and genetic summary measures. MDI in Basal, HER2-enriched, and Luminal A, all subtypes demonstrated positive associations with FGA while Luminal B— the subtype with the greatest MDI—was not significantly associated with FGA (Table 2, Fig. 2D). In contrast, we found that only MDI in HER2-enriched tumors exhibited a positive association with mutation burden, though (Table 2).
Table 2.
Linear Regression of MDI with FGA and MCB among breast cancer subtypes.
|
Fraction of the genome altered |
Mutation count |
|||
|---|---|---|---|---|
| Coefficient | P-value* | Coefficient | P-value* | |
| All Subjects (n = 636) | 8.63 | <2.2E–16** | 1.20E–03 | 0.036* |
| Clinical Subtype | ||||
| TNBC (n = 67) | 5.74 | 4.1E–03** | 1.10E–03 | 0.57 |
| HER2+ (n = 75) | 11.64 | 1.7E–07** | 1.90E–03 | 5.2E–03** |
| ER+ Other (n = 256) | 10.65 | 2.0E–10** | −3.50E–04 | 0.73 |
| PAM50 Subtype | ||||
| Basal (n = 79) | 6.21 | 3.3E–03** | 7.30E–04 | 0.72 |
| HER2-enriched (n = 30) | 16 | 5.6E–03** | 1.80E–03 | 1.6E–03** |
| Luminal A (n = 267) | 9.8 | 1.3E–09** | −2.00E-04 | 0.83 |
| Luminal B (n = 120) | −1.65 | 0.47 | −3.00E-03 | 0.41 |
| Normal-like (n = 16) | 21.2 | 0.018* | 0.014 | 0.25 |
Linear Regression adjusted for subject age, tumor stage, and TCGA estimated tumor purity.
Significant after Bonferroni correction.
Integrative analysis of somatic alteration burden types
To integrate data from all 3 somatic alteration burden measures (MDI, FGA, and MCB) and generate latent profiles of cumulative somatic alteration burden in TCGA breast tumors we employed a model-based clustering approach. We generated classes of tumors using recursively partitioned mixture modeling (RPMM) that resulted in 6 classes of tumor samples (Fig. 3A). Notably, tumors were clustered into either high molecular alteration burden (rR classes) or low molecular alteration burden (rL classes), and each of the high molecular alteration burden classes exhibited exceptionally high levels in only one of the alteration measures. Modeled high somatic alteration burden classes had distinct levels of somatic alteration, for example, tumors with high mutation burden (rRLL, n = 18), high copy number alteration (rRLR, n = 32), and high methylation dysregulation (rRR, n=287, Fig. 3). PAM50 subtype was significantly associated with somatic alteration class membership (P = 1.0E–06, Fig. 3), with the majority of Luminal B tumors (88%) residing in the rRR class, and 46% of Luminal A tumors residing in the rLR class. Additionally, Luminal A tumors account for 72% of the tumors in the high mutation-burden class (rRLL) and Basal tumors account for 44% of the tumors in the high copy number-burden class (rRLR).
Figure 3.
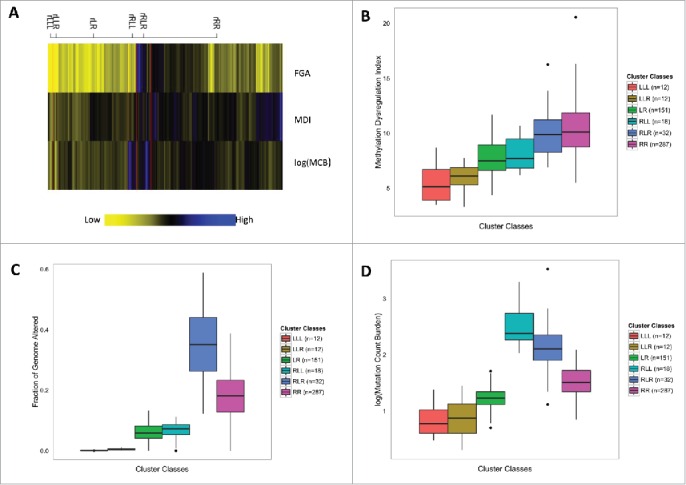
Recursively partitioned mixture model of molecular alteration burden measures in breast carcinomas. (A) The figure depicts the results of RPMM. Columns represent molecular alteration burden classes and rows represent one of the 3 burden measures (MDI, FGA, MCB). The height of each column is proportional to the number of subjects residing in the class, total n = 512. Yellow indicates low alteration burden and blue indicates high alteration burden. (B) Methylation dysregulation is significantly different among cluster classes (Kruskal-Wallis, P = 7.5E–36). (C) Fraction of the genome affected by copy number alterations is significantly different among cluster classes (Kruskal-Wallis, P = 3.4E–65). (D) Mutation Count Burden is significantly different among cluster classes (Kruskal-Wallis, P = 5.4E–51).
To understand whether the aggregate impact of epigenetic and genetic alterations varies based on other prognostic factors, we tested the relation of RPMM class membership with other patient and tumor characteristics. While age was not significantly associated with somatic alteration classes (P = 0.25, Table 3), tumor stage approached statistical significance (P=0.061, Table 3). Gene mutations frequently present in breast tumors were significantly associated with class membership, including TP53 (P = 1.0E–06, Table 3) mutation in 44% of rRLR and 40% of rRR, PIK3CA (P = 1.0E–04, Table 3) mutation in 44% of rLR and 59% of rRLL, and CDH1 (P = 1.0E–06, Table 3) in 53% of rRLL and 17% of rLLL. As the relationship between specific mutations and PAM50 subtypes are already established, we next tested the relation of each of these mutations with class membership when controlling for tumor subtype with unconditional logistic regression. Tumors with TP53 mutations were significantly more likely to be in class rRR [OR 2.35 (1.37, 4.10); P = 2.2E–03] compared with membership in any other class, while tumors with PIK3CA [OR 3.02 (1.05, 9.55); P = 0.045], and CDH1 [OR 9.6 (3.29, 29.5); P = 3.94E–05] mutations had significantly increased odds of membership in class rRLL. Although relatively low proportions of tumors had mutations in epigenetic master regulatory genes, a mutation in at least one of these genes was significantly associated with RPMM class membership (P = 0.01, Table 3) with 33% of rRLL tumors and 25% of rRLR tumors having at least one mutation in an epigenetic master regulatory gene. More specifically, somatic alteration burden class membership was significantly associated with mutations in specific genes including DNMT3A (P = 5.7E–03), TET2 (P = 9.0E–04), and IDH1 (P = 1.2E–03). However, in separate unconditional logistic regression models adjusted for MCB, the odds of class membership in the mutation burden class (rRLL), and the copy number class (rRLR), was not significantly different for tumors with a mutation in an epigenetic master regulatory gene. Lastly, we modeled somatic alteration burden, again using RPMM, stratified by PAM50 tumor subtype and observed similar relations of subject age, tumor stage, and mutation of specific genes with somatic alteration burden class membership (Supplementary Figs. 8–12).
Table 3.
RPMM alteration burden class membership by patient demographic and tumor characteristic covariates
| Class 1 (LLL) | Class 2 (LLR) | Class 3 (LR) | Class 4 (RLL) | Class 5 (RLR) | Class 6 (RR) | Permutation | |
|---|---|---|---|---|---|---|---|
| Covariates | n = 12 | n = 12 | n = 151 | n = 18 | n = 32 | n = 287 | test P-value* |
| Age (years) | 0.28 | ||||||
| Range | 31–73 | 28–71 | 30–88 | 29–77 | 36–90 | 26–90 | |
| Median | 54 | 55.5 | 58 | 61 | 58 | 58 | |
| Mean (sd) | 54.6 (12.7) | 54.3 (11.5) | 57.3 (12.7) | 58.7 (13.9) | 59.8 (14.3) | 58.1 (13.1) | |
| Stage | 0.061 | ||||||
| I | 2 (16.7) | 3 (25.0) | 32 (21.2) | 6 (33.3) | 4 (12.5) | 38 (13.2) | |
| II | 7 (58.3) | 6 (50.0) | 84 (55.6) | 7 (38.9) | 20 (62.5) | 167 (58.2) | |
| III | 3 (25.0) | 2 (16.7) | 34 (22.5) | 5 (27.8) | 5 (15.6) | 76 (26.5) | |
| IV | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) | 2 (6.3) | 3 (1.0) | |
| PAM50 Subtyoe | 1.00E–06 | ||||||
| Basal | 0 (0.0) | 1 (8.3) | 10 (6.6) | 2 (11.1) | 14 (43.8) | 52 (18.1) | |
| HER2-enriched | 0 (0.0) | 0 (0.0) | 6 (4.0) | 1 (5.6) | 3 (9.4) | 20 (7.0) | |
| Luminal A | 5 (41.7) | 10 (83.3) | 125 (82.8) | 13 (72.2) | 7 (21.9) | 107 (37.3) | |
| Luminal B | 0 (0.0) | 0 (0.0) | 6 (4.8) | 1 (5.6) | 8 (25.0) | 105 (36.6) | |
| Normal-like | 7 (58.3) | 1 (8.3) | 4 (3.2) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| TP53 Mutation | 1.00E–06 | ||||||
| No | 12 (100.0) | 11 (91.7) | 127 (84.1) | 14 (77.8) | 18 (56.3) | 171 (59.6) | |
| Yes | 0 (0.0) | 1 (8.3) | 24 (15.9) | 3 (16.6) | 14 (43.7) | 113 (39.4) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| PIK3CA Mutation | 1.00E–04 | ||||||
| No | 12 (100.0) | 10 (83.3) | 85 (56.3) | 7 (38.9) | 24 (75.0) | 203 (70.7) | |
| Yes | 0 (0.0) | 2 (16.7) | 66 (43.7) | 10 (55.5) | 8 (25.0) | 81 (28.3) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| CDH1 Mutation | 2.00E–04 | ||||||
| No | 10 (83.3) | 11 (91.7) | 130 (86.1) | 8 (44.4) | 28 (87.5) | 262 (91.3) | |
| Yes | 2 (16.7) | 1 (8.3) | 21 (13.9) | 9 (50.0) | 4 (12.5) | 22 (7.7) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| Master Regulatory Gene Mutation | 0.0104 | ||||||
| No | 10 (83.3) | 11 (91.7) | 139 (92.1) | 11 (61.1) | 24 (75.0) | 247 (86.1) | |
| Yes | 2 (16.7) | 1 (8.3) | 12 (7.9) | 6 (33.3) | 8 (25.0) | 37 (12.9) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| DNMT3B Mutation | 0.25 | ||||||
| No | 12 (100.0) | 12 (100.0) | 151 (100.0) | 17 (94.4) | 31 (96.9) | 276 (96.2) | |
| Yes | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (3.1) | 8 (2.8) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| DNMT3A Mutation | 7.70E–03 | ||||||
| No | 12 (100.0) | 11 (91.7) | 149 (98.7) | 15 (83.3) | 30 (93.8) | 282 (98.3) | |
| Yes | 0 (0.0) | 1 (8.3) | 2 (1.3) | 2 (11.1) | 2 (6.2) | 2 (0.7) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| DNMT1 Mutation | 0.2 | ||||||
| No | 12 (100.0) | 12 (100.0) | 150 (99.3) | 16 (88.8) | 31 (96.9) | 271 (94.4) | |
| Yes | 0 (0.0) | 0 (0.0) | 1 (0.7) | 1 (5.6) | 1 (3.1) | 13 (4.6) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| TET1 Mutation | 0.65 | ||||||
| No | 12 (100.0) | 12 (100.0) | 150 (99.3) | 17 (94.4) | 31 (96.9) | 279 (97.2) | |
| Yes | 0 (0.0) | 0 (0.0) | 1 (0.7) | 0 (0.0) | 1 (3.1) | 5 (1.8) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| TET2 Mutation | 6.00E–04 | ||||||
| No | 12 (100.0) | 12 (100.0) | 151 (100.0) | 14 (77.8) | 29 (90.6) | 279 (97.2) | |
| Yes | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (16.6) | 3 (9.4) | 5 (1.8) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| IDH1 Mutation | 2.40E–03 | ||||||
| No | 10 (83.3) | 12 (100.0) | 147 (97.4) | 16 (88.8) | 30 (93.8) | 283 (98.6) | |
| Yes | 2 (16.7) | 0 (0.0) | 4 (2.6) | 1 (5.6) | 2 (6.2) | 1 (0.4) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) | |
| IDH2 Mutation | 0.76 | ||||||
| No | 11 (91.7) | 12 (100.0) | 146 (96.7) | 17 (94.4) | 31 (96.9) | 275 (95.8) | |
| Yes | 1 (8.3) | 0 (0.0) | 5 (3.3) | 0 (0.0) | 1 (3.1) | 9 (3.2) | |
| Missing | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 3 (1.0) |
Fisher's exact permutation tests were performed on categorical variables and Kruskal Wallis permutation tests were performed on continuous variables 10,000 permutations were used.
Discussion
The extent of aberrant DNA methylation and genetic alterations are known to vary widely across tumors, including invasive breast cancer.15 Prior assessments of total genomic deregulation have largely been limited to studies that investigated tumor-type variation in pan cancer analyses.16-20 In this study, we quantified the cumulative burden of DNA methylation dysregulation in breast cancer using a methylation dysregulation index. We investigated the relation of MDI in combination with summary measures of genetic alteration to discover patterns of somatic alteration burden among tumors. Importantly, while we observed significant correlations between epigenetic and genetic dysregulation measures, only MDI was associated with increasing tumor stage. We also observed significant differences among breast cancer subtypes for MDI and FGA, but not for MCB. Furthermore, we integrated our novel epigenetic burden measures with other genetic burden measures via a model-based clustering method to uncover latent tumor classes with distinct patterns of somatic alteration burden.
Previous attempts to characterize genome-wide DNA methylation changes in cancer by a summary measure have not included the entire measured DNA methylome. Recently, Yang et al. investigated master epigenetic regulatory enzymes that govern hypermethylation and hypomethylation processes and focused their investigation on CpG islands in promoters and Open Sea CpGs in non-promoters. Their focused approach nicely distilled the complex relationship that exists between expression of epigenetic enzymes and hypo/hypermethylation. We extended the approach of Yang et al. to characterize the departure of DNA methylation from normal across all measured CpGs and ascribed equal weights to all genomic locations to determine cumulative DNA methylation dysregulation for each tumor. Results from our approach were strongly correlated with results from Yang et al., though including DNA methylation measurements from all CpGs resulted in stronger associations between hypomethylation and hypermethylation events. This is likely due to the inclusion CGI-shore and -shelf regions that exhibited a substantial departure from normal DNA methylation patterns. Indeed, CGI-shore regions appear to drive many gene expression differences that distinguish normal tissues from each other, and are highly dysregulated in cancer. In contrast to the work from Yang et al., our results suggest that tumors with high levels of hypermethylation also have greater levels of hypomethylation. We postulate that hypermethylation and hypomethylation that occur in the majority of genomic contexts is highly coupled, while certain more specific features of epigenetic dysregulation, as explored in the Yang et al. paper, are distinct processes.
The deregulation of both the epigenome and genome are both early events in breast carcinogenesis, and have been observed in pre-neoplastic lesions, such as ductal carcinoma in situ (DCIS).8,9 Nevertheless, there is a paucity of research that investigates epigenetic and genetic deregulation as invasive disease evolves to more advanced stages. Interestingly, our results demonstrate that the burden of genetic alterations (i.e., MCB and FGA) vary little between early and late stages of invasive disease. However, consistent with candidate gene approaches,21 our results indicate that genome-wide DNA methylation dysregulation (MDI) continues to increase over the progression of invasive disease. A possible explanation for this observation is that as a tumor progresses the increasing level of MDI may enable a tumor greater plasticity to adapt to its environment (consistent with dedifferentiation), or possibly reflect the emergence of treatment resistant cellular populations.22 Consequently, quantification of DNA methylation dysregulation of a tumor may offer a marker of disease progression and treatment response in other cancers as well.
Invasive breast carcinoma is a complex and heterogeneous disease with established tumor subtypes. Tumor subtype classification represents an approach to stratify tumor samples into potentially meaningful categories that may help guide treatment decisions.23 At the same time, tumors within a given subtype may exhibit high variability in molecular alterations not used to classify the tumors (i.e., not gene expression or receptor protein levels). In the present study, we noted that tumors with the highest burden levels of MDI, FGA, and MCB belonged to distinct clusters when we applied RPMM. Importantly, while PAM50 subtype was associated with RPMM class, there was a mixture of subtypes present in all classes. This result suggests that it may be feasible to encapsulate extensive amounts of genomic data and gain deeper biological insights by clustering the aggregate measures of molecular alterations. Nevertheless, it remains to be determined whether a propensity for high somatic alteration burden of a particular type is driven by alterations to key pathways or master regulatory genes. For example, irrespective of breast tumor subtype, the methylation dysregulation class was significantly enriched for TP53 mutations, suggesting that TP53 alterations may play an integral role in further epigenetic deregulation. Indeed, among normal-like tumors, those with a TP53 mutation were far more likely to exhibit higher MDI. This result was not present in TCGA probe-level analysis (574 probes),15 which is likely a product of a focus on CpG island probes—excluding many hypomethylation events and dysregulation in other key genomic contexts. Similarly, both PIK3CA and CDH1 mutations were enriched in the somatic alteration burden class with the highest mutation burden, suggesting that deregulation at these genes may contribute to higher levels of genetic instability compared with other gene mutations. Together, we have shown that compression of DNA methylation dysregulation data to a single, comprehensive measure and integration of global measures reveal unique characteristics about breast tumor genomes and provides etiologic information beyond standard RNA-based signatures and probe-level clustering.
While our approach to aggregate molecular alterations across distinct genomic data sets may improve tumor characterizations, our analyses have several limitations. For example, the samples involved in the present study were not collected in a population-based manner and, therefore the distribution of clinical and intrinsic tumor subtypes is skewed. Another major limitation is that the measures of MDI, FGA, and MCB are unable to account for cellular composition that may vary across tumor samples in the TCGA population.24-26 Further, while the comprehensive nature of the TCGA effort (including DNA methylation, exome sequencing, intrinsic subtyping with gene expression, and copy number alteration profiling), and its sample size is unmatched by any other studies and regarded as a strength, we were limited by the lack of a validation cohort. Finally, follow-up time in the TCGA data was insufficient to perform a survival analysis that would be necessary to test the relationship between MDI, FGA, and MCB with survival and recurrence outcomes. Future studies that directly assess genetic and epigenetic tumor burden measures in breast and other tumor types will be required to yield more robust observations.
In summary, our method to assess DNA methylation dysregulation in tumors is comprehensive and provides an intuitive interpretation of the departure from normal tissue that allows for direct comparison with genetic alteration burden measures. Our approach is also broadly applicable to other tumor types with adjacent normal samples and may highlight differences in epigenetic burden across tumors when applied in a pan-cancer analysis. Finally, an integration of epigenetic and genetic burden measures suggests that irrespective of molecular or clinical subtype, breast tumors may carry a characteristic deregulation burden profile.
Materials and methods
Population and methylation data processing
Level 3 normalized DNA methylation data and clinical information was accessed and downloaded from The Cancer Genome Atlas (TCGA) data portal.15 In the TCGA invasive breast cancer (BRCA) data set there were 841 samples for which Illumina HumanMethylation450 methylation data was available. We restricted our analyses to only those patients for whom there were both clinical information and tumor methylation from the Illumina HumanMethylation450 BeadChip (n = 700). Metastatic samples and samples with repeated measurements were removed prior to analysis. Among the 700 subjects, there were 90 subjects for which DNA methylation data on adjacent normal tissue was also available. For the DNA methylation data we removed probes on sex chromosome and analyzed autosomal CpGs available in the TCGA BRCA data set. The number of autosomal CpGs in each genomic region is summarized in Supplemental Table 1.
Definition of breast tumor subtypes
Expression of standard immunohistochemistry (IHC) biomarkers including estrogen receptor (ER), progesterone receptor (PR), and HER2 were used to define clinical subtypes. These were classified into 3 groups: triple negative (ER-, PR-, HER2-), HER2+ (ER+/ER-, PR+/PR-, HER2+), and ER+ other (ER+, PR+/PR-, HER2-). Due to its prognostic value, we also used PAM50 classification of breast tumor intrinsic subtypes: Basal-like, HER2-enriched, Luminal A, Luminal B, and Normal-like.30
DNA methylation dysregulation index construction
To summarize the genome-wide departure of DNA methylation in tumor samples from normal tissues we constructed a CpG locus-by-locus index of the mean, absolute difference in DNA methylation in tumor samples compared with adjacent normal. First, we generated a discrete matched MDI , which used only the 90 subjects with tumor and corresponding adjacent normal methylation data. For a given subject, and were the DNA methylation β values for tumor and normal, respectively, at each CpG-probe locus on the Illumina 450K array, and was the total number of CpG-probes.
To be able to extend our MDI assessment to tumors without a matched normal sample we used median CpG site-specific DNA methylation values among normal breast tissues to calculate MDI. In this case, tumor MDI was a genome-wide summary of locus-by-locus deviation in DNA methylation of a given tumor sample from median methylation of all adjacent normal samples. is the median β−value at each CpG locus among all normal samples. Median normal MDI was compared with MDIdm for the 90 matched tumor-normal subjects and then calculated for all tumor samples.
We observed high similarity of patient-matched normal and median-normal MDI values (Supplemental Fig. 13) and, as a result, we proceeded to calculate MDI for all tumor samples using the CpG site-specific median normal DNA methylation. Clinical and pathological characteristics of all subjects stratified by those with a matched normal sample (n = 90) and those without (n = 610) are also provided in Table 1. Subject and tumor characteristic distributions were similar for matched and unmatched tumors (Table 1). In addition to this genome-wide index, the same approach was used to construct methylation indices stratified by genomic context of CpG Island genomic regions including: Open Sea, North Shelf, North Shore, Island, South Shore, and South Shelf.10
Mutation and copy number alteration burden
Data for mutation count burden (MCB) and copy number alteration burden (FGA) for each subject were downloaded from the cBioPortal for the aforementioned TCGA BRCA subjects (n = 700).4 The cBioPortal accesses TCGA BRCA data to generate MCB from the total number of non-synonymous substitutions in exome sequencing while the FGA measure is calculated as the fraction of the genome affected by copy number alterations (i.e., the number of bases in segments with mean log2 greater than 0.2 or smaller than −0.2 divided by the number of bases in all segments profiled by the Affymetrix SNP arrays).4 Among the 700 TCGA BRCA subjects in our data set, 636 subjects had mutation and copy number alteration data. In addition, we accessed the mutation status of genes with a mutation frequency of at least 10% and specific epigenetic master regulatory genes (<10% prevalence), in the BRCA dataset. Genes with a high mutation frequency included: TP53, PIK3CA, and CDH1 while the epigenetic master regulatory genes included: DNMT3A, DNMT3B, DNMT1, TET1, TET2, IDH1, and IDH2.27
Statistical analysis
All analyses were performed using the R computing framework version 3.1.1 (www.r-project.org). To test for differences in the distribution of clinical and pathological characteristics between patients with matched tumor-normal methylation and subjects with unmatched tumor methylation Fisher's exact tests were used. Fisher's exact tests were also used to determine differences in clinical and pathological characteristics between the full data set (n = 700) and subjects with mutation and copy number alteration data (n = 636). Wilcoxon tests were used to test for differences in average DNA methylation between adjacent normal tissue and tumors. Kruskal-Wallis tests were implemented to test for differences in MDI, FGA, and MCB among tumor subtypes. Wilcoxon tests were used to test for differences in MDI among binary clinical and pathological characteristics. Linear regression models adjusted for age, tumor stage, and TCGA estimated tumor purity were used to test the association of MDI with MCB and FGA. Recursively partitioned mixture model (RPMM) clustering was performed using the MDI, FGA, and MCB of subjects with an identified PAM50 subtype.28 To accurately profile subjects into somatic burden classes, we normalized each burden measure before applying the RPMM clustering.29 To test the relation of somatic dysregulation RPMM class Fisher's exact permutation tests were performed on categorical variables and Kruskal Wallis permutation tests were performed on continuous variables. Permutation tests (running 10,000 permutations) were used to test for association with dysregulation class by generating a distribution of the test statistic for the null distribution for comparison with the observed distribution. All results with a P-value < 0.05 were considered statistically significant. Bonferroni correction was used and noted when multiple hypotheses were tested.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Institutes of Health (grant numbers P20GM104416/8189 to [BCC]).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5-29; PMID:25559415; http://dx.doi.org/10.3322/caac.21332 [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 3.Birkbak NJ, Kochupurakkal B, Izarzugaza JM, Eklund AC, Li Y, Liu J, Szallasi Z, Matulonis UA, Richardson AL, Iglehart JD, et al.. Tumor mutation burden forecasts outcome in ovarian cancer with BRCA1 or BRCA2 mutations. PloS One 2013; 8:e80023; PMID:24265793; http://dx.doi.org/ 10.1371/journal.pone.0080023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; http://dx.doi.org/ 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, Xiao Y, Heguy A, Huberman K, Bernstein M, Assel M, et al.. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A 2014; 111:11139-44; PMID:25024180; http://dx.doi.org/ 10.1073/pnas.1411446111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Hoesel AQ, Sato Y, Elashoff DA, Turner RR, Giuliano AE, Shamonki JM, Kuppen PJ, van de Velde CJ, Hoon DS. Assessment of DNA methylation status in early stages of breast cancer development. Br J Cancer 2013; 108:2033-8; PMID:23652305; http://dx.doi.org/ 10.1038/bjc.2013.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene 2002; 21:5462-82; PMID:12154408; http://dx.doi.org/ 10.1038/sj.onc.1205606 [DOI] [PubMed] [Google Scholar]
- 8.Fleischer T, Frigessi A, Johnson KC, Edvardsen H, Touleimat N, Klajic J, Riis ML, Haakensen VD, Warnberg F, Naume B, et al.. Genome-wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol 2014; 15:435; PMID:25146004; http://dx.doi.org/10.1186/PREACCEPT-2333349012841587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson KC, Koestler DC, Fleischer T, Chen P, Jenson EG, Marotti JD, Onega T, Kristensen VN, Christensen BC. DNA methylation in ductal carcinoma in situ related with future development of invasive breast cancer. Clin Epigenetics 2015; 7:75; PMID:26213588; http://dx.doi.org/ 10.1186/s13148-015-0094-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484-92; PMID:22641018; http://dx.doi.org/ 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- 11.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al.. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41(2):178-186; PMID:19151715; http://dx.doi.org/ 10.1038/ng.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson KC, Koestler DC, Cheng C, Christensen BC. Age-related DNA methylation in normal breast tissue and its relationship with invasive breast tumor methylation. Epigenetics 2014; 9:268-75; PMID:24196486; http://dx.doi.org/ 10.4161/epi.27015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stefansson OA, Moran S, Gomez A, Sayols S, Arribas-Jorba C, Sandoval J, Hilmarsdottir H, Olafsdottir E, Tryggvadottir L, Jonasson JG, et al.. A DNA methylation-based definition of biologically distinct breast cancer subtypes. Mol Oncol 2015; 9:555-68; PMID:25468711; http://dx.doi.org/ 10.1016/j.molonc.2014.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, et al.. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med 2011; 3:75ra25; PMID:21430268; http://dx.doi.org/ 10.1126/scitranslmed.3001875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas N . Comprehensive molecular portraits of human breast tumours. Nature 2012; 490:61-70; PMID:23000897; http://dx.doi.org/ 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Z, Jones A, Widschwendter M, Teschendorff AE. An integrative pan-cancer-wide analysis of epigenetic enzymes reveals universal patterns of epigenomic deregulation in cancer. Genome Biol 2015; 16:140; PMID:26169266; http://dx.doi.org/ 10.1186/s13059-015-0699-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 2013; 45:1127-33; PMID:24071851; http://dx.doi.org/ 10.1038/ng.2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V, et al.. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014; 158:929-44; PMID:25109877; http://dx.doi.org/ 10.1016/j.cell.2014.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al.. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499:214-8; PMID:23770567; http://dx.doi.org/ 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al.. Pan-cancer patterns of somatic copy number alteration. Nat Genet 2013; 45:1134-40; PMID:24071852; http://dx.doi.org/ 10.1038/ng.2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klajic J, Fleischer T, Dejeux E, Edvardsen H, Warnberg F, Bukholm I, Lonning PE, Solvang H, Borresen-Dale AL, Tost J, et al.. Quantitative DNA methylation analyses reveal stage dependent DNA methylation and association to clinico-pathological factors in breast tumors. BMC Cancer 2013; 13:456; PMID:24093668; http://dx.doi.org/ 10.1186/1471-2407-13-456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stone A, Zotenko E, Locke WJ, Korbie D, Millar EK, Pidsley R, Stirzaker C, Graham P, Trau M, Musgrove EA, et al.. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat Commun 2015; 6:7758; PMID:26169690; http://dx.doi.org/ 10.1038/ncomms8758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al.. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009; 27:1160-7; PMID:19204204; http://dx.doi.org/ 10.1200/JCO.2008.18.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houseman EA, Ince TA. Normal cell-type epigenetics and breast cancer classification: a case study of cell mixture-adjusted analysis of DNA methylation data from tumors. Cancer Inform 2014; 13(Suppl 4):53-64; PMID:25574126; http://dx.doi.org/ 10.4137/CIN.S13980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 2014; 30(10):1431-1439; PMID:24451622; http://dx.doi.org/ 10.1093/bioinformatics/btu029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houseman EA, Kelsey KT, Wiencke JK, Marsit CJ. Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinformatics 2015; 16(1):95; PMID:25887114; http://dx.doi.org/ 10.1186/s12859-015-0527-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy DM, Walsh LA, Chan TA. Driver mutations of cancer epigenomes. Protein Cell 2014; 5:265-96; PMID:24622842; http://dx.doi.org/ 10.1007/s13238-014-0031-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Houseman EA, Christensen BC, Yeh RF, Marsit CJ, Karagas MR, Wrensch M, Nelson HH, Wiemels J, Zheng S, Wiencke JK, et al.. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of β distributions. BMC Bioinformatics 2008; 9:365; PMID:18782434; http://dx.doi.org/ 10.1186/1471-2105-9-365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Berg RA, Hoefsloot HC, Westerhuis JA, Smilde AK, van der Werf MJ. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics 2006; 7:142; PMID:16762068; http://dx.doi.org/ 10.1186/1471-2164-7-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.