

ABSTRACT
Human monocytes are a heterogeneous cell population consisting of 3 subsets: classical CD14++CD16-, intermediate CD14++CD16+ and nonclassical CD14+CD16++ monocytes. Via poorly characterized mechanisms, intermediate monocyte counts rise in chronic inflammatory diseases, among which chronic kidney disease is of particular epidemiologic importance. DNA methylation is a central epigenetic feature that controls hematopoiesis. By applying next-generation Methyl-Sequencing we now tested how far the 3 monocyte subsets differ in their DNA methylome and whether uremia induces DNA methylation changes in differentiating monocytes. We found that each monocyte subset displays a unique phenotype with regards to DNA methylation. Genes with differentially methylated promoter regions in intermediate monocytes were linked to distinct immunological processes, which is in line with results from recent gene expression analyses. In vitro, uremia induced dysregulation of DNA methylation in differentiating monocytes, which affected several transcription regulators important for monocyte differentiation (e.g., FLT3, HDAC1, MNT) and led to enhanced generation of intermediate monocytes. As potential mediator, the uremic toxin and methylation inhibitor S-adenosylhomocysteine induced shifts in monocyte subsets in vitro, and associated with monocyte subset counts in vivo. Our data support the concept of monocyte trichotomy and the distinct role of intermediate monocytes in human immunity. The shift in monocyte subsets that occurs in chronic kidney disease, a proinflammatory condition of substantial epidemiological impact, may be induced by accumulation of uremic toxins that mediate epigenetic dysregulation.
KEYWORDS: CD14, CD16, chronic kidney disease, DNA methylation, monocyte subsets, S-adenosylhomocysteine
Introduction
The circulating pool of human monocytes is composed of 3 subsets that are specialized to fulfill distinct tasks in the immune system. Based on the classification of the International Consensus Statement on Monocyte Nomenclature, these subsets are defined as classical CD14++CD16- monocytes, intermediate CD14++CD16+ monocytes, and nonclassical CD14+CD16++ monocytes.1
Following this official nomenclature, several studies characterized monocyte subsets in genome-wide gene expression approaches.2-5 Although these studies did not analyze the mechanisms of transcriptional regulation, they broadened our understanding of monocyte heterogeneity and specifically acknowledged the importance of intermediate monocytes. So far, intermediate monocytes were either neglected, or they were merely considered as a transient cell population in the differentiation process from classical into nonclassical monocytes with no notable immunological functions. In contrast, these recent gene expression analyses, and other functional studies, identified intermediate monocytes as distinct mediators of proinflammatory responses.6
This is in line with clinical studies on monocyte heterogeneity. Counts of intermediate monocytes are increased in several proinflammatory conditions, among which chronic kidney disease (CKD) is of particular epidemiological importance, as it affects one tenth of the global adult population.7 Declining renal function is associated with a shift in monocyte subsets, and CKD patients with highest cell counts of intermediate monocytes have worst cardiovascular outcome.8-11 However, the underlying mechanism for the shift of monocyte subsets in CKD and other proinflammatory diseases remains enigmatic.
Pioneering animal studies found the one carbon (C1) metabolism to be directly linked to monocyte heterogeneity and characterized the C1 metabolite homocysteine as a central mediator causing shifts in monocyte subsets toward intermediate monocytes.12,13 Importantly, the C1 metabolism is closely connected to epigenetic gene regulation, particularly to DNA methylation; the C1 metabolite S-adenosylmethionine (SAM) is the universal methyl group donor, whereas its derivate S-adenosylhomocysteine (SAH) is a strong inhibitor of cellular methyltransferases.
Despite the evidence for a direct link between the C1 metabolism and monocyte heterogeneity and the central role of DNA methylation in the differentiation of haematopoietic stem cells to monocytes,14 no data on the impact of DNA methylation on the regulation of monocyte heterogeneity have been provided so far. This impact might be of particular interest in CKD patients, who have tremendously increased plasma levels of homocysteine, SAM and SAH.
By applying the next-generation sequencing Methyl-Sequencing (Methyl-Seq) method, we now characterized genome-wide DNA methylation in the 3 monocyte subsets and analyzed the impact of uremia on DNA methylation in differentiating monocytes.
Results
Generation of Methyl-Seq libraries for the 3 monocyte subsets
To analyze whether the 3 monocyte subsets differ in their genome-wide DNA methylation profiles, we isolated classical, intermediate, and nonclassical monocytes from 7 healthy volunteers (Fig. S1) and generated 3 independent Methyl-Seq libraries. After removal of duplicates, low-quality reads, and reads that could not be mapped to the genome, the total number of reads was 70,168,933, comprising 28,259,303 from classical, 22,871,218 from intermediate, and 19,038,412 from nonclassical monocytes. In total, Methyl-Seq analysis allowed us to analyze 746,801 genomic loci. Of these 746,801 loci, 79,988 are located in upstream regions of genes (47,971 in CpG islands), on which our following analyses are focused given that those regions are primarily involved in the regulation of gene expression. The distribution of mapping regions is given in Figure 1A.
Figure 1.
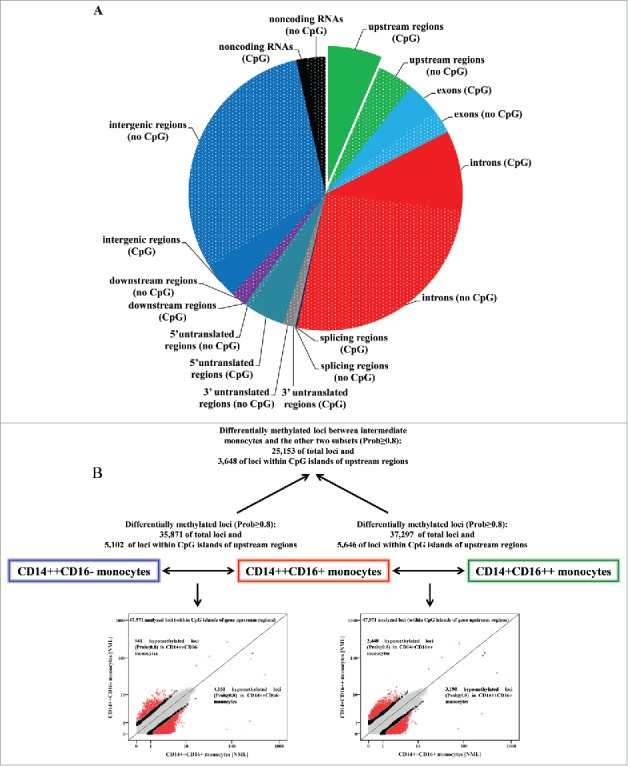
Methyl-Seq analysis of isolated monocyte subsets. (A) Distribution of mapping regions of Methyl-Seq analysis. Plain sections show analyzed regions in CpG islands, patterned sections show analyzed regions that are not located in CpG islands. CpG islands within gene upstream regions are highlighted, as analyses are focused on these regions throughout the manuscript. (B) Schematic representation of differences in DNA methylation between the 3 monocyte subsets. The total number of differentially methylated loci (Prob≥0.8) is given for total loci and for loci within CpG islands of gene upstream regions. The scatter plots illustrate differences in DNA methylation between intermediate and the 2 other monocyte subsets. Presented are loci within CpG islands of gene upstream regions; loci with differential methylation are given as circles (Prob≥0.9 in red, Prob between 0.8 and 0.9 in black) and all other loci are given as gray dots.
Differentially methylated loci in intermediate monocytes
First, we aimed to characterize differentially methylated genomic loci in intermediate monocytes given the recent acknowledgment of these cells as a distinct cell population with particular proinflammatory potential.1-6,15 When comparing genome-wide DNA methylation between intermediate monocytes and the 2 other monocyte subsets, we found 35,871 differentially methylated loci between intermediate and classical monocytes and 37,297 differentially methylated loci between intermediate and nonclassical monocytes (Prob≥0.8; Fig. 1B). When focusing on CpG islands in gene upstream regions, we found 5,102 differentially methylated loci between intermediate and classical and 5,646 differentially methylated loci between intermediate and nonclassical monocytes. Interestingly, of those differentially methylated loci, a high amount of loci were hypomethylated in intermediate monocytes as compared to classical monocytes (4161 hypomethylated vs. 941 hypermethylated loci) and to nonclassical monocytes (3198 hypomethylated vs. 2448 hypermethylated loci; Fig. 1B).
The distinct nature of intermediate monocytes is underscored by the observation that a substantial number of loci differed both between intermediate and classical monocytes, as well as between intermediate and nonclassical monocytes (25,153 of total loci, and 3,648 loci in CpG islands of gene upstream regions, of which the 50 with strongest differences in DNA methylation are given in Table 1). Characterizing these 50 genes by using the Human Gene Database (GeneCards®) (http://www.genecards.org/) we could categorize these genes to distinct cellular processes such as cell differentiation and cell cycle progression (e.g., NOTCH2NL, MXD4, RFWD2), regulation of gene expression (e.g., ZNF787, ELOF1, SETX), intracellular transport processes (e.g., CSNK1G2, FYCO1, GCC1), lipid metabolism and oxidation (e.g., GPX4, TMEM97, ACOT7), as well as immune system processes, such as T cell activation, inflammation, and adhesion and migration (e.g., FYN, CXCL12, STK10).
Table 1.
Top differentially methylated loci (in CpG islands of gene upstream regions) in CD14++CD16+ monocytes as compared to CD14++CD16- and CD14+CD16++ monocytes.
| Position | Gene | Description | CD14++CD16- NML | CD14++CD16+ NML | CD14+CD16++ NML | Prob CD14++CD16+ vs. CD14++CD16- / CD14+CD16++ | log2FC CD14++CD16+ vs. CD14++CD16- / CD14+CD16++ |
|---|---|---|---|---|---|---|---|
| chr1:25757294-25757299 | TMEM57 | transmembrane protein 57 | 0.1 | 5.1 | 0.1 | 0.999 / 0.999 | 5.2 / 5.6 |
| chr1:145208992-145208997 | NOTCH2NL | notch 2 N-terminal like | 0.2 | 5.2 | 0.4 | 0.999 / 0.998 | 4.9 / 3.8 |
| chr2:43823285-43823290 | THADA | thyroid adenoma associated | 0.2 | 5.0 | 0.1 | 0.999 / 0.999 | 4.3 / 6.6 |
| chr19:1267318-1267323 | CIRBP | cold inducible RNA binding protein | 0.4 | 5.5 | 0.5 | 0.999 / 0.998 | 4.0 / 3.5 |
| chr17:43324893-43324898 | MAP3K14-AS1 | MAP3K14 antisense RNA 1 | 0.3 | 5.0 | 0.3 | 0.999 / 0.998 | 4.2 / 4.3 |
| chr1:148556381-148556386 | NBPF15 | neuroblastoma breakpoint family, member 15 | 1.0 | 9.1 | 0.8 | 0.998 / 0.999 | 3.2 / 3.5 |
| chr10:46993229-46993234 | GPRIN2 | G protein regulated inducer of neurite outgrowth 2 | 0.6 | 8.4 | 0.9 | 1.000 / 0.997 | 3.8 / 3.2 |
| chr7:143534549-143534554 | LOC154761 | family with sequence similarity 115, member C pseudogene | 0.2 | 5.5 | 0.5 | 0.999 / 0.997 | 4.9 / 3.4 |
| chr17:7819587-7819592 | LOC284023 | NA | 0.4 | 5.3 | 0.4 | 0.999 / 0.998 | 3.6 / 3.7 |
| chr2:128785597-128785602 | SAP130 | Sin3A-associated protein, 130kDa | 0.4 | 5.0 | 0.3 | 0.998 / 0.998 | 3.7 / 4.2 |
| chr19:56632656-56632661 | ZNF787 | zinc finger protein 787 | 0.4 | 5.0 | 0.3 | 0.998 / 0.998 | 3.6 / 4.3 |
| chr1:36916162-36916167 | OSCP1 | organic solute carrier partner 1 | 0.2 | 5.4 | 0.5 | 0.999 / 0.997 | 4.4 / 3.4 |
| chr11:76092381-76092386 | PRKRIR | protein-kinase, interferon-inducible double stranded RNA dependent inhibitor, repressor of (P58 repressor) | 0.2 | 4.7 | 0.2 | 0.999 / 0.998 | 4.5 / 4.5 |
| chr17:74070713-74070718 | GALR2 | galanin receptor 2 | 0.2 | 4.7 | 0.2 | 0.999 / 0.998 | 4.7 / 4.5 |
| chr19:11670214-11670219 | ELOF1 | elongation factor 1 homolog (S. cerevisiae) | 0.4 | 4.9 | 0.3 | 0.998 / 0.998 | 3.6 / 4.2 |
| chr19:1940458-1940463 | CSNK1G2 | casein kinase 1, gamma 2 | 0.4 | 4.8 | 0.3 | 0.998 / 0.998 | 3.8 / 4.2 |
| chr7:102213259-102213264 | POLR2J3 | polymerase (RNA) II (DNA directed) polypeptide J3 | 7.5 | 0.8 | 7.6 | 0.998 / 0.998 | −3.2 / −3.3 |
| chr17:8534325-8534330 | MYH10 | myosin, heavy chain 10, non-muscle | 0.2 | 4.6 | 0.3 | 0.998 / 0.997 | 4.7 / 3.9 |
| chr6:112194667-112194672 | FYN | FYN proto-oncogene, Src family tyrosine kinase | 0.5 | 5.1 | 0.4 | 0.997 / 0.998 | 3.4 / 3.8 |
| chr12:49246065-49246070 | DDX23 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 23 | 0.1 | 4.5 | 0.3 | 0.998 / 0.997 | 5.4 / 4.1 |
| chr16:686860-686865 | C16orf13 | chromosome 16 open reading frame 13 | 0.5 | 5.2 | 0.5 | 0.998 / 0.997 | 3.5 / 3.4 |
| chr4:2264327-2264332 | MXD4 | MAX dimerization protein 4 | 0.3 | 4.7 | 0.4 | 0.998 / 0.997 | 4.1 / 3.7 |
| chr1:120838772-120838777 | FAM72B | family with sequence similarity 72, member B | 0.2 | 4.8 | 0.4 | 0.999 / 0.996 | 4.5 / 3.5 |
| chr17:34957991-34957996 | MRM1 | mitochondrial rRNA methyltransferase 1 homolog (S. cerevisiae) | 0.1 | 4.4 | 0.1 | 0.998 / 0.997 | 4.9 / 5.4 |
| chr4:13486515-13486520 | RAB28 | RAB28, member RAS oncogene family | 0.3 | 4.5 | 0.2 | 0.998 / 0.997 | 4.0 / 4.8 |
| chr11:75236631-75236636 | GDPD5 | glycerophosphodiester phosphodiesterase domain containing 5 | 0.2 | 4.4 | 0.1 | 0.998 / 0.997 | 4.4 / 5.4 |
| chr10:44880740-44880745 | CXCL12 | chemokine (C-X-C motif) ligand 12 | 0.4 | 4.7 | 0.4 | 0.998 / 0.997 | 3.7 / 3.7 |
| chr3:46037337-46037342 | FYCO1 | FYVE and coiled-coil domain containing 1 | 0.3 | 4.5 | 0.3 | 0.998 / 0.997 | 3.8 / 4.1 |
| chr19:1103841-1103846 | GPX4 | glutathione peroxidase 4 | 0.2 | 4.4 | 0.2 | 0.998 / 0.997 | 4.1 / 4.8 |
| chr4:2264184-2264189 | MXD4 | MAX dimerization protein 4 | 0.2 | 4.9 | 0.5 | 0.999 / 0.995 | 4.3 / 3.2 |
| chr5:171615865-171615870 | STK10 | serine/threonine kinase 10 | 0.2 | 4.3 | 0.2 | 0.998 / 0.996 | 4.6 / 4.4 |
| chr16:770721-770726 | FAM173A | family with sequence similarity 173, member A | 0.1 | 4.2 | 0.2 | 0.998 / 0.996 | 5.9 / 4.7 |
| chr2:87303947-87303952 | LOC285074 | anaphase promoting complex subunit 1 pseudogene | 6.1 | 0.7 | 6.5 | 0.996 / 0.997 | −3.1 / −3.2 |
| chr10:44882133-44882138 | CXCL12 | chemokine (C-X-C motif) ligand | 0.4 | 4.4 | 0.2 | 0.997 / 0.997 | 3.6 / 4.4 |
| chr2:73340397-73340402 | RAB11FIP5 | RAB11 family interacting protein 5 (class I) | 0.4 | 4.5 | 0.4 | 0.997 / 0.996 | 3.7 / 3.6 |
| chr6:34217221-34217226 | C6orf1 | chromosome 6 open reading frame 1 | 0.1 | 4.3 | 0.3 | 0.998 / 0.995 | 4.9 / 3.8 |
| chr7:127225841-127225846 | GCC1 | GRIP and coiled-coil domain containing 1 | 0.2 | 5.5 | 0.7 | 0.999 / 0.994 | 4.5 / 3.0 |
| chr5:612156-612161 | CEP72 | centrosomal protein 72kDa | 0.3 | 4.4 | 0.3 | 0.997 / 0.996 | 3.8 / 4.1 |
| chr17:26645494-26645499 | TMEM97 | transmembrane protein 97 | 0.4 | 4.4 | 0.2 | 0.997 / 0.997 | 3.6 / 4.4 |
| chr4:152330105-152330110 | FAM160A1 | family with sequence similarity 160, member A1 | 0.5 | 4.9 | 0.4 | 0.996 / 0.997 | 3.2 / 3.6 |
| chr16:67678949-67678954 | RLTPR | RGD motif, leucine rich repeats, tropomodulin domain and proline-rich containing | 0.2 | 4.3 | 0.3 | 0.997 / 0.996 | 4.3 / 4.0 |
| chr5:137090687-137090692 | HNRNPA0 | heterogeneous nuclear ribonucleoprotein A0 | 0.2 | 4.9 | 0.6 | 0.999 / 0.994 | 4.5 / 3.1 |
| chr4:1858552-1858557 | LETM1 | leucine zipper-EF-hand containing transmembrane protein 1 | 0.2 | 4.5 | 0.4 | 0.998 / 0.995 | 4.4 / 3.4 |
| chr4:53525726-53525731 | USP46 | ubiquitin specific peptidase 46 | 0.3 | 4.2 | 0.1 | 0.996 / 0.996 | 3.7 / 6.3 |
| chr16:686756-686761 | C16orf13 | chromosome 16 open reading frame 13 | 5.5 | 0.7 | 6.4 | 0.995 / 0.998 | −3.1 / −3.3 |
| chr1:176176803-176176808 | RFWD2 | ring finger and WD repeat domain 2, E3 ubiquitin protein ligase | 0.3 | 4.8 | 0.5 | 0.998 / 0.994 | 3.9 / 3.2 |
| chr16:3550165-3550170 | CLUAP1 | clusterin associated protein 1 | 0.3 | 4.2 | 0.2 | 0.997 / 0.996 | 4.1 / 4.7 |
| chr1:6453838-6453843 | ACOT7 | acyl-CoA thioesterase 7 | 0.2 | 4.3 | 0.4 | 0.998 / 0.995 | 4.4 / 3.6 |
| chr9:135230442-135230447 | SETX | Senataxin | 0.1 | 4.2 | 0.3 | 0.997 / 0.995 | 5.3 / 4.0 |
| chr19:11592808-11592813 | ELAVL3 | ELAV like neuron-specific RNA binding protein 3 | 0.3 | 4.2 | 0.3 | 0.997 / 0.995 | 4.1 / 3.8 |
NML: normalized tag count (tags per million); Prob: probability; FC: fold change; NA: not available
We next performed pathway analysis with genes showing differential methylated upstream regions in intermediate monocytes as compared to both classical and nonclassical monocytes, in order to characterize differentially regulated pathways in intermediate monocytes (Fig. S2). Using the string database (http://string-db.org/)16 we identified differentially regulated biological processes in intermediate monocytes, most of which were associated with cell differentiation and cellular biosynthesis processes (e.g., JUNB, TGFB1, ERF), regulation of gene expression (e.g., KLF13, EGR3, POU3F2), and immune system processes (e.g., FASN, ELMO2, ITGA2). Genes located at central positions within this pathway analysis are linked to NF-κB signaling (e.g., RELA, NFKB1, NFKB2) and other transcription factor dependent processes that regulate gene expression within immune system processes (e.g., STAT1, CEBPB, FOS). These results were confirmed by an enrichment analysis performed with the WEBGESTALT platform (http://bioinfo.vanderbilt.edu/webgestalt/), which linked genes with differentially methylated promoter regions in intermediate monocytes to distinct processes that include immune system processes, cell differentiation and cell cycle control, TNF receptor signaling, as well as lipid and lipoprotein metabolism. Genes enriched in these pathways were subsequently used as input to the GeneMANIA database (http://www.genemania.org/) in order to construct networks of interactions (Fig. S3).
Interestingly, 214 loci in CpG islands of gene upstream regions that were differentially methylated in intermediate monocytes could be linked to regions coding for microRNAs (Table S1 for the 50 most differentially methylated loci). Pathway analysis (http://mirsystem.cgm.ntu.edu.tw/)17 of these miRNAs confirmed an enrichment of pathways for transcriptional regulation and for pathways associated with regulation of immune system processes, comprising toll-like receptor cascades, cytokine signaling, and angiopoietin receptor Tie2 mediated signaling (e.g., miR-126, miR-148a, miR-210).
Finally, we analyzed whether these differentially methylated loci are associated with differential expression of the corresponding genes. Among 97 genes that were recently identified to be specifically expressed in intermediate monocytes,5 those 61 genes defined by a 26-hit tag (perfect match in annotation) were now analyzed with regard to DNA methylation. We identified 55 loci (10 in CpG islands of gene upstream regions) linked to these genes that showed lower DNA methylation in intermediate monocytes as compared to both classical and nonclassical monocytes (Prob≥0.8; Table S2).
Differentiation of monocyte subsets under uremic conditions
We next addressed the question of whether the shift of monocyte subsets from classical toward intermediate and nonclassical monocytes that is observed in several proinflammatory diseases may be mediated via DNA methylation. We focused this analysis on patients with chronic kidney disease (CKD) because of its high prevalence7 and its robust association with pronounced shifts in monocyte subset distribution.8,9
Therefore, we recruited patients with advanced CKD who were treated with standard hemodialysis treatment as well as age and gender matched controls. As expected, patients had higher counts of intermediate (P<0.05) and nonclassical (P<0.01) monocytes compared to control subjects (Fig. 2A for a representative example).
Figure 2.
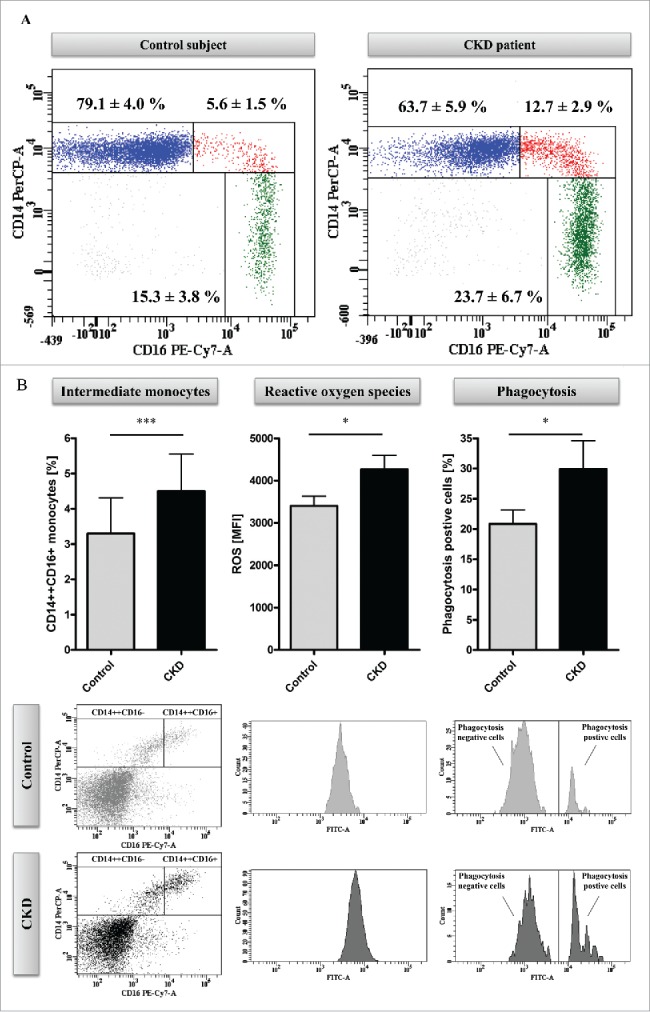
Methyl-Seq analysis of uremic monocytes. (A) Representative dot plots of monocyte subsets of a hemodialysis patient (right panel) and an age and gender matched control subject (left panel). Given are mean percentages (±SD) for monocyte subsets of 5 patients and controls, respectively. (B) Analysis of in vitro differentiated intermediate monocytes. Percentages of intermediate monocytes (left panel) as well as the capacity of intermediate monocytes to produce reactive oxygen species (middle panel) and to phagocyte (right panel) were determined with flow-cytometry. Data are presented as mean±SEM and compared by Student t test. Representative dot plots and histograms are shown. * P<0.05, ***P<0.001. ROS indicates reactive oxygen species. (C) Distribution of mapping regions of Methyl-Seq analysis. Plain sections show analyzed regions in CpG islands, patterned sections show analyzed regions that are not located in CpG islands. CpG islands within gene upstream regions are highlighted, as analyses are focused on these regions throughout the manuscript. (D) Schematic representation of differences in DNA methylation between monocytes that were differentiated under control or under uremic conditions. Presented are total loci (left panel) as well as loci within CpG islands of gene upstream regions (right panel). Loci with differential methylation are given as circles (Prob≥0.9 in red, Prob between 0.8 and 0.9 in black) and all other loci are given as gray dots. To allow better illustration, axes are cut at NML counts of 100, as no differences in DNA methylation were observed in loci with higher NML count. Complete scatter plots are given in Figure S5.
Figure 2.

(Continued)
We first tested whether uremia per se induces enhanced generation of intermediate monocytes. Therefore, we collected serum samples from patients and controls and differentiated intermediate monocytes from haematopoietic stem cells isolated from healthy donors.18 Upon stimulation with uremic serum, more intermediate monocytes were generated as compared to control conditions (P<0.001; Fig. 2B). Moreover, intermediate monocytes that were generated under uremic conditions were characterized by a higher ROS production and higher phagocytosis capacity compared to monocytes generated under control conditions (P<0.05; Fig. 2B). Similarly, classical monocytes generated under uremic conditions produced higher ROS levels (P<0.05) and had higher phagocytosis capacity than classical monocytes generated under control conditions, although the latter finding did not reach statistical significance (Fig. S4).
Generation of Methyl-Seq libraries of differentiated monocytes
In order to analyze whether uremia affects DNA methylation in differentiating monocytes, we performed Methyl-Seq analysis of monocytes that were differentiated under control and under uremic conditions. Thus, we generated 2 independent libraries (“control monocytes” and “uremic monocytes”), which consisted of 38,707,311 reads in total, comprising 18,611,539 reads from control monocytes and 20,095,772 reads from uremic monocytes. Methyl-Seq analysis allowed us to analyze 813,267 genomic loci. Of these 813,267 loci, 69,615 are located in upstream regions of genes (39,295 in CpG islands), on which we focused our next analyses. The distribution of mapping regions is given in Fig. 2C.
Differentially methylated loci in uremic monocytes
Genome-wide DNA methylation analysis revealed 6,618 differentially methylated loci between monocytes that were differentiated under control or uremic conditions, respectively (Prob≥0.8). When focusing on CpG islands in gene upstream regions, we found 283 differentially methylated loci, of which 136 were hypomethylated and 147 hypermethylated in uremic monocytes (Fig. 2D, Fig. S5). Interestingly, when focusing on those loci with highest differences in DNA methylation (Prob≥0.9; indicated in red in Fig. 2D), we found a strong enrichment of hypomethylated loci in uremic monocytes (total loci: 408 hypomethylated vs. 79 hypermethylated; loci in CpG islands of upstream regions: 54 hypomethylated vs. 2 hypermethylated). Those 50 loci with strongest differences in DNA methylation are presented in Table 2. Importantly, within these 50 loci that differed most pronouncedly in DNA methylation, we identified several loci linked to transcription factors (e.g., ATF4, SALL1, MNT).
Table 2.
Top differentially methylated loci (in CpG islands of gene upstream regions) in uremic monocytes as compared to control monocytes.
| Position | Gene | Description | Control monocytes NML | Uremic monocytes NML | Prob | log2FC |
|---|---|---|---|---|---|---|
| chr17:42298408-42298413 | UBTF | upstream binding transcription factor, RNA polymerase I | 0.2 | 8.6 | 1.000 | 5.3 |
| chr11:43702119-43702124 | HSD17B12 | hydroxysteroid (17-β) dehydrogenase 12 | 0.1 | 7.8 | 1.000 | 7.2 |
| chr4:166128588-166128593 | KLHL2 | kelch-like family member 2 | 0.4 | 8.1 | 1.000 | 4.4 |
| chr2:86116229-86116234 | ST3GAL5 | ST3 β-galactoside α-2,3-sialyltransferase 5 | 0.5 | 7.8 | 1.000 | 4.0 |
| chr9:140118246-140118251 | C9orf169 | chromosome 9 open reading frame 169 | 0.1 | 6.2 | 1.000 | 6.9 |
| chr2:45878887-45878892 | PRKCE | protein kinase C, epsilon | 0.3 | 6.4 | 1.000 | 4.6 |
| chr16:51185257-51185262 | SALL1 | spalt-like transcription factor 1 | 0.2 | 6.2 | 1.000 | 4.9 |
| chr12:7053171-7053176 | C12orf57 | chromosome 12 open reading frame 57 | 0.0 | 5.5 | 1.000 | 7.7 |
| chr9:127703512-127703517 | GOLGA1 | golgin A1 | 0.2 | 5.6 | 1.000 | 5.1 |
| chr10:94449085-94449090 | HHEX | hematopoietically expressed homeobox | 0.6 | 7.3 | 0.999 | 3.6 |
| chr15:101835544-101835549 | SNRPA1 | small nuclear ribonucleoprotein polypeptide A' | 0.3 | 5.2 | 0.999 | 4.3 |
| chr22:50683474-50683479 | TUBGCP6 | tubulin, gamma complex associated protein 6 | 0.1 | 4.2 | 0.998 | 6.3 |
| chr1:861097-861102 | SAMD11 | sterile α motif domain containing 11 | 0.1 | 4.2 | 0.998 | 6.3 |
| chr9:90112524-90112529 | DAPK1 | death-associated protein kinase 1 | 0.0 | 3.4 | 0.994 | 7.0 |
| chr21:37692207-37692212 | MORC3 | MORC family CW-type zinc finger 3 | 1.4 | 10.2 | 0.994 | 2.9 |
| chr14:103524263-103524268 | CDC42BPB | CDC42 binding protein kinase β (DMPK-like) | 0.1 | 3.2 | 0.993 | 5.9 |
| chr5:10761675-10761680 | DAP | death-associated protein | 0.7 | 5.1 | 0.992 | 2.9 |
| chr17:1533226-1533231 | SLC43A2 | solute carrier family 43 (amino acid system L transporter), member 2 | 0.1 | 2.3 | 0.980 | 5.4 |
| chr3:48723486-48723491 | NCKIPSD | NCK interacting protein with SH3 domain | 0.1 | 2.4 | 0.980 | 4.5 |
| chr1:91183789-91183794 | BARHL2 | BarH-like homeobox 2 | 2.1 | 10.5 | 0.978 | 2.3 |
| chr1:33430402-33430407 | RNF19B | ring finger protein 19B | 0.2 | 2.3 | 0.978 | 3.9 |
| chr12:65153256-65153261 | GNS | glucosamine (N-acetyl)-6-sulfatase | 0.2 | 2.4 | 0.977 | 3.5 |
| chr17:1613840-1613845 | TLCD2 | TLC domain containing 2 | 0.0 | 2.1 | 0.976 | 6.3 |
| chr1:2458037-2458042 | PANK4 | pantothenate kinase 4 | 1.5 | 7.0 | 0.973 | 2.2 |
| chr9:140116222-140116227 | RNF208 | ring finger protein 208 | 0.5 | 2.9 | 0.972 | 2.6 |
| chr3:56591104-56591109 | CCDC66 | coiled-coil domain containing 66 | 0.0 | 2.0 | 0.972 | 6.2 |
| chr4:1872228-1872233 | WHSC1 | Wolf-Hirschhorn syndrome candidate 1 | 0.0 | 2.0 | 0.972 | 6.2 |
| chr1:20512525-20512530 | UBXN10 | UBX domain protein 10 | 0.4 | 2.6 | 0.966 | 2.6 |
| chr22:39916141-39916146 | ATF4 | activating transcription factor 4 | 0.3 | 2.3 | 0.965 | 2.8 |
| chr1:2344070-2344075 | PEX10 | peroxisomal biogenesis factor 10 | 0.1 | 1.9 | 0.965 | 4.1 |
| chr22:39916233-39916238 | ATF4 | activating transcription factor 4 | 0.1 | 1.8 | 0.963 | 4.1 |
| chr1:14026636-14026641 | PRDM2 | PR domain containing 2, with ZNF domain | 0.0 | 1.7 | 0.962 | 6.0 |
| chr3:48723384-48723389 | NCKIPSD | NCK interacting protein with SH3 domain | 0.6 | 3.0 | 0.960 | 2.2 |
| chr19:58897792-58897797 | RPS5 | ribosomal protein S5 | 0.1 | 1.7 | 0.958 | 5.0 |
| chr6:36355988-36355993 | ETV7 | ets variant 7 | 0.4 | 2.3 | 0.958 | 2.6 |
| chr17:1613937-1613942 | TLCD2 | TLC domain containing 2 | 0.1 | 1.6 | 0.956 | 4.9 |
| chr17:74379544-74379549 | SPHK1 | sphingosine kinase 1 | 0.1 | 1.5 | 0.947 | 4.8 |
| chr17:2305318-2305323 | MNT | MAX network transcriptional repressor | 0.2 | 1.6 | 0.945 | 3.3 |
| chr7:45961262-45961267 | IGFBP3 | insulin-like growth factor binding protein 3 | 2.2 | 8.3 | 0.932 | 1.9 |
| chr19:531530-531535 | CDC34 | cell division cycle 34 | 0.1 | 1.3 | 0.932 | 3.6 |
| chr8:42010204-42010209 | AP3M2 | adaptor-related protein complex 3, mu 2 subunit | 0.1 | 1.2 | 0.922 | 4.5 |
| chr2:133174115-133174120 | GPR39 | G protein-coupled receptor 39 | 3.2 | 10.5 | 0.920 | 1.7 |
| chr7:112430503-112430508 | TMEM168 | transmembrane protein 168 | 1.4 | 0.2 | 0.918 | −2.5 |
| chr12:56652328-56652333 | ANKRD52 | ankyrin repeat domain 52 | 0.1 | 1.1 | 0.916 | 4.4 |
| chr20:19997703-19997708 | NAA20 | N(α)-acetyltransferase 20, NatB catalytic subunit | 3.2 | 10.0 | 0.915 | 1.6 |
| chr19:47551909-47551914 | TMEM160 | transmembrane protein 160 | 0.1 | 1.1 | 0.911 | 3.4 |
| chr1:32757466-32757471 | HDAC1 | histone deacetylase 1 | 1.5 | 0.3 | 0.907 | −2.2 |
| chr21:35747557-35747562 | SMIM11 | small integral membrane protein 11 | 0.0 | 1.0 | 0.903 | 5.2 |
| chr8:17780349-17780354 | PCM1 | pericentriolar material 1 | 1.8 | 5.0 | 0.903 | 1.5 |
| chr21:28339999-28340004 | ADAMTS5 | ADAM metallopeptidase with thrombospondin type 1 motif, 5 | 0.2 | 1.2 | 0.900 | 2.5 |
NML: normalized tag count (tags per million); Prob: probability; FC: fold change.
String and gene ontology analysis of genes with differentially methylated upstream regions in uremic monocytes (Fig. S6) confirmed an enrichment of GO terms connected to transcriptional regulation [e.g., “negative regulation of gene expression” (SAP130, LIMS1, UBTF, in biological process), “sequence-specific DNA binding transcription factor activity” (e.g., HDAC1, ZNF224, BTAF1, in molecular function)]. Of particular interest, the upstream region of FLT3, which plays an important role in the differentiation of haematopoietic stem cells, was found to be differentially methylated in uremic monocytes. Genes with differentially methylated upstream regions in uremic monocytes could additionally be linked to GO terms associated with atherogenesis and cholesterol metabolism such as “regulation of macrophage derived foam cell differentiation,” “regulation of cholesterol storage,” and “regulation of lipid metabolic process” (e.g., NR1H2, MAPK9, PRKCE).
To further analyze the impact of uremia on the induction of proatherogenic pathways in differentiating monocytes, we analyzed whether those genes with differentially methylated upstream regions can be associated with cardiovascular disease (CVD) and with underlying pathological pathways of atherosclerosis, namely infections and immune disease. By using the Genetic Association Database (accessible from the National Institutes of Health; http://geneticassociationdb.nih.gov/) we found that numerous genes with differentially methylated upstream regions (64 out of 283) are associated with CVD, infections and/or immune diseases (Table S3).
Finally, we aimed to analyze whether monocytes generated under uremic conditions are closer related to circulating intermediate monocytes than monocytes generated under control conditions. Therefore, we performed a hierarchical cluster analysis with those 10,000 loci that have the highest tag count in the combined analysis comprising monocyte subsets and in vitro generated monocytes. Indeed, we confirmed that monocytes generated under uremic conditions are closer related to circulating intermediate monocytes than monocytes generated under control conditions (Fig. S7). In line, most of those loci that were identified to be hypomethylated in uremic monocytes as compared to control monocytes were also hypomethylated in circulating intermediate as compared to circulating classical and to circulating nonclassical monocytes (88% of those loci presented in Table 2).
Impact of C1 metabolites on monocyte subsets
Finally, we aimed to analyze whether central uremic toxins that are linked to epigenetic dysregulation—homocysteine and S-adenosylhomocysteine (SAH)—may account for shifts in monocyte subsets. Stimulation of monocytes with increasing concentrations (30 µM, 100 µM, and 500 µM) of homocysteine for 5 h induced a significant rise in intermediate monocytes (Fig. 3A, Fig. S8). As the incubation with homocysteine also increases the intracellular concentrations of SAH, we next stimulated monocytes with different concentrations (10 µM and 50 µM) of Adenosine-2′,3′-dialdehyde (Adox), which is an inhibitor of the SAH hydrolase and thus selectively increases intracellular SAH concentrations without increasing homocysteine levels.19 We found that Adox also induced a significant increase of intermediate monocytes after 5 h stimulation (Fig. 3A, Fig. S8). Interestingly, stimulation of monocytes with the DNA hypomethylating drug 5-aza-2′-deoxycytidine induced a similar shift in monocyte subsets as homocysteine and Adox stimulation (Fig. 3B).
Figure 3.
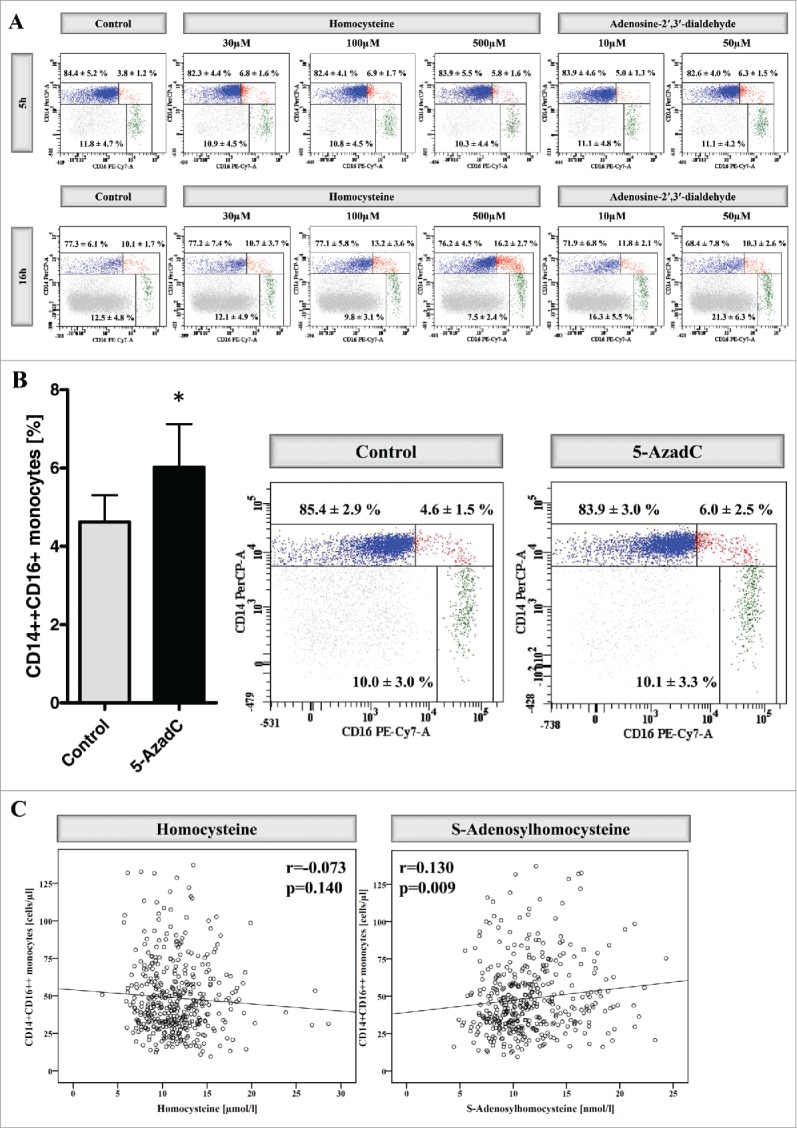
Impact of C1 metabolites on monocyte subsets. (A) Distribution of monocyte subsets after stimulation of whole blood for 5 h (upper panel) and 16 h (lower panel) with increasing concentrations of homocysteine (30 µM, 100 µM, and 500 µM) or Adenosine-2′,3′-dialdehyde (10 µM and 50 µM). Data are presented as mean±SD of 6 (for 5 h) and 7 (16 h) independent experiments, and one representative example is illustrated. (B) Distribution of monocyte subsets after stimulation of whole blood for 5 h with 5-aza-2′-deoxycytidine (0.5 µM). Data are presented as mean±SEM of 5 independent experiments (left panel). One representative example is illustrated and the mean percentages (±SD) of the 3 monocyte subsets from 5 independent experiments are presented (right panel). (C) Correlation between nonclassical monocytes and homocysteine (left panel) as well as between nonclassical monocytes and S-adenosylhomocysteine (right panel) within the I Like HOMe study. To allow better visualization, data from 2 participants (participant 1: plasma homocysteine 46.7 mmol/l; participant 2: plasma SAH 43.0 nmol/l) are not depicted in the figure, but included in all calculations. Correlation coefficients were calculated by Pearson test.
We subsequently aimed to verify whether the increase of intermediate monocyte percentages after homocysteine and Adox stimulation represents a true conversion of classical monocytes into intermediate monocytes or a mere upregulation of CD16 on classical monocytes, so that our gating strategies would identify bona fide classical monocytes as intermediate monocytes. We therefore analyzed the expression of distinct surface markers on total monocytes, without further gating for monocyte subsets. Compared to unstimulated cells, homocysteine and Adox stimulation induced an upregulation of CD16 and CD86 (Fig. S9) as well as CCR5 and CX3CR1 (Figure S10) on total monocytes, even though the expression within each predefined monocyte subset remained stable. As all 4 antigens are less strongly expressed on classical monocytes than on other monocyte subsets, their upregulation points toward true monocyte subset conversion in the context of homocysteine and Adox stimulation. The stable expression of CCR5 expression on those intermediate monocytes in closest vicinity to classical monocytes after their stimulation with homocysteine and Adox further points against a shift of bona fide classical monocytes into the intermediate monocyte gate (Figure S11).
Upon more prolonged incubation of 16 h, the increase in intermediate monocytes after homocysteine stimulation persists, whereas stimulation with Adox drives an increase in nonclassical monocytes (Fig. 3A).
We finally reanalyzed data from the I Like HOMe study, in which cell counts of monocyte subsets and plasma levels of C1 metabolites were analyzed in 420 subjects from the general population. In line with the in vitro data, we found a significant association between nonclassical monocytes and SAH levels (Fig. 3C, Figure S12). No association was found between nonclassical monocytes and homocysteine as well as between intermediate monocytes and the 2 C1 metabolites (Fig. 3C, Figure S12, Figure S13).
Discussion
As a central epigenetic feature, DNA methylation controls differentiation of haematopoietic stem cells toward specialized lineage-committed cell types. Distinct cell populations differ in their DNA methylation profiles, which allow these cells to adhere to their specific gene expression programs.14
Although monocytes have been acknowledged as a heterogeneous cell population with subset-specific gene expression profiles1-6,15, data on transcriptional regulation of the 3 monocyte subsets have been lacking so far. Gene expression is centrally regulated by epigenetic mechanisms, which can broadly be subdivided into the 3 categories: DNA methylation, histone modifications, and microRNAs (miRNAs). Within the FANTOM5 (Functional Annotation of Mammalian Genome 5) project, which aims to characterize transcriptional regulation within the majority of human cells, significant differences in histone modifications were found between classical and nonclassical monocytes, which correlated with their gene expression profiles.20
Using the Methyl-Seq next-generation sequencing method, we now performed for the first time a genome-wide DNA methylation analysis of the 3 human monocyte subsets. By sequencing 70,168,933 reads and analyzing 746,801 loci, we generated high-resolution DNA methylation profiles of the 3 monocyte subsets. Demonstrating that intermediate monocytes show a distinct phenotype with regard to DNA methylation, our data further supports the trichotomy of monocytes.2-5 Particularly, we found a higher number of hypomethylated than hypermethylated loci when comparing intermediate monocytes to classical and nonclassical monocytes.
Intermediate monocytes are centrally involved in distinct immune system processes, such as antigen processing and presentation.4,5,20-22 Additionally, they were characterized as a proinflammatory monocyte subset, as they produce high levels of reactive oxygen species5 and of proinflammatory cytokines such as TNFα and IL1β after LPS stimulation.2 The present DNA methylation analysis extends these previous findings and shows that the 3 monocyte subsets differ in their DNA methylation profiles. Next, pathway analyses support the concept that intermediate monocytes may have a distinct role in immunity with regards to T cell activation, inflammation as well as adhesion and migration. Moreover, these analyses linked differentially methylated genes to further cellular processes such as gene regulation, cell differentiation, and cellular biosynthesis, in line with the idea that intermediate monocytes are cells in transition, originating from classical monocytes and differentiating to nonclassical monocytes.
In clinical studies, counts of intermediate monocytes were found to be elevated in many pathological conditions,6 particularly in chronic kidney disease (CKD), which affects one tenth of the human population.7 In line with their proinflammatory potential, intermediate monocyte counts predict future cardiovascular events both in patients with8-11 and without23 overt CKD. Thus, monocyte subsets have been proposed as future targets in cardiovascular and nephrological medicine.24 However, the development of pharmaceutics that will affect monocyte subset biology requires a more detailed understanding of the mechanisms that underlie the shift of monocyte subsets in human disease.
The plasticity of epigenetics is an important tool for immune cells to react upon changes in the environment. Failure in epigenetic regulation may therefore induce immune cell dysfunction and thus lead to changes in the immune cell composition, as seen in CKD patients.25 Importantly, the non-physiologic uremic milieu may directly induce epigenetic dysregulation in circulating immune cells and may thus contribute to immune dysfunction in CKD.26-29
By applying Methyl-Seq next-generation sequencing analysis we now show for the first time that uremia per se induces differential DNA methylation in differentiating monocytes. By sequencing 38,707,311 reads, we analyzed DNA methylation of 813,267 genomic loci. Among 6,618 loci (283 in CpG upstream regions) that were found to be differentially methylated between control and uremic monocytes, we identified several loci linked to transcriptional regulators and to monocyte differentiation. Moreover, several differentially methylated loci were connected to proatherogenic pathways, which is of particular interest in CKD, as many patients have premature cardiovascular disease caused by accelerated atherosclerosis, to which monocytes centrally contribute.30
In line with previous reports,27,31 we found that uremic monocytes displayed a high number of strongly hypomethylated loci. Importantly, declining renal function is associated with a strong increase in S-adenosylhomocysteine (SAH),32,33 the derivate of the universal methyl group donor S-Adenosylmethionine (SAM). SAH itself is a strong inhibitor of cellular methyltransferases and is thus directly linked to epigenetic regulation.26
As SAH is rapidly converted into homocysteine, homocysteine and SAH levels are closely correlated. Notably, SAH is currently discussed as the real culprit for the high cardiovascular disease risk in patients with hyperhomocysteinemia.33-36 In these patients, lowering of homocysteine with B vitamins did not affect cardiovascular outcome, which questions a pathophysiological role of homocysteine in cardiovascular disease. Instead, B vitamin supplementation does not lower SAH levels at all,37,38 which leaves SAH as the potential culprit in hyperhomocysteinemia.
Experimental studies that analyzed the pathophysiological effects of SAH found that the increase of SAH—either by a specific diet or by supplementation of the SAH hydrolase inhibitor adenosine-2,3-dialdehyde (Adox)—accelerated the development of atherosclerosis, which was associated with DNA methylation, without a contribution of homocysteine.39,40 However, data on the implications of C1 metabolites on monocyte biology focused on homocysteine rather than on SAH so far. First, animal studies with cystathionine β-synthase deficient mice have shown that hyperhomocysteinemia promotes the generation of intermediate (and classical) monocytes.12,13 Moreover, these studies demonstrated that the addition of homocysteine to in vitro cultures of mouse primary splenocytes induced the intermediate monocyte subset. However, as the supplementation of homocysteine also increases the intracellular SAH levels,19 the exact mediator for the enhanced generation of intermediate monocytes cannot fully be identified from these studies. Notably, in the same animal model, intermediate monocytes negatively correlated with the SAM/SAH ratio, which is an indicator for DNA methylation capacity.41 Moreover, a recent clinical study demonstrated that DNA hypomethylating drugs induce the generation of intermediate and nonclassical monocytes in patients with chronic myelomonocytic leukemia.42 In line, in vitro stimulation with the DNA hypomethylating drug 5-aza-2′-deoxycytidine induced a shift of monocyte subsets toward intermediate monocytes in the present study. Further studies are needed in order to analyze whether the induction of DNA hypermethylation in monocyte subsets, e.g., by supplementation of S-Adenosylmethionine, may be an option to reduce intermediate monocyte cell counts in patients.
Our data support the notion that SAH rather than homocysteine itself may mediate the enhanced generation of intermediate monocytes via epigenetic mechanisms. Of note, beyond C1 metabolites, also other uremic toxins may trigger epigenetic dysregulation in circulating immune cells and may thus contribute to immune dysfunction in CKD. Particularly, chronic inflammation—which is a common feature in CKD43—has been associated with changes in DNA methylation. First, proinflammatory cytokines were shown to regulate a DNA methyltransferase gene.44 Second, an association between the inflammation status and global DNA methylation was shown in CKD patients.28 Finally, we previously demonstrated that the p66Shc (SHC1) gene, which plays a central role in the stress response and reactive oxygen species metabolism, is hypomethylated in dialysis patients.45
In summary, we demonstrated for the first time that the 3 human monocyte subsets differ in their DNA methylation profiles, which may account for their different immunological functions. Next, we suggest that the accumulation of epigenetically active uremic toxins in patients with CKD may induce a reprogramming of differentiating monocytes, an enhanced generation of intermediate monocytes and an activation of their proinflammatory signaling cascades.
Materials and methods
Isolation of monocyte subsets
The three monocyte subsets were isolated from 7 healthy volunteers according to our established method.5 In brief, peripheral blood mononuclear cells (PBMCs) were isolated from EDTA-anticoagulated blood by density gradient centrifugation (Ficoll-Paque PLUS, GE Healthcare [17-1440-03]). Subsequently, CD16 expressing non-monocytes, i.e., NK cells and neutrophils, were depleted by using a cocktail of CD15 and CD56 MicroBeads (Non-monocyte Depletion Cocktail, CD16+ Monocyte Isolation Kit, Miltenyi Biotec [130-091-765]). Next, cells were incubated with anti-CD14 FITC antibody (clone TÜK4 Miltenyi Biotec [130-080-701]) and anti-FITC MultiSort MicroBeads (Anti-FITC MultiSort Kit; Miltenyi Biotec [130-058-701]), in order to separate classical and intermediate from nonclassical and non-monocytes. Finally, both cell fractions (classical/intermediate monocytes and nonclassical/non-monocytes) were incubated with CD16 MicroBeads to separate classical from intermediate monocytes and to separate nonclassical from non-monocytes. A representative example of isolated monocyte subsets is depicted in Figure S1. All steps were performed at 4°C and purity was analyzed flow-cytometrically after staining cells with anti-CD86 PE (clone HA5.2B7, Beckman-Coulter [IM2729U]), anti-CD14 PerCP (clone MΦP9, BD Biosciences [345786]) and anti-CD16 PE-Cy7 (clone 3G8, BD Biosciences [557744]) antibodies. For all flow-cytometry experiments, we applied our validated CD86-based gating strategy.5,11
All participants recruited for this study gave informed consent. The study protocol was approved by the local Ethics Committee.
Generation of monocytes from haematopoietic stem cells
Monocytes were generated from haematopoietic stem cells as described previously.18 In brief, haematopoietic stem cells were first isolated from PBMCs of healthy subjects using the CD34 MicroBead Kit (Miltenyi Biotec [130-046-702]). Purity of isolated haematopoietic stem cells was analyzed flow-cytometrically after staining cells with anti-CD34 APC (clone 581; BD Biosciences [555824]) and anti-CD45 PE (clone HI30; BD Biosciences [555483]).
Differentiation of monocytes from haematopoietic stem cells was performed in a 2-step culture. First, haematopoietic stem cells were expanded in the Haematopoietic Progenitor Cell Expansion Medium DXF (PromoCell [C-28021]). For this purpose, isolated cells were cultured in 6-well plates in the presence of the recombinant human growth factors TPO, SCF, flt3-ligand and IL-3 (Cytokine Mix E, PromoCell [C-39890]) for 13 d. Second, after initial expansion, cells were transferred into the Haematopoietic Progenitor Medium (PromoCell [C-28020]) and cultured for further 7 d. Success of differentiation was flow-cytometrically analyzed at day 7 of differentiation after anti-CD86, anti-CD14 and anti-CD16 staining. For Methyl-Seq analysis, classical and intermediate monocytes were isolated with CD14 MicroBeads (Miltenyi Biotec [130-050-201]) according to manufacturer's instructions.
In order to analyze the impact of uremia on monocyte differentiation, Haematopoietic Progenitor Cell Expansion Medium DXF and the Haematopoietic Progenitor Medium were supplemented with 10% serum of hemodialysis patients or of age and gender matched controls. Clinically stable patients (n = 5), undergoing standard hemodialysis therapy 3 times a week, were recruited from the Department of Internal Medicine IV of the Saarland University Medical Center. Blood was drawn before a dialysis session after the long interdialytic interval. To circumvent age- and sex-specific impact on our epigenetic analyses, recruitment was confined to male subjects aged between 60 and 70 y. Mean age of dialysis patients was 64.2 ± 2.8 y. Control subjects (n = 5) were recruited from the outpatient department of the Department of Internal Medicine IV. Control subjects had normal kidney function (eGFR = 77.1 ± 10.3 ml/min/1.73m2), no albuminuria, and a mean age of 66.6±4.3 y.
Methyl-Seq analysis
Isolated monocyte subsets and in vitro differentiated monocytes were frozen in liquid nitrogen and stored at −80°C until DNA isolation. DNA was isolated by using the DNeasy Blood & Tissue Kit (Qiagen [69504]). Genome-wide analysis of CpG DNA methylation was performed by a modified Methyl-Seq method46 at GenXPro GmbH. In brief, HpaII was used as the methylation-sensitive enzyme, recognizing non-CpG-methylated CCGG sites. After digestion with HpaII, “TrueQuant” Y-adapters (GenXPro) suitable for direct Illumina Sequencing were ligated to the fraction of molecules between 100 and 500 bps, which was prior separated from larger DNA fragments using AMpureBeads (Beckman-Coulter [A63882]). Subsequently, the larger DNA was randomly sheared using a bioruptor (Diagenode) to a size of 300–500 bps and adapters containing Illumina P7 priming sites were ligated. The products were PCR amplified using P5 and P7 primers (Illumina) with 10 cycles and sequenced on an Illumina Hiseq2000 machine with 50 cycles.
Bioinformatics
After removal of PCR-duplicates identified by TrueQuant technology, reads starting with the HpaII-specific recognition sequence (CCGG) were quality-trimmed and mapped to the human genome (hg19) using novoalign (http://www.novocraft.com/products/novoalign/). For each genomic HpaII site, reads starting at this position were quantified in each library. The RefSeq track as well as the CpG island track from the UCSC table browser (https://genome.ucsc.edu/cgi-bin/hgTables) were used to annotate genomic restriction sites. Regions up to 2 kb upstream of the transcription start site (TSS) of genes contained in the RefSeq track were defined as promoter regions. Accordingly, downstream regions were defined as genomic regions up to 2 kb downstream of the 3′UTR. In order to annotate miRNA promoter regions, miRNA TSS information was retrieved from the miRStart web service (http://mirstart.mbc.nctu.edu.tw/).
To test for differential methylation, quantification results for all restriction sites were normalized to tags per million (NML) and a statistical test was carried out using NOISeq.47 Significant differential methylation was indicated by a q-value of 0.8 indicating odds of 4:1 that the region is truly differentially methylated (Prob≥0.8).47,48
All Methyl-Seq data are available in the Gene Express Omnibus (GEO) under accession number GSE73788.
Characterization of in vitro generated monocytes
Phagocytosis capacity was measured by using the Fluoresbrite yellow green carboxylate microspheres (0.75 μm; Polysciences [07766-10]), which were first opsonized for 30 min at 37°C with serum from healthy subjects (diluted to 50% with Krebs Ringers PBS) and adjusted to 108 particles/ml. For the phagocytosis assay, 10 μl of opsonized microspheres were mixed with 1 × 104 cells (day 7 of differentiation) in 100 μl culture medium and incubated for 30 min at 37°C with gentle shaking. Finally, samples were stained with anti-CD86, anti-CD14, and anti-CD16, and counts of FITC positive cells were determined flow-cytometrically.
Reactive oxygen species (ROS) were measured flow-cytometrically in monocyte subsets with the cell permanent 5-(and-6)-carboxy-2′,7′-difluorodihydrofluorescein diacetate (H2DFFDA, Life Technologies [C-13293]). Therefore, 1 × 104 cells (day 7 of differentiation) were incubated with H2DFFDA in a concentration of 10 μM for 15 min at 37°C and 5% CO2. Finally, cells were stained with anti-CD86, anti-CD14, and anti-CD16, and ROS levels were determined as MFI in monocyte subsets. Background fluorescence, which was measured in negative controls, was subtracted.
Stimulation of monocytes
In a whole-blood assay, monocytes were stimulated with different concentrations of homocysteine, Adenosine-2′,3′-dialdehyde and 5-aza-2′-deoxycytidine (Sigma Aldrich [H4628,A9384 and A3656]) for 5 or 16 h at 37°C and 5% CO2. Afterwards, cells were washed, stained and analyzed by flow-cytometry according our validated protocol.5,11
Association of monocyte heterogeneity and C1 metabolites in the I Like HOMe study
To prove the epidemiological relevance of our in vitro findings, we analyzed C1 metabolites and monocyte subsets among 420 healthy individuals, who were recruited into our I Like HOMe study (Inflammation, Lipoprotein Metabolism and Kidney Damage in early atherogenesis—The Homburg Evaluation). Study details were published before.33 Briefly, the I Like HOMe study recruited health-care workers to analyze the association of inflammatory and metabolic risk factors with subclinical atherosclerosis. In all participants, monocyte subsets were analyzed and C1 metabolites were measured as described before.33 For technical reasons, data on monocyte subsets, homocysteine, and SAH are missing in 2, 5, and 12 individuals, respectively.
All study participants provided written consent. The study protocol was approved by the local Ethics Committee.
Statistics
Clinical and flow-cytometry data are presented as mean ± SEM, or as numbers (percentages), unless stated differently, and compared with the Student's t-test or one-way analysis of variance (ANOVA), followed by Dunnett's multiple comparison test as post hoc test, as appropriate. Associations between continuous variables were analyzed using Pearson correlation testing. Detailed information on statistical analyses of DNA methylation data are provided above.
Two-sided P-values<0.05 were considered significant. Data were analyzed with IBM SPSS Statistics 23 (SPSS, Inc.) and GraphPad Prism4 (GraphPad).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The present work was supported by a grant from the Else Kröner-Fresenius-Stiftung.
References
- 1.Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, et al.. Nomenclature of monocytes and dendritic cells in blood. Blood 2010; 116:e74-80; PMID:20628149; http://dx.doi.org/ 10.1182/blood-2010-02-258558 [DOI] [PubMed] [Google Scholar]
- 2.Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, Puel A, Biswas SK, Moshous D, Picard C, et al.. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 2010; 33:375-86; PMID:20832340; http://dx.doi.org/ 10.1016/j.immuni.2010.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hofer TP, Zawada AM, Frankenberger M, Skokann K, Satzl AA, Gesierich W, Schuberth M, Levin J, Danek A, Rotter B, et al.. Characterization of subsets of the CD16-positive monocytes: impact of granulomatous inflammation and M-CSF-receptor mutation. Blood 2015; 126:2601-10; PMID:26443621; http://dx.doi.org/ 10.1182/blood-2015-06-651331 [DOI] [PubMed] [Google Scholar]
- 4.Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, Kourilsky P, Wong SC. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011; 118:e16-31; PMID:21653326; http://dx.doi.org/ 10.1182/blood-2010-12-326355 [DOI] [PubMed] [Google Scholar]
- 5.Zawada AM, Rogacev KS, Rotter B, Winter P, Marell RR, Fliser D, Heine GH. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 2011; 118:e50-61; PMID:21803849; http://dx.doi.org/ 10.1182/blood-2011-01-326827 [DOI] [PubMed] [Google Scholar]
- 6.Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res 2012; 53:41-57; PMID:22430559; http://dx.doi.org/ 10.1007/s12026-012-8297-3 [DOI] [PubMed] [Google Scholar]
- 7.Mills KT, Xu Y, Zhang W, Bundy JD, Chen CS, Kelly TN, Chen J, He J. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int 2015; 88:950-7; PMID:26221752; http://dx.doi.org/ 10.1038/ki.2015.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heine GH, Ulrich C, Seibert E, Seiler S, Marell J, Reichart B, Krause M, Schlitt A, Kohler H, Girndt M. CD14(++)CD16+ monocytes but not total monocyte numbers predict cardiovascular events in dialysis patients. Kidney Int 2008; 73:622-9; PMID:18160960; http://dx.doi.org/ 10.1038/sj.ki.5002744 [DOI] [PubMed] [Google Scholar]
- 9.Rogacev KS, Seiler S, Zawada AM, Reichart B, Herath E, Roth D, Ulrich C, Fliser D, Heine GH. CD14++CD16+ monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur Heart J 2011; 32:84-92; PMID:20943670; http://dx.doi.org/ 10.1093/eurheartj/ehq371 [DOI] [PubMed] [Google Scholar]
- 10.Rogacev KS, Zawada AM, Emrich I, Seiler S, Bohm M, Fliser D, Woollard KJ, Heine GH. Lower Apo A-I and lower HDL-C levels are associated with higher intermediate CD14++CD16+ monocyte counts that predict cardiovascular events in chronic kidney disease. Arterioscler Thromb Vasc Biol 2014; 34:2120-7; PMID:25060791; http://dx.doi.org/ 10.1161/ATVBAHA.114.304172 [DOI] [PubMed] [Google Scholar]
- 11.Zawada AM, Fell LH, Untersteller K, Seiler S, Rogacev KS, Fliser D, Ziegler-Heitbrock L, Heine GH. Comparison of two different strategies for human monocyte subsets gating within the large-scale prospective CARE FOR HOMe Study. Cytometry A 2015; 87:750-8; PMID:26062127; http://dx.doi.org/ 10.1002/cyto.a.22703 [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Jiang X, Fang P, Yan Y, Song J, Gupta S, Schafer AI, Durante W, Kruger WD, Yang X, et al.. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation 2009; 120:1893-902; PMID:19858416; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.109.866889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang D, Fang P, Jiang X, Nelson J, Moore JK, Kruger WD, Berretta RM, Houser SR, Yang X, Wang H. Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice. Circ Res 2012; 111:37-49; PMID:22628578; http://dx.doi.org/ 10.1161/CIRCRESAHA.112.269472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bocker MT, Hellwig I, Breiling A, Eckstein V, Ho AD, Lyko F. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood 2011; 117:e182-9; PMID:21427290; http://dx.doi.org/ 10.1182/blood-2011-01-331926 [DOI] [PubMed] [Google Scholar]
- 15.Shantsila E, Wrigley B, Tapp L, Apostolakis S, Montoro-Garcia S, Drayson MT, Lip GY. Immunophenotypic characterization of human monocyte subsets: possible implications for cardiovascular disease pathophysiology. J Thromb Haemost 2011; 9:1056-66; PMID:21342432; http://dx.doi.org/ 10.1111/j.1538-7836.2011.04244.x [DOI] [PubMed] [Google Scholar]
- 16.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al.. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015; 43:D447-52; PMID:25352553; http://dx.doi.org/ 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu TP, Lee CY, Tsai MH, Chiu YC, Hsiao CK, Lai LC, Chuang EY. miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS One 2012; 7:e42390; PMID:22870325; http://dx.doi.org/ 10.1371/journal.pone.0042390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogacev KS, Zawada AM, Hundsdorfer J, Achenbach M, Held G, Fliser D, Heine GH. Immunosuppression and monocyte subsets. Nephrol Dial Transplant 2015; 30:143-53; PMID:25313167; http://dx.doi.org/ 10.1093/ndt/gfu315 [DOI] [PubMed] [Google Scholar]
- 19.Sipkens JA, Hahn NE, Blom HJ, Lougheed SM, Stehouwer CD, Rauwerda JA, Krijnen PA, van Hinsbergh VW, Niessen HW. S-Adenosylhomocysteine induces apoptosis and phosphatidylserine exposure in endothelial cells independent of homocysteine. Atherosclerosis 2012; 221:48-54; PMID:22204864; http://dx.doi.org/ 10.1016/j.atherosclerosis.2011.11.032 [DOI] [PubMed] [Google Scholar]
- 20.Schmidl C, Renner K, Peter K, Eder R, Lassmann T, Balwierz PJ, Itoh M, Nagao-Sato S, Kawaji H, Carninci P, et al.. Transcription and enhancer profiling in human monocyte subsets. Blood 2014; 123:e90-9; PMID:24671955; http://dx.doi.org/ 10.1182/blood-2013-02-484188 [DOI] [PubMed] [Google Scholar]
- 21.Qu C, Edwards EW, Tacke F, Angeli V, Llodra J, Sanchez-Schmitz G, Garin A, Haque NS, Peters W, van Rooijen N, et al.. Role of CCR8 and other chemokine pathways in the migration of monocyte-derived dendritic cells to lymph nodes. J Exp Med 2004; 200:1231-41; PMID:15534368; http://dx.doi.org/ 10.1084/jem.20032152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum 2012; 64:671-7; PMID:22006178; http://dx.doi.org/ 10.1002/art.33418 [DOI] [PubMed] [Google Scholar]
- 23.Rogacev KS, Cremers B, Zawada AM, Seiler S, Binder N, Ege P, Grosse-Dunker G, Heisel I, Hornof F, Jeken J, et al.. CD14++CD16+ monocytes independently predict cardiovascular events: a cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol 2012; 60:1512-20; PMID:22999728; http://dx.doi.org/ 10.1016/j.jacc.2012.07.019 [DOI] [PubMed] [Google Scholar]
- 24.Hristov M, Weber C. Differential role of monocyte subsets in atherosclerosis. Thromb Haemost 2011; 106:757-62; PMID:21901241; http://dx.doi.org/ 10.1160/TH11-07-0500 [DOI] [PubMed] [Google Scholar]
- 25.Betjes MG. Immune cell dysfunction and inflammation in end-stage renal disease. Nat Rev Nephrol 2013; 9:255-65; PMID:23507826; http://dx.doi.org/ 10.1038/nrneph.2013.44 [DOI] [PubMed] [Google Scholar]
- 26.Dwivedi RS, Herman JG, McCaffrey TA, Raj DS. Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int 2011; 79:23-32; PMID:20881938; http://dx.doi.org/ 10.1038/ki.2010.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ingrosso D, Cimmino A, Perna AF, Masella L, De Santo NG, De Bonis ML, Vacca M, D'Esposito M, D'Urso M, Galletti P, et al.. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet 2003; 361:1693-9; PMID:12767735; http://dx.doi.org/ 10.1016/S0140-6736(03)13372-7 [DOI] [PubMed] [Google Scholar]
- 28.Stenvinkel P, Karimi M, Johansson S, Axelsson J, Suliman M, Lindholm B, Heimburger O, Barany P, Alvestrand A, Nordfors L, et al.. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease? J Intern Med 2007; 261:488-99; PMID:17444888; http://dx.doi.org/ 10.1111/j.1365-2796.2007.01777.x [DOI] [PubMed] [Google Scholar]
- 29.Zawada AM, Rogacev KS, Muller S, Rotter B, Winter P, Fliser D, Heine GH. Massive analysis of cDNA Ends (MACE) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics 2014; 9:161-72; PMID:24184689; http://dx.doi.org/ 10.4161/epi.26931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med 1999; 340:115-26; PMID:9887164; http://dx.doi.org/ 10.1056/NEJM199901143400207 [DOI] [PubMed] [Google Scholar]
- 31.Zawada AM, Rogacev KS, Hummel B, Grun OS, Friedrich A, Rotter B, Winter P, Geisel J, Fliser D, Heine GH. SuperTAG methylation-specific digital karyotyping reveals uremia-induced epigenetic dysregulation of atherosclerosis-related genes. Circ Cardiovasc Genet 2012; 5:611-20; PMID:23074332; http://dx.doi.org/ 10.1161/CIRCGENETICS.112.963207 [DOI] [PubMed] [Google Scholar]
- 32.Garibotto G, Valli A, Anderstam B, Eriksson M, Suliman ME, Balbi M, Rollando D, Vigo E, Lindholm B. The kidney is the major site of S-adenosylhomocysteine disposal in humans. Kidney Int 2009; 76:293-6; PMID:19357721; http://dx.doi.org/ 10.1038/ki.2009.117 [DOI] [PubMed] [Google Scholar]
- 33.Zawada AM, Rogacev KS, Hummel B, Berg JT, Friedrich A, Roth HJ, Obeid R, Geisel J, Fliser D, Heine GH. S-adenosylhomocysteine is associated with subclinical atherosclerosis and renal function in a cardiovascular low-risk population. Atherosclerosis 2014; 234:17-22; PMID:24589563; http://dx.doi.org/ 10.1016/j.atherosclerosis.2014.02.002 [DOI] [PubMed] [Google Scholar]
- 34.Wagner C, Koury MJ. S-Adenosylhomocysteine: a better indicator of vascular disease than homocysteine? Am J Clin Nutr 2007; 86:1581-5; PMID:18065573 [DOI] [PubMed] [Google Scholar]
- 35.Xiao Y, Su X, Huang W, Zhang J, Peng C, Huang H, Wu X, Huang H, Xia M, Ling W. Role of S-adenosylhomocysteine in cardiovascular disease and its potential epigenetic mechanism. Int J Biochem Cell Biol 2015; 67:158-66; PMID:26117455; http://dx.doi.org/ 10.1016/j.biocel.2015.06.015 [DOI] [PubMed] [Google Scholar]
- 36.Xiao Y, Zhang Y, Wang M, Li X, Su D, Qiu J, Li D, Yang Y, Xia M, Ling W. Plasma S-adenosylhomocysteine is associated with the risk of cardiovascular events in patients undergoing coronary angiography: a cohort study. Am J Clin Nutr 2013; 98:1162-9; PMID:24004894; http://dx.doi.org/ 10.3945/ajcn.113.058727 [DOI] [PubMed] [Google Scholar]
- 37.Green TJ, Skeaff CM, McMahon JA, Venn BJ, Williams SM, Devlin AM, Innis SM. Homocysteine-lowering vitamins do not lower plasma S-adenosylhomocysteine in older people with elevated homocysteine concentrations. Br J Nutr 2010; 103:1629-34; PMID:20089204; http://dx.doi.org/ 10.1017/S0007114509993552 [DOI] [PubMed] [Google Scholar]
- 38.Hubner U, Geisel J, Kirsch SH, Kruse V, Bodis M, Klein C, Herrmann W, Obeid R. Effect of 1 year B and D vitamin supplementation on LINE-1 repetitive element methylation in older subjects. Clin Chem Lab Med 2013:1-7; PMID:23495396 [DOI] [PubMed] [Google Scholar]
- 39.Liu C, Wang Q, Guo H, Xia M, Yuan Q, Hu Y, Zhu H, Hou M, Ma J, Tang Z, et al.. Plasma S-adenosylhomocysteine is a better biomarker of atherosclerosis than homocysteine in apolipoprotein E-deficient mice fed high dietary methionine. J Nutr 2008; 138:311-5; PMID:18203897 [DOI] [PubMed] [Google Scholar]
- 40.Luo X, Xiao Y, Song F, Yang Y, Xia M, Ling W. Increased plasma S-adenosyl-homocysteine levels induce the proliferation and migration of VSMCs through an oxidative stress-ERK1/2 pathway in apoE(−/−) mice. Cardiovasc Res 2012; 95:241-50; PMID:22492673; http://dx.doi.org/ 10.1093/cvr/cvs130 [DOI] [PubMed] [Google Scholar]
- 41.Fang P, Zhang D, Cheng Z, Yan C, Jiang X, Kruger WD, Meng S, Arning E, Bottiglieri T, Choi ET, et al.. Hyperhomocysteinemia potentiates hyperglycemia-induced inflammatory monocyte differentiation and atherosclerosis. Diabetes 2014; 63:4275-90; PMID:25008174; http://dx.doi.org/ 10.2337/db14-0809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Selimoglu-Buet D, Wagner-Ballon O, Saada V, Bardet V, Itzykson R, Bencheikh L, Morabito M, Met E, Debord C, Benayoun E, et al.. Characteristic repartition of monocyte subsets as a diagnostic signature of chronic myelomonocytic leukemia. Blood 2015; 125:3618-26; PMID:25852055; http://dx.doi.org/ 10.1182/blood-2015-01-620781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Machowska A, Carrero JJ, Lindholm B, Stenvinkel P. Therapeutics targeting persistent inflammation in chronic kidney disease. Transl Res 2016; 167:204-13; PMID:26173187; http://dx.doi.org/ 10.1016/j.trsl.2015.06.012 [DOI] [PubMed] [Google Scholar]
- 44.Hodge DR, Xiao W, Clausen PA, Heidecker G, Szyf M, Farrar WL. Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J Biol Chem 2001; 276:39508-11; PMID:11551897; http://dx.doi.org/ 10.1074/jbc.C100343200 [DOI] [PubMed] [Google Scholar]
- 45.Geisel J, Schorr H, Heine GH, Bodis M, Hubner U, Knapp JP, Herrmann W. Decreased p66Shc promoter methylation in patients with end-stage renal disease. Clin Chem Lab Med 2007; 45:1764-70; PMID:18067454; http://dx.doi.org/ 10.1515/CCLM.2007.357 [DOI] [PubMed] [Google Scholar]
- 46.Brunner AL, Johnson DS, Kim SW, Valouev A, Reddy TE, Neff NF, Anton E, Medina C, Nguyen L, Chiao E, et al.. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res 2009; 19:1044-56; PMID:19273619; http://dx.doi.org/ 10.1101/gr.088773.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res 2011; 21:2213-23; PMID:21903743; http://dx.doi.org/ 10.1101/gr.124321.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valencia-Morales Mdel P, Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramirez-Ruz J, Gomez A, Moran S, et al.. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genomics 2015; 8:7; PMID:25881171; http://dx.doi.org/ 10.1186/s12920-015-0085-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.