

The “immuno-logical” recognition of cell death as a non-inflammatory (tolerogenic or null) or an inflammatory (immunogenic) event has a crucial role in tumor progression and the response to antineoplastic therapy. Harnessing the immune system to achieve clinical efficacy is realistic in the context of conventional anticancer agents whose outcome mainly relies on the ability to (re)instate tumor-host interaction, thus translating cell death into an immune response. Several factors work in concert to determine whether cell death is immunogenic or not, including the intrinsic antigenicity of cancer cells, the host immunocompetence, the initiating stress stimulus and the cell death pathways engaged.1
Cell death events have been historically dichotomized into either of 2 mutually exclusive and diametrically opposed morphotypes: accidental/pathological cell death (i.e., necrosis) and programmed/physiological cell death (i.e., apoptosis), being considered immunogenic and tolerogenic, respectively. Nevertheless, the panel of cell death morphologies is much broader than originally thought. Hence, although at operational level the dichotomy accidental cell death (ACD) versus regulated, genetically-encoded, pharmacologic/genetic modulable cell death (RCD) is still employed, novel systematic classifications based on measurable biochemical, molecular and functional parameters have been recently proposed.2 Four major aspects have emerged. First, apoptotic cell demises may follow distinct signaling cascades all converging toward caspase activation. Second, some instances of necrotic cell death are tightly regulated (e.g., necroptosis). Third, mitochondria are central regulators of both apoptotic and necrotic RCD. Fourth, depending on the upstream triggers, apoptosis can be highly immunogenic hence alerting the innate immune system and instructing it to stimulate a cognate response. Thus, a limited array of antineoplastic agents activates immunogenic cell death (ICD) inducing tumoricidal immune responses potentially erasing residual neoplastic cells. ICD involves (i) the release of soluble chemotactic and chemotropic (find-me) mediators, such as type I interferons, and (ii) the surface exposure of engulfement (eat-me) signals, such as calreticulin (CALR), which respectively recall and activate phagocytes thus tipping the “decision” toward an inflammatory event.3
Cationic antimicrobial peptides (CAPs) are small synthetic molecules able to selectively kill tumor cells due to the electrostatic attraction with the anionic nature of cancer cell membranes.4 LTX-315 is a helical CAP optimized for membrane destabilization, which has been recently shown to induce cell lysis when administered intralesionally. This led to the release of immunomediators into the tumor microenvironment and unleashed an inflammatory response whereby specific cytotoxic T cells eradicated residual cancer cells.5
These discoveries prompted Forveille and colleagues to study the nature of such an ICD based on the notion that apoptotic and necrotic subroutes both are immunogenic.6 Through morphometric and ultrastructural analyses they showed that the appearance of human U2OS osteosarcoma cells exposed to LTX-315 lacks features of apoptosis, such as pyknosis and karyorhexis. Instead, these cells acquired a necrotic, "ghost-like" morphotype manifesting with plasma membrane integrity loss, vacuolated cytoplasm and mitochondria dilatation. This is in line with previous evidence showing mitochondriotoxicity of LTX-315 coupled to mitochondrial outer membrane permeabilization (MOMP), release of pro-apoptotic factors and a modest activation of mitophagy.7 Despite inducing MOMP, LTX-315 fails to activate caspase-3, and hence regulated apoptosis, at any dosage. To define whether LTX-315 treatment dooms tumor cells to regulated necrosis, the authors analyzed the impact of necrostatin-1 and cyclosporine A - 2 necroptosis inhibitors acting on the death domain receptor-associated adaptor kinase receptor-interacting protein 1 (RIP1) and the mitochondrial permeability transition, respectively - demonstrating that they did not impact on LTX-315-induced cell demise. This suggests that LTX-315 ignites unregulated necrosis (Fig. 1). The authors surmise that necrosis-related plasma membrane collapse and the resultant release of danger associated molecules in the local microenvironment may be responsible for LTX-315 pro-inflammatory and pro-immunogenic properties.
Figure 1.
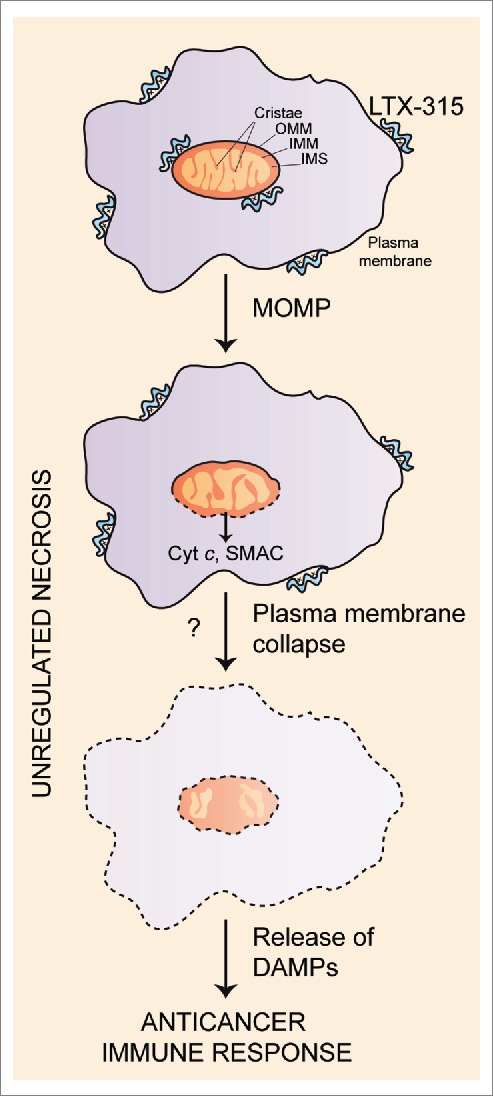
Schematic representation of the mechanism of unregulated necrosis induced by LTX-315, which involves the mitochondrial enrichment of LTX-315 followed by mitochondrial outer membrane permeabilization (MOMP), release of cytochrome c (Cyt c) and second mitochondria-derived activator of caspase (SMAC) into the cytoplasm, and final plasma membrane collapse. This may result in the release of damage-associated molecular patterns (DAMPs) in the tumor microenvironment, and the activation of tumoricidal immune responses.
These results unveil an unexpected and unconventional central role of unregulated necrosis in LTX-315-related immunogenicity since antimicrobial peptides are known to mainly induce MOMP- and caspase-dependent apoptosis.2 Further work is required to elucidate the precise molecular mechanisms underlying this new modality of ICD as well as the nature of released pro-inflammatory factors (Fig. 1). Nonetheless, it is reasonable to speculate from a clinical point of view that LTX-315 epitomizes an oncolytic agent that, due to its immunogenic potential, may represent a new cancer immunotherapeutic approach for triggering tumoricidal immune responses at will.
Abbreviations
- ACD
accidental cell death
- CALR
calreticulin
- CAPs
cationic antimicrobial peptides
- ICD
immunogenic cell death
- MOMP
mitochondrial outer membrane permeabilization
- RCD
regulated cell death
- RIP1
receptor-interacting protein 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Green DR, et al. Nat Rev Immunol 2009; 9:353-63; PMID:19365408; http://dx.doi.org/ 10.1038/nri2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Galluzzi L, et al. Cell Death Differ 2012; 19:107-20; PMID:21760595; http://dx.doi.org/ 10.1038/cdd.2011.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sistigu A, et al. Nat Med 2014; 20:1301-9; PMID:25344738; http://dx.doi.org/ 10.1038/nm.3708 [DOI] [PubMed] [Google Scholar]
- [4].Mader JS, et al. Mol Cancer Ther 2005; 4:612-24; PMID:15827335; http://dx.doi.org/ 10.1158/1535-7163.MCT-04-0077 [DOI] [PubMed] [Google Scholar]
- [5].Camilio KA, et al. Cancer Immunol Immunother 2014; 63:601-13; PMID:24676901; http://dx.doi.org/ 10.1007/s00262-014-1540-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Forveille S, et al. Cell Cycle 2015; 14:3506-12; PMID:26566869; http://dx.doi.org/ 10.1080/15384101.2015.1093710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhou H, et al. Oncotarget 2015; 6:26599-614; PMID:26378049; http://dx.doi.org/ 10.18632/oncotarget.5613 [DOI] [PMC free article] [PubMed] [Google Scholar]