

ABSTRACT
Cyclin Dependent Kinases (CDKs) are important regulators of DNA replication. In this work we have investigated the consequences of increasing or decreasing the CDK activity in S phase. To this end we identified S-phase regulators of the fission yeast CDK, Cdc2, and used appropriate mutants to modulate Cdc2 activity. In fission yeast Mik1 has been thought to be the main regulator of Cdc2 activity in S phase. However, we find that Wee1 has a major function in S phase and thus we used wee1 mutants to investigate the consequences of increased Cdc2 activity. These wee1 mutants display increased replication stress and, particularly in the absence of the S-phase checkpoint, accumulate DNA damage. Notably, more cells incorporate EdU in a wee1− strain as compared to wildtype, suggesting altered regulation of DNA replication. In addition, a higher number of cells contain chromatin-bound Cdc45, an indicator of active replication forks. In addition, we found that Cdc25 is required to activate Cdc2 in S phase and used a cdc25 mutant to explore a situation where Cdc2 activity is reduced. Interestingly, a cdc25 mutant has a higher tolerance for replication stress than wild-type cells, suggesting that reduced CDK activity in S phase confers resistance to at least some forms of replication stress.
KEYWORDS: CDK, Cell cycle, Cdc25, fission yeast, Mik1, replication stress, S phase, Wee1
Introduction
Cyclin Dependent Kinases (CDKs) are crucial regulators of cell-cycle progression, regulating both entry into mitosis and S phase. The regulation of CDK activity at entry into mitosis is well understood, but the regulation of CDKs in S phase is less well described. CDK activity is required to control replication initiation, both at entry into and through S phase. In eukaryotes replication initiation occurs at multiple origins, allowing complete replication of all chromosomes within a relatively short S phase. To avoid repeated firing of the same origins within a single S phase, loading of the replicative helicase and its activation are separated in time. The core of the helicase is loaded onto replication origins in G1 phase as part of the pre-Replicative Complex (pre-RC).1 Origin firing requires activation of the helicase, which involves the loading of a number of other proteins, including Sld3S.p./Sld3S.c./ TreslinH.s., and culminating in the loading of Cdc45,2 a cofactor of the helicase. The loading of Cdc45 is carefully regulated and requires phosphorylation of Sld3S.p./Sld3S.c./ TreslinH.s. by the S-phase CDK 3-6 implying that origin firing in S phase is strictly regulated by the activity of the CDK. CDK activity, in turn, is regulated by inhibitory phoshorylations on the T14 and Y15 residues by the Wee kinases.7 There are two kinases to perform these phosphorylations in mammalian cells, Wee1 and Myt1. Recent work has shown a role for Wee1 in regulating CDK activity during S phase.8
The only CDK in fission yeast, Cdc2, can also be phosphorylated and thereby inhibited by two kinases, namely Wee1 and Mik1. Y15 can be phosphorylated by either kinase,9-11 whereas T14 is phosphorylated by Wee1.12 Wee1 is known as the major Cdc2 kinase in G2, since wee1− mutants enter mitosis at a reduced cell size.13,14 Mik1 has been considered the main regulator of S phase, based on the observations that mik1 is expressed shortly before and during S phase, due to transcriptional regulation by the Cdc10 transcription factor.15,16 Furthermore, Mik1 is required to prevent entry into mitosis when the S-phase checkpoint is activated.17 However, the involvement of Mik1 in the regulation of the S-phase CDK in an undisturbed cell cycle has not been rigorously tested.
There are some indications in the literature that Wee1 might have roles outside of G2/M. In particular, cells lacking Wee1 need Rad3 for survival.18 Rad3 (ATRH. s.) is a checkpoint kinase that is activated by a wide range of DNA-damaging agents in both S phase and G2 phase. If Wee1 is merely regulating entry into mitosis, the reason for the lethality of rad3 wee1 mutants is not obvious since (i) Rad3 has no known role in an unperturbed G2/M transition and (ii) Wee1 is thought to ensure that the cells have attained the appropriate size at entry into mitosis19 and this mechanism does not involve the G2/M checkpoint.
Here we show that fission yeast Wee1 is a major kinase regulating Cdc2 activity in S phase and thus we used wee1− mutants to investigate the consequences of increased Cdc2 activity. The increased Cdc2 activity results in increased replication stress and DNA damage, particularly in the absence of the S-phase checkpoint. In addition, we identify Cdc25 as a regulator of CDK activity in S phase and show that a cdc25 mutant is resistant to hydroxyurea (HU). This observation suggests that reducing CDK activity renders the cells resistant to some agents that confer replication stress.
Results
In order to study the role and regulation of Cdc2 at entry into and progression through S phase we identified mutants that alter the activity of Cdc2 in S phase. The lack of Wee1 function renders the cells dependent on a checkpoint, since wee1 and rad3 mutations are synthetic lethal.18
The synthetic lethality of wee1− rad3− double mutants led us to speculate that Wee1 has a role in regulating Cdc2 in cell-cycle phases other than G2/M. We compared the consequences of removing Wee1 or Mik1 function, both in the presence and absence of Rad3 function. The wee1-50 rad3Δ, wee1Δ rad3ts and mik1Δ rad3ts double mutants are viable at the permissive temperature of 25°C, but when Wee1 and Rad3 functions are both compromised, at the semi-restrictive or restrictive temperatures, the cells do not grow (Fig 1A). In contrast, mik1Δ rad3ts cells grow at the restrictive temperature (Fig 1A). Thus, cells lacking Wee1 but not Mik1 depend on an intact checkpoint for survival.
Figure 1.

wee1− and cdc2-1w but not mik1Δ and cdc2-3w cells depend on Rad3 for survival. Exponentially growing cells of the indicated genotypes were streaked out onto YES plates and incubated at the temperatures shown.
A known substrate of Wee1 is Cdc2, but other substrates most likely exist. In order to investigate whether the requirement for the checkpoint in wee1− cells is due to impaired regulation of Cdc2 activity or to that of other substrates, we made use of the wee alleles of cdc2. These cdc2 mutants display the wee phenotype in that they enter mitosis prematurely, giving rise to small cells. The reason for their wee phenotype is due to aberrant regulation by inhibitors or activators of Cdc2. Cdc2-1w (G146D) is insensitive to regulation by Wee1 20 while Cdc2-3w (C67Y) is insensitive to regulation by the phosphatase Cdc25.21 We constructed cdc2-1w rad3ts and cdc2-3w rad3ts double mutants and examined their survival at the restrictive temperature for rad3ts. Cells carrying the cdc2-1w but not those carrying the cdc2-3w allele are inviable in the absence of Rad3 function (Fig 1B). These results suggest that at least one of the reasons why the checkpoint is essential in the absence of Wee1 function is the aberrant regulation of Cdc2 activity.
The synthetic lethality of wee1− rad3− cells derives from problems in S phase
Next, we wished to identify the cell-cycle phase where checkpoint function is required in the absence of Wee1. We synchronized wee1-50 rad3Δ cells grown at the permissive temperature in early G2 by size selection and shifted them to the restrictive temperature at different times, so that they entered mitosis with or without Wee1 activity. We then examined the kinetics of mitosis and the microscopic appearance of the cells going through mitosis.
In the first experiment, we shifted the wee1-50 rad3Δ cells to the restrictive temperature for 100 minutes after synchronization to allow them to undergo the first mitosis in the absence of Wee1 function. The cells started to enter mitosis at 50 minutes after shift-up (Fig 2A). They entered mitosis at a reduced size compared to wild-type cells, as expected in the absence of Wee1. Otherwise, the first mitosis appeared normal and no uneven segregation of chromosomes (“cutting”) was observed (Fig 2A, right panel). After 100 minutes half the culture was kept at the restrictive temperature and the other half was shifted to the permissive temperature. The cells that were kept at the restrictive temperature rapidly underwent another round of mitosis, which was abnormal, generating a high proportion of cut cells (Fig 2A, right panel). In contrast, the cells shifted to the permissive temperature did not enter a second mitosis (Fig 2A) within the time frame of the experiment (220 min).
Figure 2.
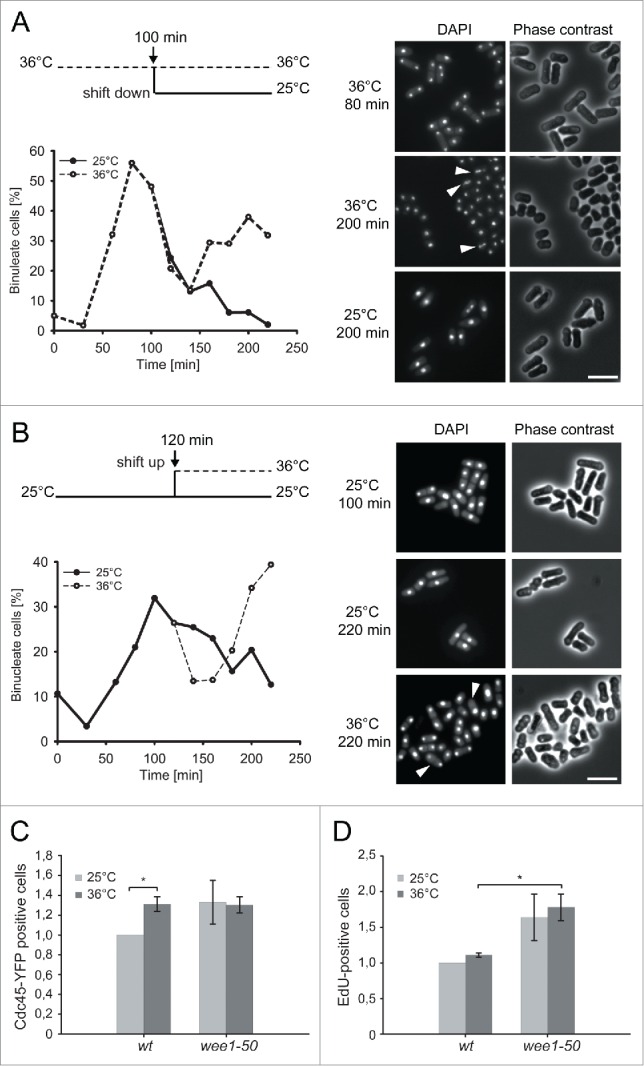
The lethality of wee1− rad3− cells derives from problems in S phase A and B The experimental regime is shown in the upper panel. Binucleate index is shown as a function of time and microscopic images of the cells at the indicated time points are shown on the right. Cells displaying the cut phenotype are marked with arrowheads. C Quantification of Cdc45:YFP-positive cells in wt and wee1-50 strain (mean ± SD, n = 3, *p < 0,05). Cells were counted after extraction with a low-salt buffer and detergent. The data are normalized to the values in the wild-type at 25°C. D EdU-positive cells in wt strain compared to wee1-50 strain (mean ± SD, n = 3, *p < 0,005)Cells were pulse-labeled with EdU for min.
In the second experiment, after synchronization we incubated the wee1-50 rad3Δ cells at the permissive temperature until they completed the first mitosis in the presence of Wee1 activity. Then we left half of the culture at the permissive temperature and shifted the other half to the restrictive temperature to inactivate Wee1. At the permissive temperature they did not enter the second mitosis during the course of the experiment (Fig 2B), which corresponds to normal cell-cycle kinetics, and they did not display the cut phenotype (Fig 2B, right panel). The cells shifted to the restrictive temperature entered mitosis soon after they exited the first one and in this mitosis they displayed the cut phenotype.
The above results show that the rad3Δ cells enter a second, abnormal mitosis only if they lack Wee1 after the first mitosis and this is regardless of whether or not Wee1 was functional in the first mitosis. We conclude that the second abnormal mitosis is not a consequence of an aberrant entry into or progression through the first mitosis. In contrast, wee1-50 cells do not cut at the restrictive temperature when Rad3 is functional19 (Fig. 3B). Thus, these findings demonstrate that the synthetic lethality of wee1− rad3− double mutants does not stem from the loss of the Wee1 function in G2/M, but rather that the cells enter a lethal second mitosis very soon after the first one. The time of entry into the aberrant mitosis corresponds to G1 or S phase.
Figure 3.

Replication stress in wee1 mutants. A Serial dilutions of cells exponentially growing in YES were spotted onto YES media containing HU. B Microscopic images of cells incubated in the absence and presence of HU. C Cells exponentially growing in YES were streaked out onto YES plates and incubated at 25 or 29°C. D Immunoblot analysis of Mcm4: HA levels in wee1 rad3 mcm4:HA:aid cells treated with auxin at the indicated concentrations for 1 h. The detection of tubulin is used as loading control. E wee1 rad3 mcm4:HA:aid cells were incubated in the presence of auxin for 1 h at the permissive temperature before being shifted to the restrictive temperature for 4 hours. Then the cells were plated at appropriate dilutions in triplicates onto YES plates and incubated at the permissive temperature for 3 days before colonies were counted. The graph shows relative survival normalized to the survival without auxin (mean ± SD, n≥3, *p < 0,05).
Wee1 regulates DNA replication
Cdc45 is a cofactor of the replicative helicase and its loading onto chromatin immediately precedes activation of origins. To further clarify the role of Wee1, we measured the number of cells containing active origins by monitoring Cdc45:YFP loading in the presence or absence of Wee1 function. Exponentially growing cells were shifted to the permissive or restrictive temperature for 1 hour, then the proteins not bound to chromatin were extracted with a low-salt buffer and detergent, and the fraction of Cdc45:YFP positive cells was scored. At the permissive temperature a higher proportion of cells contained Cdc45:YFP foci in the wee1-50 mutant than in wild-type cells (Fig 2C), suggesting that wee1− cells spend a longer time in S phase. An alternative explanation is that wee1− cells have more active and therefore more Cdc45-containing replication origins and forks and thus more cells are scored as positive with our settings. At the restrictive temperature the wild-type strain had significantly higher proportion of Cdc45-positive cells than it had at the permissive temperature, suggesting that at 36°C origin firing was increased. Interestingly, there was no difference between wild-type and wee1-50, suggesting that at 36°C a factor other than CDK activity became limiting.
In order to quantify DNA synthesis in the absence or presence of Wee1, we constructed a wee1-50 strain that also carries the hsv thymidine kinase and the human transporter hENT1, which allows fission yeast cells to take up and incorporate thymidine analogs into the DNA.22 Wild-type and wee1-50 cells were grown at the permissive temperature and then shifted to the restrictive temperature for 0 or 30 minutes, pulse-labeled with EdU for 6 minutes and EdU incorporation was measured. Significantly more cells incorporated EdU in the absence of Wee1 than in its presence (Fig 2C). This might be due to a prolonged S phase and/or to a higher rate of DNA synthesis leading to more cells incorporating detectable amounts of EdU. In order to explore these alternatives further we attempted to quantify the intensity of the EdU signal in the EdU-positive cells but failed to obtain reliable, reproducible measurements. In agreement with the higher proportion of Cdc45-positive cells, the increased EdU-incorporation in the wee1-50 strain was observable at the permissive temperature, suggesting that the S-phase function of Wee1 is compromised in the wee1-50 mutant even at this temperature.
The increased Cdc2 activity results in replication stress
The above results suggest aberrant regulation of DNA replication in wee1 mutants, presumably due to increased Cdc2 activity. To explore how cells with increased Cdc2 activity respond to replication stress, we tested the sensitivity of the cells to reduced nucleotide pools. We plated serial dilutions of wild-type, wee1−, mik1− and rad3− cells on YE plates containing 0, 4, or 6 mM hydroxyurea (HU), an inhibitor of ribonucleotide reductase. As expected, rad3− cells were HU-sensitive and did not grow even on 4 mM HU (Fig 3A). mik1− cells were also HU-sensitive (Fig 3A) as expected when an effector of the S-phase checkpoint is missing.9,23 Cells lacking Wee1 function were much more sensitive to HU than mik1− cells, although not as much as rad3− cells (Fig 3A). To test whether the increased HU-sensitivity of wee1− cells is due to a checkpoint defect, we investigated the microscopic appearance of the cells in the presence of HU. Like wild-type cells, wee1− cells became elongated in the presence of HU (Fig 3B), suggesting a functional S-phase checkpoint. Thus, we conclude that in the absence of Wee1 increased Cdc2 activity in S phase leads to a faster depletion of nucleotide pools, making the cells sensitive to HU, even though the S-phase checkpoint is functional. A prediction of the above model is that the viability of wee1− rad3− cells should be higher with higher nucleotide pools. We employed a strain that lacks Spd1, the inhibitor of ribonucleotide reductase, and has about 50% larger nucleotide pools than wild-type.24 We compared the viability of wee1− rad3− cells to that of wee1− rad3− spd1Δ and found that at the semirestrictive temperature of 30°C the lack of Spd1, and presumably the elevated dNTP levels, partially restored the viability of wee1− rad3− cells (Fig 3C).
If the increased Cdc2 activity in the absence of Wee1 leads to more origins being fired in each S phase, reducing the number of pre-RC-containing origins is expected to alleviate the effects of the lack of Wee1 function. We have constructed a wee1− rad3− strain that also carries an auxin-degradable Mcm4,25 a core component of the pre-RC. In the presence of auxin the amount of Mcm4 is reduced (Fig 3D), which would result in a lower number of pre-RCs. Exponentially growing wee1− rad3− mcm4:aid cells were treated with 0, 200 or 300 μM auxin for 1 hour at the permissive temperature, then half of each culture was shifted to the restrictive temperature for 5 hours before the cells were plated onto YE plates and incubated at the permissive temperature to assess survival. We compared the viability of wee1-50 and wee1-50 rad3Δ mutants that were incubated in the presence and absence of auxin before the temperature shift. We observed a small but reproducible effect so that wee1-50 rad3Δ cells survived significantly better when the level of Mcm4 was reduced (Fig 3E). Increasing auxin concentrations and thus reducing Mcm4 levels further lead to a G1 arrest.25 The partial rescue of the wee1-50 rad3Δ mutant indicates that the primary effect of loss of Wee1 is increased replication initiation, which can be counteracted by a reduced number of pre-RCs.
The absence of Wee1 leads to DNA damage
The above results suggest that Wee1 function is important for a normal S phase and suggests that the S-phase checkpoint is activated in the absence of Wee1. Rad3 is involved not only in the S-phase checkpoint but also in the G2/M DNA-damage checkpoint and therefore we investigated the requirement for Cds1, which is specifically needed for the former but not for the latter.26 We constructed a wee1-50 cds1Δ double mutant and investigated its viability at the permissive and restrictive temperatures. Similarly to the wee1− rad3− double mutants, wee1-50 cds1Δ cells grow very poorly at the restrictive temperature for wee1, although the synthetic lethal effect in the wee1-50 cds1Δ double mutant is not quite as severe as in the wee1− rad3− double mutant (Fig 4A). In order to directly assess the activation of Cds1 we monitored the phosphorylation of Mus81, a known substrate of Cds1.27,28 Phosphorylation can be observed as a Cds1-dependent bandshift by immunoblotting.27 In the absence of Wee1 a slower migrating band of Mus81 was observed (Fig 4B) even at the permissive temperature and this band shift was even more pronounced when the cells were incubated for 1 hour at the semirestrictive temperature. We conclude that the requirement for Rad3 in wee1 mutants reflects a requirement for activation of Cds1 and the S-phase checkpoint.
Figure 4.
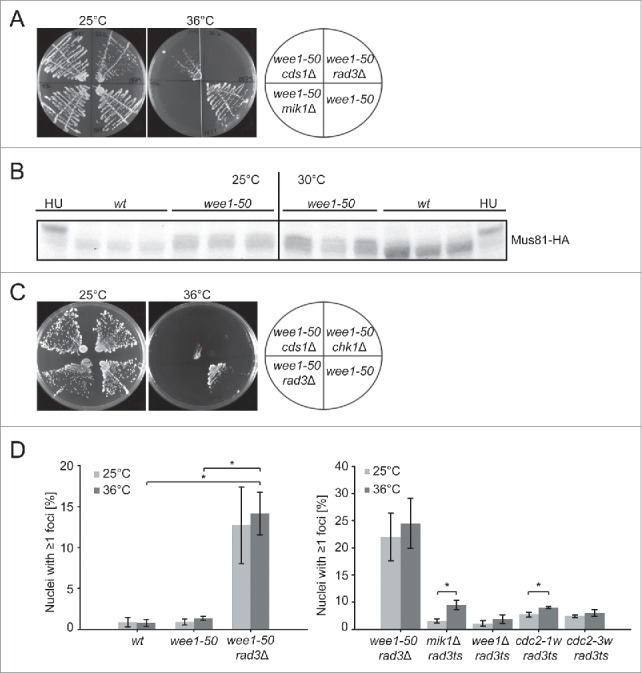
Replication stress in wee1 mutants leads to DNA damage A and C Cells exponentially growing in YES were streaked out onto YES plates and incubated at the temperatures shown. B Immunoblot analysis of Mus81:3HA in the indicated strains incubated at the temperatures shown. HU-treated samples were used as control. D The percentage of cells containing Rad22:YFP foci in the indicated strains is shown. At least 200 cells were counted for each sample (mean ± SD, n = 3, *p < 0,05).
It was previously reported that chk1 and wee1 mutations are synthetic lethal.29 We compared the viability of wee1-50 cds1Δ and wee1-50 chk1Δ and found that they both die at 36°C (Fig 3C). Thus, these data collectively suggest that the increased Cdc2 activity in the wee1 mutant leads to increased replication stress, which challenges the S-phase checkpoint (Cds1) as well as Chk1 function.
Chk1 is known to be activated primarily in response to DNA strand breaks and therefore the requirement for Chk1 in wee1− cells indicates the presence of double-strand breaks. In order to explore this further, we determined the frequency of DNA double strand breaks in wild-type, wee1-50 and wee1-50 rad3Δ cells. To this end, we used a marker of double-strand breaks, Rad22 foci, which arise during repair of double-strand breaks by homologous recombination.30 Exponentially growing wild-type, wee1-50 and wee1-50 rad3Δ cells were incubated at the permissive or restrictive temperature for 1 hour and the frequency of Rad22:YFP-foci was scored (Fig 4D). We observed very few Rad22 foci in wild-type and wee1− cells at either temperature. However, we observed a significantly higher number of Rad22 foci in the wee1-50 rad3Δ cells at the permissive temperature, and even more foci were observed at the restrictive temperature. These results are consistent with a model where wee1− mutants depend on the S-phase checkpoint to avoid DNA double-strand breaks. To further explore the extent of DNA damage when Cdc2 activity is increased, we scored the frequency of Rad22:YFP foci in wee1Δ rad3ts, mik1Δ rad3ts, cdc2-1w rad3ts and cdc2-3w rad3ts cells incubated at the permissive and restrictive temperatures (Fig 4D). Surprisingly, none of these double mutants had a large number of cells with ≥1 Rad22 foci, unlike wee1-50 rad3Δ cells shown above. It should be noted that a previous study compared the phenotypes of rad3Δ and rad3ts (see Fig 3 in ref 4) and found that the checkpoint deficiency of the temperature-sensitive mutant is not as severe as that of the deletion mutant, which might explain the low number of cells with Rad22 foci in the current experiments. Nevertheless, we compared the number of cells displaying Rad22 foci at the permissive and restrictive temperatures and found a small but significant increase at the restrictive temperature in mik1Δ rad3ts and cdc2-1w rad3ts.
Cdc25 is involved in the regulation of Cdc2 during S phase
The above data suggest that an increase in Cdc2 activity during S phase can have grave consequences for genomic stability. This led us to speculate that Wee1 activity is normally counteracted by a phosphatase to regulate Cdc2 activity and/or that Wee1 activity itself is carefully regulated. There are two tyrosine-phosphatases that can dephosphorylate Cdc2 on Y15: Cdc25 and Pyp3. Cdc25 is essential and mutants arrest at the G2/M transition. Pyp3 can rescue a cdc25 mutant when overexpressed,31 but its role in the cell cycle is not understood. At the G2/M transition Wee1 is known to be inhibited by Cdr1 and Cdr2, which in turn are regulated by Pom132-35 but no upstream regulator of Wee1 in S phase has been identified. In order to investigate whether any of these factors is involved in the regulation of Cdc2 activity in S phase, we investigated whether DNA replication is affected in strains deficient in Cdc25, Pyp3 or Cdr2.
The cdc25-22 mutant was grown at the permissive temperature, then shifted to the semi-restrictive temperature of 28°C for 0 or 30 minutes and pulse-labeled with EdU for 10 minutes. Interestingly, significantly fewer cdc25-22 cells incorporated EdU when Cdc25 function was impaired at the semi-permissive temperature (Fig 5A), suggesting that Cdc25 has a role in regulating Cdc2 activity in S phase. To test whether the lack of Cdc25 can rescue the synthetic lethality of wee1 rad3 double mutants, we introduced a cdc25-22 mutation in a wee1-50 rad3Δ background and incubated the triple mutant at permissive, semirestrictive and restrictive temperatures. The triple mutant, but not the wee1 rad3 double mutant, was able to grow at the semirestrictive temperature of 30°C, but not at 36°C, demonstrating that cdc25-22 can partially rescue the lethality of the wee1-50 rad3Δ double mutant (Fig 5B). This observation is best explained by Cdc25 counteracting Wee1 activity and activating Cdc2 also in S phase.
Figure 5.
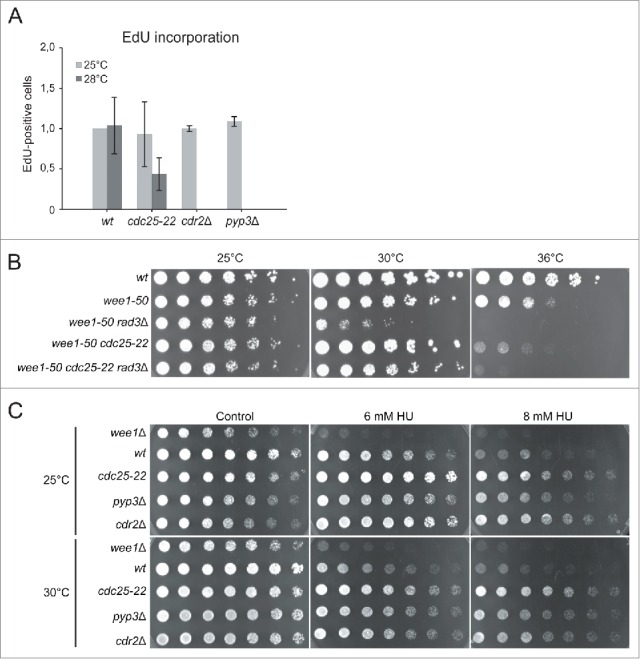
Cdc25 is involved in the regulation of S phase A Cells were incubated at 25°C, shifted to 28°C for 30 min and pulse-labeled with EdU for 6 min. EdU-positive cells were counted and the average of three experiments is shown. Error bars represent standard deviation. B Cells exponentially growing in YES were streaked out onto YES plates and incubated at the temperatures shown. C Serial dilutions of cells exponentially growing in YES were spotted onto YES media containing HU.
To test the possible involvement of Pyp3 and Cdr2 in the regulation of S phase, wild-type, cdr2Δ and pyp3Δ cells were grown at 25°C, pulse-labeled with EdU for 6 minutes and fixed to measure EdU-incorporation. The number of EdU-positive cells in the pyp3Δ and the cdr2Δ mutants was similar to that in wild-type cells, suggesting that the respective proteins are not major regulators of Cdc2 activity during S phase (Fig 5A).
To investigate the roles of Cdc25, Cdr2 and Pyp3 in the regulation of Cdc2 in S phase further, we tested the HU sensitivity of the appropriate mutants. Serial dilutions of wild-type, wee1Δ, cdc25-22, cdr2Δ and pyp3Δ cells were spotted onto YE plates containing 0, 6, or 8 mM HU and the plates were incubated at 25 or 30°C. As shown above, wee1Δ cells were HU-sensitive and did not grow on plates containing 6 or 8 mM HU (Fig 5C). The viability of pyp3Δ and cdr2Δ cells was similar to that of wild-type cells. Interestingly, the cdc25-22 mutant had a higher viability than wild-type even at the permissive temperature and even more so at the semi-restrictive temperature. These observations suggest that decreased Cdc2 activity leads to resistance to HU, presumably due to fewer origins being fired and a slower exhaustion of nucleotide pools.
Discussion
In this work we investigated the regulation of CDKs at entry into and progression through S phase and the consequences of increased and decreased CDK activity. We show that in fission yeast Wee1 is the major inhibitory kinase regulating CDK activity in S phase. An important clue leading to this conclusion was the finding that wee1− mutants enter an abnormal mitosis in the absence of Rad3 soon after exit from a normal mitosis. The timing suggests that the mutants enter an abnormal mitosis either from G1 or from S phase. G1 is short under standard laboratory growth conditions, but since wee1− cells enter mitosis at a reduced cell size, they prolong G1 to allow for sufficient growth.14 Thus, if Rad3 was required to prolong G1 in wee1− cells, one might expect that wee1− cells would depend on Rad3 for survival. However, no such role for Rad3 has been described and thus it appears more plausible that the S-phase checkpoint is challenged in wee1− mutants. Consistently, we show that a cds1Δ mutation is synthetically lethal with wee1-50 and that Cds1 is activated in the absence of Wee1. Cds1 is specifically required for the S-phase checkpoint, supporting the argument that the cells depend on the S-phase checkpoint in the absence of Wee1.
We suggest the following model (Fig 6) that is consistent with all our findings: increasing Cdc2 activity by using wee1 mutants leads to increased replication stress, which challenges the S-phase checkpoint (Cds1). The increased replication stress is due to the presence of an increased number of replication forks, since increased Cdc2 activity leads to Cdc45 loading at more origins and thus more origins are fired. This reasoning is supported by the finding that reducing the amount of Mcm4 and thereby the number of pre-RCs can partially rescue the viability of the wee1 rad3 mutant. If replication is initiated at more origins than the number of forks the cell can support, replication factors and/or nucleotides become limiting, leading to fork stalling. The requirement for Chk1 suggests that the S-phase checkpoint cannot stabilize all the stalled forks, some forks collapse and strand breaks are generated, leading to activation of Chk1. Consistently, we have shown an increased number of Rad22 foci in the absence of Wee1. Our findings mirror observations in human cells, where Wee1 inhibition leads to high CDK activity during S phase, which in turn results in unscheduled origin firing and depletion of nucleotide pools, which ultimately causes fork stalling and DNA damage.8
Figure 6.
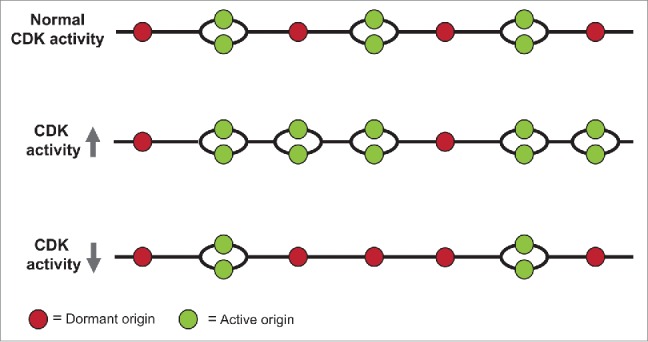
A model of the effects of increasing and decreasing CDK activity on origin firing. See text for explanation.
CDKs are the best-known substrates of Wee1, but the existence of other substrates that could affect progression through S phase is quite possible. However, the phenotypes of wee1− cells shown here are most likely caused by aberrant regulation of Cdc2. We show here that the checkpoint dependence of wee1 mutants as well as the increased number of strand breaks in the absence of Rad3 is shared by the cdc2-1w mutant, which is specifically deficient in the Wee1-dependent regulation of Cdc2. Furthermore, the lack of Wee1 can be partially compensated by the lack of Cdc25, which again points to altered Cdc2 regulation as the decisive factor for the observed effects. It should be noted that cdc2-3w cells, which are specifically deficient in the Cdc25-dependent regulation of Cdc2, do not depend on the S-phase checkpoint for survival. This could be due to the extent of how deregulated Cdc2 function is in each cdc2-w mutant. Furthermore, additional substrates of Wee1 might contribute to the S-phase-related phenotypes observed in wee1 mutants. One candidate is the Mus81-Eme1 endonuclease, which was shown to interact with and possibly is phosphorylated by Wee1 in vivo.36,37 Furthermore, depletion of Wee1 results in a lower speed of the replication forks in mammalian cells and this could result from altered regulation of Mus81-Eme1. In addition, Wee1 phosphorylates Histone 2B upstream of histone cluster 1, providing an epigenetic modulation that contributes to ensuring appropriate histone abundance,38 which is particularly important during DNA replication.
Wee1 inhibitors are gaining increasing attention as therapeutic agents in cancer therapy and are known to potentiate the DNA-damaging effect of cytotoxic chemotherapies. These effects have been attributed to the function of Wee1 in the G2/M checkpoint.39 Interestingly, there have been a number of reports in the literature suggesting that fission yeast wee1 mutants are sensitive to DNA-damaging agents, including ionizing radiation,40 MMS, camptothecin and the UV-mimetic drug 4NQO41 and HU41 and current work. The reason for this sensitivity is not clear, since the role of Wee1 in G2/M checkpoint regulation was debated23,42 and the HU sensitivity of Wee1 cells is not due to a lack of checkpoint function (this work). Another possible mechanism to affect survival is regulating the efficiency of DNA repair. A recent study showed that Crb2-T215 phosphorylation by Cdc2 regulates repair-pathway choice41 and thus might contribute to efficient repair of DNA damage and thereby to survival.
We suggest that the increased CDK-activity in S phase might well be the main reason for the increased sensitivity to DNA-damaging agents. Increased CDK activity in S phase and the accompanying replication stress engages the cellular mechanisms responding to DNA damage. As the checkpoint and repair mechanisms have a limited capacity, further damage by the DNA-damaging agents will lead to cell death at lower doses than in cells not exposed to prior replications stress. Such synthetic lethal effects might be exploited in cancer therapy if Wee1 inhibitors were combined with DNA-damaging treatments and explains why Wee1 inhibitors potentiate the DNA-damaging effect of cytotoxic chemotherapies.43
Materials and methods
Yeast strains and growth conditions
Strain construction and maintenance were as described.44 The cells were grown in Yeast Extract medium (YES) or Edinburgh Minimal Medium (EMM) at 25°C.
Survival assays
Two types of survival assays were performed. 1) Cells were streaked onto YES plates and incubated at the permissive or restrictive temperature for 3 days. 2) Log-phase cells grown in YES were plated in a two-fold serial dilution onto YES plates and incubated for 3 days at the permissive or semirestrictive temperature.
Protein extracts and western blots
Protein extracts for protein gel blotting were made by TCA extraction, as described previously.45 For western blot analysis the following antibodies were used: anti-HA (Santa Cruz sc-7392 ) at a dilution of 1:500) and anti-α-tubulin (Sigma T5168) at a dilution of 1:30,000. The secondary antibodies were HRP conjugated anti-mouse IgG (Cell Signaling #7076), used at a dilution of 1/5000. Detection was performed using the enhanced chemiluminescence procedure ( ECL kit).
Immunofluorescence microscopy
For analyses by immunoflourescence microscopy, cells were resuspended in phosphate-buffered saline (PBS), mounted on poly-L-lysine microscope slides, dried, and viewed in the presence of 0.2 µg/ml 4′,6-diamidino-2-phenylindole (DAPI). Imaging was performed on a Leica CTR DM6000 microscope equipped with a Leica DFC350 FX monochrome digital camera and a 63X/1.4 NA objective. Images were processed and analyzed in Fiji.
Cdc45 chromatin binding assay
Extractions of Cdc45 protein not bound to chromatin was performed as described,46 except that Cdc45:YFP was detected instead of MCMs.
Lactose gradient centrifugation
Cells grown in EMM were synchronized in G2 by lactose gradient centrifugation, performed as described.47 The cells were released into G2 phase, treated as described in Results and fixed in ice–cold, 70 % ethanol at the time points indicated on the figures.
EdU incorporation and detection
Cells growing exponentially in YES were shifted to 36°C for 0 or 30 minutes and then pulse-labeled for 6 minutes with 10 µM EdU. The cells were fixed in 70% ethanol, washed once with PBS and treated with 1 mg/ml zymolyase 20T (Sunrise Science Products, https://www.sunrisescience.com/pages/prexpyeast_zymo.html) for 20 minutes at 36°C. The cells were washed once with PBS and permeabilized with 1% triton for one minute. For EdU detection, the Click-iT EdU Alexa Flour 555 kit (Life Technologies https://www.lifetechnologies.com/order/catalog/product/C10338) was used as described by the manufacturer.
Detection of Rad22: YFP foci
Cells were grown exponentially in YES and shifted to the restrictive (36°C) or kept at the permissive (25°C) temperature for 1 hour. Cells were collected and fixed in 100% methanol for 5 minutes and kept in acetone. A Python-based script was used for automated analysis to count the number of cells containing Rad22:YFP foci. The script uses the DAPI channel to extract the area of the nucleus and employs the function “Find Maxima” in the YFP-channel to quantify the number of rad22:YFP foci within each nucleus.
Statistics
Significance was assessed with Students t-test. A p-value of 0,05 or lower was regarded as statistically significant.
Table 1.
Complete genotypes of each strain used in the experiments shown in the indicated figures.
| Figure 1A | ||
|---|---|---|
| 1374 | h− | wee1::ura4+ ura4-D18 |
| 1863 | h+ | mik1::ura4+ ura4-D18 |
| 1865 | h− | rad3ts wee1::ura4+ ura4-D18 leu1-32 |
| 1868 | rad3ts mik1::ura4+ ura4-D18 | |
| Figure 1B | ||
| 404 | h− | cdc2-3w ade6-M210 |
| 551 | h− | cdc2-1w |
| 1374 | h− | wee1::ura4+ ura4-D18 |
| 1787 | h+ | wee1-50 rad3::ura4+ ura4-(D18?) |
| 1863 | h+ | mik1::ura4+ ura4-D18 |
| 1865 | h- | rad3ts wee1::ura4+ ura4-D18 leu1-32 |
| 1868 | rad3ts mik1::ura4+ ura4-D18 | |
| 1877 | h+ | wee1-50 ura4(294 or D) |
| 1900 | h− | cdc2-1w rad3ts |
| 1901 | h− | cdc2-3w rad3ts ade6−M210?704? |
| Figure 2A, B | ||
| 1787 | h+ | wee1-;50 rad3::ura4+ ura4-(D18?) |
| Figure 2C | ||
| 1925 | sna41(cdc45):YFP:ura4+ ura4-(294 or D18) | |
| 1926 | wee1-50 ura4(294 or D18) sna41(cdc45):YFP:ura4+ | |
| Figure 2D | ||
| 1849 | h− | leu1-32::hENT1-leu1+ (pJAH29) ura4-294::hsv-tk-ura4+ (pJK210-tk+) |
| 1871 | h− | wee1-50 leu1-32::hENT1-leu1+ (pJAH29) ura4-294::hsv-tk-ura4+ (pJK210-tk+) ade6 |
| Figure 3A | ||
| 19 | h− | L972 |
| 1153 | h− | rad3::ura4+ leu1-32 ura4-D18 ade6-704 |
| 1374 | h− | wee1::ura4+ ura4-D18 |
| 1863 | h+ | mik1::ura4+ ura4-D18 |
| Figure 3B | ||
| 38 | h+ | ura4-D18 leu1-32 |
| 1877 | h+ | wee1-50 ura4(294 or D) |
| 2025 | h+ | ura4-D18 leu1-32 rad3::hphMX6 |
| Figure 3C | ||
| 1787 | h+ | wee1-50 rad3::ura4+ ura4-(D18?) |
| 1876 | h− | rad3::natMX6 spd1::ura4+ ura4-D18 wee1-50 |
| 1877 | h+ | wee1-50 ura4(294 or D) |
| Figure 3D, E | ||
| 1858 | rad3::ura4+ wee1-50 ade6+:pADH15.skp1:AtTIR1:2NLS:9myc mcm4:2HA:aid:ura4+ | |
| ura4-D18 | ||
| Figure 4A | ||
| 968 | h+ | wee1-50 mik1::ura4+ ura4-D18 ade1-D25 |
| 1787 | h+ | wee1-50 rad3::ura4+ ura4-(D18?) |
| 1877 | h+ | wee1-50 ura4(294 or D) |
| 1941 | wee1-50 cds1::ura4+ ura4(?) | |
| Figure 4B | ||
| 2116 | h+ | mus81:3HA:kanMX6 ura4-D18 leu1-32 |
| 2118 | h+ | mus81:3HA:kanMX6 wee-50 |
| Figure 4C | ||
| 1787 | h+ | wee1-50 rad3::ura4+ ura4-(D18?) |
| 1877 | h+ | wee1-50 ura4(294 or D) |
| 1941 | wee1-50 cds1::ura4+ ura4(?) | |
| 1980 | chk1::ura4+ wee1-50 ura4 | |
| Figure 4D | ||
| 1433 | h+ | rad22:YFP:kanMX4 ade6-M210 ura4-D18 leu1-32 |
| 1973 | wee1-50 rad22:YFP:kanMX4 ura4 | |
| 1974 | rad3::natMX6 wee1-50 rad22:YFP:kanMX4 ura4 | |
| 2097 | rad22:YFP:kanMX4 rad3ts mik1::ura4+ ura4-D18 | |
| 2100 | h− | rad22:YFP:kanMX4 rad3ts wee1::ura4+ ura4-D18 leu1-32 |
| 2101 | h− | rad22:YFP:kanMX4 cdc2-1w rad3ts |
| 2102 | h− | rad22:YFP:kanMX4 cdc2-3w rad3ts ade6-M210?704? |
| Figure 5A | ||
| 1402 | h− | leu1-32::hENT1-leu1+ (pJAH29) ura4-294::hsv-tk-ura4+ (pJK210-tk+) ade6-704 |
| 2031 | cdr2::kanMX4 ade6-(704 or M210) leu1-32::hENT1-leu1+ (pJAH29) ura4-294::hsv-tk- | |
| ura4+ (pJK210-tk+) | ||
| 2032 | pyp3::kanMX4 ade6-(704 or M210) leu1-32::hENT1-leu1+ (pJAH29) ura4-294::hsv-tk- | |
| ura4+ (pJK210-tk+) | ||
| 2055 | cdc25-22 leu1-32::hENT1-leu1+ (pJAH29) ura4-294::hsv-tk-ura4+ (pJK210-tk+) | |
| Figure 5B | ||
| 19 | h− | L972 |
| 1787 | h+ | wee1-50 rad3::ura4+ ura4-(D18?) |
| 1877 | h+ | wee1-50 ura4(294 or D) |
| 1932 | cdc25-22 wee1-50 | |
| 1939 | h− | cdc25-22 wee1-50 rad3::natMX6 ura4 |
| Figure 5C | ||
| 19 | h− | L972 |
| 137 | h− | cdc25-22 |
| 1374 | h− | wee1::ura4+ ura4-D18 |
| 2044 | h− | cdr2::kanMX6 |
| 2030 | pyp3::kanMX4 ade6-M210 ura4-D18 leu1-32 | |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Kay Oliver Schink for his help with analyzing the Rad22 foci, Thomas Caspari, Olaf Nielsen and James Moseley for strains and Marit Osland Haugli for technical assistance.
Funding
This work was funded by The South-Eastern Norwegian Health Authorities and the Norwegian Cancer Society.
References
- [1].Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem 2002; 71:333-74; PMID:12045100; http://dx.doi.org/ 10.1146/annurev.biochem.71.110601.135425 [DOI] [PubMed] [Google Scholar]
- [2].Zou L, Stillman B. Formation of a preinitiation complex by S-phase cyclin CDK-dependent loading of Cdc45p onto chromatin. Science 1998; 280:593-6; PMID:9554851; http://dx.doi.org/ 10.1126/science.280.5363.593 [DOI] [PubMed] [Google Scholar]
- [3].Nakajima R, Masukata H. SpSld3 is required for loading and maintenance of SpCdc45 on chromatin in DNA replication in fission yeast. Mol Biol Cell 2002; 13:1462-72; PMID:12006645; http://dx.doi.org/ 10.1091/mbc.02-01-0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kamimura Y, Tak YS, Sugino A, Araki H. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J 2001; 20:2097-107; PMID:11296242; http://dx.doi.org/ 10.1093/emboj/20.8.2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Masumoto H, Muramatsu S, Kamimura Y, Araki H. S-Cdk-dependent phosphorylation of Sld2 essential for chromosomal DNA replication in budding yeast. Nature 2002; 415:651-5; PMID:11807498; http://dx.doi.org/ 10.1038/nature713 [DOI] [PubMed] [Google Scholar]
- [6].Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J Cell Biol 2011; 193:995-1007; PMID:21646402; http://dx.doi.org/ 10.1083/jcb.201102003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Berry LD, Gould KL. Regulation of Cdc2 activity by phosphorylation at T14/Y15. Prog Cell Cycle Res 1996; 2:99-105; PMID:9552387; http://dx.doi.org/ 10.1007/978-1-4615-5873-6_10 [DOI] [PubMed] [Google Scholar]
- [8].Beck H, Nahse-Kumpf V, Larsen MS, O'Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuasen RG, et al.. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol 2012; 32:4226-36; PMID:22907750; http://dx.doi.org/ 10.1128/MCB.00412-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Christensen PU, Bentley NJ, Martinho RG, Nielsen O, Carr AM. Mik1 levels accumulate in S phase and may mediate an intrinsic link between S phase and mitosis. Proc Natl Acad Sci USA 2000; 97:2579-84; PMID:10716991; http://dx.doi.org/ 10.1073/pnas.97.6.2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gould KL, Feoktistova A. Characterization of novel mutations at the Schizosaccharomyces pombe cdc2 regulatory phosphorylation site, tyrosine 15. Mol Biol Cell 1996; 7:1573-86; PMID:8898363; http://dx.doi.org/ 10.1091/mbc.7.10.1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lundgren K, Walworth N, Booher R, Dembski M, Kirschner M, Beach D. mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell 1991; 64:1111-22; PMID:1706223; http://dx.doi.org/ 10.1016/0092-8674(91)90266-2 [DOI] [PubMed] [Google Scholar]
- [12].Den Haese GJ, Walworth N, Carr AM, Gould KL. The Wee1 protein kinase regulates T14 phosphorylation of fission yeast Cdc2. Mol Biol Cell 1995; 6:371-85; PMID:7626804; http://dx.doi.org/ 10.1091/mbc.6.4.371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fantes PA, Nurse P. Control of the timing of cell division in fission yeast. Cell size mutants reveal a second control pathway. Exp Cell Res 1978; 115:317-29; PMID:689088; http://dx.doi.org/ 10.1016/0014-4827(78)90286-0 [DOI] [PubMed] [Google Scholar]
- [14].Nurse P. Genetic control of cell size at cell division in yeast. Nature 1975; 256:547-51; PMID:1165770; http://dx.doi.org/ 10.1038/256547a0 [DOI] [PubMed] [Google Scholar]
- [15].Ng SS, Anderson M, White S, McInerny CJ. mik1(+) G1-S transcription regulates mitotic entry in fission yeast. FEBS Lett 2001; 503:131-4; PMID:11513869; http://dx.doi.org/ 10.1016/S0014-5793(01)02720-X [DOI] [PubMed] [Google Scholar]
- [16].Lowndes NF, McInerny CJ, Johnson AL, Fantes PA, Johnston LH. Control of DNA synthesis genes in fission yeast by the cell-cycle gene cdc10+. Nature 1992; 355:449-53; PMID:1734281; http://dx.doi.org/ 10.1038/355449a0 [DOI] [PubMed] [Google Scholar]
- [17].Rhind N, Russell P. Tyrosine phosphorylation of cdc2 is required for the replication checkpoint in Schizosaccharomyces pombe. Mol Cell Biol 1998; 18:3782-7; PMID:9632761; http://dx.doi.org/ 10.1128/MCB.18.7.3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].al-Khodairy F, Carr AM. DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. Embo J 1992; 11:1343-50; PMID:1563350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Russell P, Nurse P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987; 49:559-67; PMID:3032459; http://dx.doi.org/ 10.1016/0092-8674(87)90458-2 [DOI] [PubMed] [Google Scholar]
- [20].Nurse P, Thuriaux P. Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. Genetics 1980; 96:627-37; PMID:7262540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fantes PA. Isolation of cell size mutants of a fission yeast by a new selective method: characterization of mutants and implications for division control mechanisms. J Bacteriol 1981; 146:746-54; PMID:7217015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hodson JA, Bailis JM, Forsburg SL. Efficient labeling of fission yeast Schizosaccharomyces pombe with thymidine and BUdR. Nucleic Acids Res 2003; 31:e134; PMID:14576334; http://dx.doi.org/ 10.1093/nar/gng134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rhind N, Russell P. Roles of the mitotic inhibitors Wee1 and Mik1 in the G(2) DNA damage and replication checkpoints. Mol Cell Biol 2001; 21:1499-508; PMID:11238887; http://dx.doi.org/ 10.1128/MCB.21.5.1499-1508.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fleck O, Vejrup-Hansen R, Watson A, Carr AM, Nielsen O, Holmberg C. Spd1 accumulation causes genome instability independently of ribonucleotide reductase activity but functions to protect the genome when deoxynucleotide pools are elevated. J Cell Sci 2013; 126:4985-94; PMID:23986475; http://dx.doi.org/ 10.1242/jcs.132837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kanke M, Nishimura K, Kanemaki M, Kakimoto T, Takahashi TS, Nakagawa T, Masukata H. Auxin-inducible protein depletion system in fission yeast. BMC Cell Biol 2011; 12:8; PMID:21314938; http://dx.doi.org/ 10.1186/1471-2121-12-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lindsay HD, Griffiths DJ, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev 1998; 12:382-95; PMID:9450932; http://dx.doi.org/ 10.1101/gad.12.3.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Boddy MN, Lopez-Girona A, Shanahan P, Interthal H, Heyer WD, Russell P. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol 2000; 20:8758-66; PMID:11073977; http://dx.doi.org/ 10.1128/MCB.20.23.8758-8766.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kai M, Boddy MN, Russell P, Wang TS. Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev 2005; 19:919-32; PMID:15805465; http://dx.doi.org/ 10.1101/gad.1304305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Walworth N, Davey S, Beach D. Fission yeast chkl protein kinase links the rad checkpoint pathway to cdc2. Nature 1993; 363:368-71; PMID:8497322; http://dx.doi.org/ 10.1038/363368a0 [DOI] [PubMed] [Google Scholar]
- [30].Kim WJ, Lee S, Park MS, Jang YK, Kim JB, Park SD. Rad22 Protein, a Rad52 Homologue inSchizosaccharomyces pombe, Binds to DNA Double-strand Breaks. J Biol Chem 2000; 275:35607-11; PMID:10956666; http://dx.doi.org/ 10.1074/jbc.M007060200 [DOI] [PubMed] [Google Scholar]
- [31].Millar JB, Lenaers G, Russell P. Pyp3 PTPase acts as a mitotic inducer in fission yeast. EMBO J 1992; 11:4933-41; PMID:1464318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Martin SG, Berthelot-Grosjean M. Polar gradients of the DYRK-family kinase Pom1 couple cell length with the cell cycle. Nature 2009; 459:852-6; PMID:19474792; http://dx.doi.org/ 10.1038/nature08054 [DOI] [PubMed] [Google Scholar]
- [33].Moseley JB, Mayeux A, Paoletti A, Nurse P. A spatial gradient coordinates cell size and mitotic entry in fission yeast. Nature 2009; 459:857-60; PMID:19474789; http://dx.doi.org/ 10.1038/nature08074 [DOI] [PubMed] [Google Scholar]
- [34].Grallert A, Connolly Y, Smith DL, Simanis V, Hagan IM. The S. pombe cytokinesis NDR kinase Sid2 activates Fin1 NIMA kinase to control mitotic commitment through Pom1/Wee1. Nat Cell Biol 2012; 14(7):738-45; PMID:22684255; http://dx.doi.org/doi:10.1038/ncb2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Deng L, Moseley JB. Compartmentalized nodes control mitotic entry signaling in fission yeast. Mol Biol Cell 2013; 24:1872-81; PMID:23615447; http://dx.doi.org/ 10.1091/mbc.E13-02-0104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dominguez-Kelly R, Martin Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, Debatisse M, Freire R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol 2011; 194:567-79; PMID:21859861; http://dx.doi.org/ 10.1083/jcb.201101047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Martin Y, Dominguez-Kelly R, Freire R. Novel insights into maintaining genomic integrity: Wee1 regulating Mus81/Eme1. Cell Div 2011; 6:21; PMID:22152133; http://dx.doi.org/ 10.1186/1747-1028-6-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mahajan K, Fang B, Koomen JM, Mahajan NP. H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat Struct Mol Biol 2012; 19:930-7; PMID:22885324; http://dx.doi.org/ 10.1038/nsmb.2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013; 12:3159-64; PMID:24013427; http://dx.doi.org/ 10.4161/cc.26062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barbet NC, Carr AM. Fission yeast wee1 protein kinase is not required for DNA damage- dependent mitotic arrest. Nature 1993; 364:824-7; PMID:8355807; http://dx.doi.org/ 10.1038/364824a0 [DOI] [PubMed] [Google Scholar]
- [41].Mahyous SA, Ewert-Krzemieniewska K, Liu B, Caspari T. Hyperactive Cdc2 kinase interferes with the response to broken replication forks by trapping S.pombe Crb2 in its mitotic T215 phosphorylated state. Nucleic Acids Res 2014; 42:7734-47; PMID:24861625; http://dx.doi.org/ 10.1093/nar/gku452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].O'Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J 1997; 16:545-54; PMID:9034337; http://dx.doi.org/ 10.1093/emboj/16.3.545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res 2012; 40:477-86; PMID:21937510; http://dx.doi.org/ 10.1093/nar/gkr697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol 1991; 194:795-823; PMID:2005825; http://dx.doi.org/ 10.1016/0076-6879(91)94059-L [DOI] [PubMed] [Google Scholar]
- [45].Caspari T, Dahlen M, Kanter-Smoler G, Lindsay HD, Hofmann K, Papadimitriou K, Sunnerhagen P, Carr AM. Characterization of Schizosaccharomyces pombe Hus1: a PCNA-related protein that associates with Rad1 and Rad9. Mol Cell Biol 2000; 20:1254-62; PMID:10648611; http://dx.doi.org/ 10.1128/MCB.20.4.1254-1262.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kearsey SE, Montgomery S, Labib K, Lindner K. Chromatin binding of the fission yeast replication factor mcm4 occurs during anaphase and requires ORC and cdc18. EMBO J 2000; 19:1681-90; PMID:10747035; http://dx.doi.org/ 10.1093/emboj/19.7.1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Carr AM, Moudjou M, Bentley NJ, Hagan IM. The chk1 pathway is required to prevent mitosis following cell-cycle arrest at ‘start’. Curr Biol 1995; 5:1179-90; PMID:8548290; http://dx.doi.org/ 10.1016/S0960-9822(95)00234-X [DOI] [PubMed] [Google Scholar]