

With the increased prevalence of obesity, non-alcoholic fatty liver disease (NAFLD) is now the most common chronic liver disease. At present, NAFLD has no approved pharmacotherapy, likely due to incomplete understanding of its molecular mediators, exacerbated by its complex hormonal regulation. Insulin represses hepatic glucose production (HGP), but simultaneously acts as a permissive signal for increased de novo lipogenesis (DNL).1 Thus, obesity-induced insulin resistance and compensatory hyperinsulinemia increases DNL, exacerbating hepatic steatosis.2 Identification of selective molecular regulators of DNL is necessary to uncouple these insulin-regulated processes to develop novel therapeutics to combat the burgeoning public health crisis of Type 2 Diabetes (T2D) and NAFLD.
One potential regulator of DNL is the mechanistic target of rapamycin (mTOR), a phosphoinositide 3-kinase-like serine/threonine protein kinase that comprises the catalytic core of 2 distinct protein complexes – mTOR complex 1 (mTORC1) and 2 (mTORC2) – that integrate anabolic and catabolic processes to cellular or environmental cues. For instance, mTORC1 links cell growth to nutrient availability,3 through substrate identification mediated by Raptor.4 The current prevailing hypothesis, based largely on hepatocyte-specific Raptor knockout mice, is that mTORC1 activity is necessary for DNL.4 This conclusion, however, is inconsistent with data from mTOR inhibitor-treated mice, and assumes the unproven hypotheses that Raptor has no extra-mTORC1 activity and that Raptor-mTOR interaction is constitutive, incompatible with data that mTORC1 complex stability is affected by hormonal and other signals.
We have recently discovered that the established role of Raptor as a scaffold for mTOR catalytic activity is an oversimplification, that mTORC1-independent Raptor (or, Raptorfree) stabilizes the Akt phosphatase, PHLPP2 (PH domain leucine-rich repeat-containing protein-phosphatase 2), to prevent fatty liver.5 In this work, through complementary biochemical methods, we found abundant Raptorfree in livers isolated from young, healthy mice, but increased mTOR-Raptor association with aging and obesity, leading to the progressive disappearance of Raptorfree. Strikingly, replacement of the Raptorfree deficit reduced feeding-associated Akt Ser473 phosphorylation (with no effects on PI3K-induced Akt Thr308 phosphorylation or other upstream stimuli) by preventing proteosomal degradation of PHLPP2, with end result of reduced DNL and hepatic triglyceride accumulation.5 As such, hepatic PHLPP2 levels are also lower in aged or obese mice lacking Raptorfree, and PHLPP2 knockdown reversed Raptorfree-mediated improvements in DNL/hepatic triglyceride. Consistently, adenoviral PHLPP2 overexpression abrogated obesity-induced fatty liver, surprisingly without adverse effects on glucose homeostasis. These data suggest 2 novel and complementary aspects of insulin action in liver: 1) the existing bifurcation model of hepatic insulin action6 is missing a critical temporal element – post-prandial Akt activation is necessary to inhibit HGP, but prolonged Akt activity increases DNL if the insulin signal is sustained, without beneficial effects on glucose; and 2) in aging/obesity-induced insulin resistance, loss of hepatocyte Raptorfree destabilizes PHLPP2, which allows compensatory hyperinsulinemia to stimulate DNL, that exacerbates NAFLD (Fig. 1).
Figure 1.
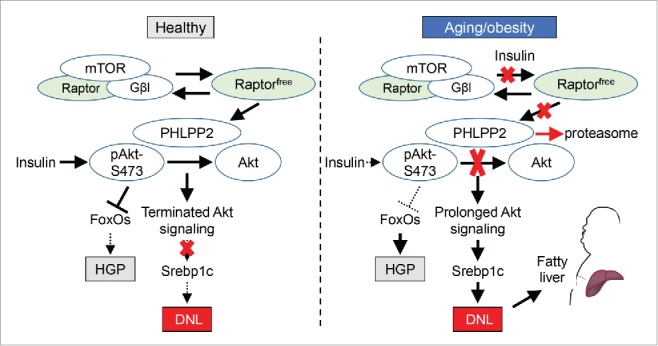
Proposed model of Raptor-PHLPP2 signaling. In normal/healthy liver, mTORC1-independent (”free”) Raptor stabilizes PHLPP2 to terminate Akt action. Aging or obesity reduces Raptorfree, leading to PHLPP2 degradation and prolonged Akt action, increasing de novo lipogenesis (DNL) and fatty liver. Hepatic glucose production (HGP) is also increased by insulin resistance, but is unaffected by the Raptorfree-PHLPP2 axis.
Although negative feedback of mTOR-potentiated insulin action by Raptorfree is conceptually attractive and paradigm-altering, we are not naïve to the unanswered questions raised by this work. For instance, although we have shown that chronic hyperinsulinemia is sufficient to reduce Raptorfree in hepatocytes, it is likely that mTOR-Raptor interaction is affected by other growth factors/hormones and/or nutrients, and just as Raptorfree places a brake on mTORC1-induced DNL, it may have a role in context-dependent modulation of other mTORC1-regulated processes including protein synthesis, autophagy and others. Further, we have yet to prove the mechanism by which Raptorfree exists. Although free- and mTORC1-bound Raptor populations may have pre-ordained and separate fates, we hypothesize that these pools may interchange in the presence of a biochemical signal, such as a post-translational modification on an mTORC1 complex member, possibly Raptor itself. There exists some evidence for this hypothesis – Raptor ubiquitination, mediated in part by competing influences of the DDB1-CUL4 ubiquitin ligase and the ubiquitin hydrolase UCH-L1, has been shown to determine mTORC1 stability and activity.7 Alternatively, undefined molecules recruited to the mTORC1 complex may affect Raptor-mTOR interaction. A higher-throughput screening tool than the cumbersome biochemical methods (size-exclusion chromatography, chemical cross-linking) we used, such as a Raptorfree-ELISA, will be helpful to evaluate these possibilities in a systematic fashion.
Despite these remaining questions, our findings indicate that the Raptorfree/PHLPP2 axis regulates lipid homeostasis, and contributes to growing evidence that mTORC1 stability influences mTORC1-dependent and potentially –independent processes. Understanding the crosstalk between mTORC1-bound and Raptorfree may aid in the development of novel therapeutic strategies to treat NAFLD and T2D, but we are also cognizant of the broader ramifications of these data. First, as alluded to earlier, the role of mTORC1 in a variety of cellular processes has been cemented by study of tissue-specific Raptor knockout mice – it is possible that these phenotypes may be confounded by the absence of Raptorfree, and thus, these data may require re-examination. Additionally, as PHLPPs act as tumor suppressors, and mTORC1 inhibitors are approved for cancer, we are studying whether Raptorfree determines the NAFLD transition to non-alcoholic steatohepatitis (NASH) and hepatocellular carcinoma, or influence risk of other obesity-potentiated cancers, and whether these actions are PHLPP-dependent. This latter point is important, as DNL has emerged as a cancer target in its own right. Finally, we predict that a mTORC1-bound:Raptorfree equilibrium exists in other tissues, and may be differentially affected by obesity or other pathology – this intriguing possibility may open several areas of biological inquiry.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Haas JT, et al.. Cell Metab 2012; 15:873-84; PMID:22682225; http://dx.doi/org/ 10.1016/j.cmet.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Savage DB, et al.. Curr Opin Lipidol 2010; 21:329-36; PMID:20581678; http://dx.doi/org/ 10.1097/MOL.0b013e32833b7782 [DOI] [PubMed] [Google Scholar]
- [3].Howell JJ, et al.. Trends Endocrinol Metab 2011; 22:94-102; PMID:21269838; http://dx.doi/org/ 10.1016/j.tem.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Laplante M, et al.. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi/org/ 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim K, et al.. Nat Commun 2016; 7:10255; PMID:26743335; http://dx.doi/org/ 10.1038/ncomms10255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li S, et al.. Proc Natl Acad Sci U S A 2010; 107:3441-6; PMID:20133650; http://dx.doi/org/ 10.1073/pnas.0914798107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hussain S, et al.. Mol Cell Biol 2013; 33:1188-97; PMID:23297343;http://dx.doi/org/ 10.1128/mcb.01389-12 [DOI] [PMC free article] [PubMed] [Google Scholar]