

Abstract
Long-term angiotensin II (ANG II) infusion significantly increases ANG II levels in the kidney through two major mechanisms: AT1 receptor-mediated augmentation of angiotensinogen (AGT) expression and uptake of circulating ANG II by the proximal tubules. However, it is not known whether intracellular ANG II stimulates AGT expression in the proximal tubule. In the present study, we overexpressed an intracellular cyan fluorescent ANG II fusion protein (Ad-sglt2-ECFP/ANG II) selectively in the proximal tubule of rats and mice using the sodium and glucose cotransporter 2 (sglt2) promoter. AGT mRNA and protein expression in the renal cortex and 24-h urinary AGT excretion were determined 4 wk following overexpression of ECFP/ANG II in the proximal tubule. Systolic blood pressure was significantly increased with a small antinatriuretic effect in rats and mice with proximal tubule-selective expression of ECFP/ANG II (P < 0.01). AGT mRNA and protein expression in the cortex were increased by >1.5-fold and 61 ± 16% (P < 0.05), whereas urinary AGT excretion was increased from 48.7 ± 5.7 (n = 13) to 102 ± 13.5 (n = 13) ng/24 h (P < 0.05). However, plasma AGT, renin activity, and ANG II levels remained unaltered by ECFP/ANG II. The increased AGT mRNA and protein expressions in the cortex by ECFP/ANG II were blocked in AT1a-knockout (KO) mice. Studies in cultured mouse proximal tubule cells demonstrated involvement of AT1a receptor/MAP kinases/NF-кB signaling pathways. These results indicate that intracellular ANG II stimulates AGT expression in the proximal tubules, leading to increased AGT formation and secretion into the tubular fluid, which contributes to ANG II-dependent hypertension.
Keywords: angiotensin II, angiotensinogen, kidney, NF-кB, proximal tubule
the important role of the renin-angiotensin system (RAS), especially its major effector peptide, angiotensin II (ANG II), in the development and maintenance of hypertension has been well established in all ANG II-dependent hypertension models, including two-kidney, one-clip (2K1C) (5, 30, 46), Ren-2 gene transgenic (40, 42, 59), and ANG II-infused animals (7, 56, 61, 63). One important and consistent feature of these ANG II-dependent hypertension models is that ANG II levels in the kidneys are increased to a greater extent than can be explained on the basis of elevated plasma ANG II which suppresses renin expression in the juxtaglomerular apparatus (JGA) of the kidney (12, 29, 44, 61). The mechanisms underlying this phenomenon remain incompletely understood. Currently, there are at least two potential mechanisms contributing to augmentation of ANG II levels in the kidney: AT1 receptor-mediated angiotensinogen (AGT) expression and uptake of circulating ANG II by the proximal tubules. We and others have shown that circulating ANG II or systemically infused ANG II is taken up and accumulated in the proximal tubule of the kidney, including endosomal, mitochondrial, and nuclear compartments (1, 15, 29, 47, 61). The uptake of circulating ANG II by the proximal tubule of the kidney appears to be mediated primarily by AT1 (AT1a) receptors (29, 32, 55, 61, 64), but also partially by the multiligand endocytic receptor megalin (10, 37)- and caveolin 1-dependent mechanisms (26). Our studies further suggest that internalized (or intracellular) ANG II may act on cytoplasmic and nuclear AT1 receptors to induce long-term transcriptional effects on the RAS (33, 34). Second, there is increasing evidence suggesting that ANG II may be generated intrarenally, especially in the proximal tubules and collecting ducts during ANG II-induced hypertension (11, 16, 22, 44). Indeed, we and others have shown that chronic infusion of ANG II is associated with marked increases in angiotensinogen (AGT) expression in the renal cortex and urinary AGT excretion, which is also AT1 receptor dependent (11, 16, 22, 44). The upregulation of AGT, along with angiotensin-converting enzyme (ACE) (14) and AT1 receptor expression in the proximal tubules by ANG II (14, 59), may therefore serve as a powerful feed-forward mechanism to promote local ANG II production and actions (44).
While there is a general consensus on AT1 (AT1a) receptor-mediated uptake of circulating and/or extracellular ANG II by the proximal tubule of the kidney, the major source of augmented AGT mRNA and protein expression in the kidneys by ANG II remains uncertain. Recently, Matsusaka et al. (39) reported that mice with kidney-specific knockout of AGT maintained similar levels of AGT and ANG II proteins in the kidneys, compared with wild-type control mice. These authors further showed that liver-specific AGT knockout (AGT-KO) in mice almost completely abolished plasma and kidney AGT and ANG II proteins (4). The findings of this study suggest that liver-derived AGT is the primary source of AGT and ANG II proteins in the kidneys under normal conditions. In contrast, previous studies have demonstrated that 1) AGT levels in the proximal tubule fluid are greater than in plasma (43); 2) proximal tubules secrete ANG II and/or AGT (3); and 3) there is augmented AGT mRNA and protein expression in ANG II-induced hypertension in rats and mice (11, 16, 17). Although it is possible that some liver-derived AGT may be filtered through the glomeruli to enter the proximal tubules, where AGT may be taken up by the multiligand endocytic receptor megalin (48), our studies also support an important role for augmented proximal tubule AGT expression, contributing to higher levels of intrarenal ANG II in ANG II-dependent hypertension.
In the present study, we tested the hypothesis that tissue-specific overexpression of a cyan fluorescent protein-tagged intracellular ANG II fusion protein, ECFP/ANG II, directly stimulates AGT mRNA and protein expression in the proximal tubule cells, independently of liver-derived AGT in rats and mice. We chose to overexpress the intracellular ANG II fusion protein selectively in the proximal tubule instead of systemic infusion of ANG II to exclude the direct stimulatory effect of circulating ANG II on AGT biosynthesis and release in the liver. Since overexpression of ECFP/ANG II selectively in the proximal tubules did not increase plasma AGT, renin activity (PRA), and ANG II levels, our approach provides evidence that the expression of AGT mRNA and protein in the proximal tubules is augmented by intracellular ANG II independently of liver-derived AGT and circulating ANG II and independently of cell surface AT1 receptor activation.
METHODS
Animals.
Forty-eight adult male Sprague-Dawley rats (SD; ∼250 g body wt), purchased from Charles River, 24 adult male C57BL/6J mice, purchased from Jackson Laboratories, and 24 adult male AT1a-KO mice (∼25 g body wt), bred in our laboratory, were used in the present study. Upon arrival, all rats were maintained on a normal rodent chow and had free access to tap water. Furthermore, all animals were trained with the tail-cuff measurement of blood pressure daily for 1 wk before basal systolic blood pressure (SBP) and 24-h urine and urinary sodium excretion were determined. All animal surgical and experimental protocols were approved by the Institutional Animal Care and Use Committees of the University of Mississippi Medical Center and Tulane University School of Medicine.
Overexpression of ECFP-tagged intracellular ANG II fusion protein, ECFP/ANG II, or its scrambled control, ECFP/ANG IIc, selectively in proximal tubules.
Construction of adenovirus-mediated sglt2 promoter-directed ECFP/ANG II or ECFP/ANG IIc constructs and adenovirus-mediated ECFP/ANG II or ECFP/ANG IIc expression selectively in the proximal tubules of the kidney have been described previously (25, 28, 36). The constructs of ECFP/ANG II and ECFP/ANG IIc were provided by Dr. Julia Cook of the Ochsner Clinic Foundation (6), whereas the sglt2 promoter was provided by Dr. I. Rubera of the University of Nice-Sophia Antipolis (France), respectively (49). SD rats and C57BL/6J and AT1a-KO mice were divided into three groups with 8 animals in each group: control, Ad-sglt2-ECFP/ANG II, or Ad-sglt2-ECFP/ANG IIc (a scrambled control of ECFP/ANG II). Briefly, the rats or mice were anesthetized with pentobarbital sodium (50 mg/kg ip), and both kidneys were exposed via left and right flank incisions, respectively. The left renal artery was first temporarily clamped with a fine vessel clip to briefly stop blood flow to the left kidney for ∼5 min to transfer the target gene. Ad-sglt2-ECFP/ANG II (1.1 × 1011 pfu/ml) or its scrambled control, Ad-sglt2-ECFP/ANG IIc (5.5 × 1010 pfu/ml), was diluted 1:5 in phosphate-buffered saline and directly injected into the superficial cortex evenly at six to eight different locations (20 μl/injection) (25, 28, 36). The injection procedure took ∼1 min/kidney. Blood flow to the kidney was restored ∼5 min after injection of Ad-sglt2-ECFP/ANG II or Ad-sglt2-ECFP/ANG IIc. With this brief duration of blood flow interruption, no renal injury was observed while effective infection of the proximal tubules with the transgene was ensured (25, 28, 36). The same procedure for intrarenal adenovirus-mediated ECFP/ANG II was then repeated to infect the proximal tubules of the contralateral right kidney.
Measurement of systolic blood pressure and 24-h urinary excretion of water and electrolytes.
Systolic blood pressure responses in rats and mice were measured weekly for 4 wk using the tail-cuff Visitech System (Visitech, Apex, NC) as described previously (25, 36, 61). Basal and weekly 24-h urine and urinary sodium and potassium excretion were measured using a metabolic cage and NOVA 13 electrolyte analyzer (Nova Biomedical) (25, 36, 61).
Measurement of AGT mRNA expression in the renal cortex by semiquantitative RT-PCR.
AGT mRNA expression in the renal cortex, primarily in the proximal tubule of the kidney, was determined as described by Kobori et al. (17, 19). Briefly, total RNA was extracted from the renal cortex using a RNeasy minikit (Qiagen, Chatsworth, CA), and the integrity and quality of the purified RNA were verified by the presence of the 28S and 18S rRNA bands after agarose gel electrophoresis. The extracted and purified RNA from each renal cortical sample was then reverse-transcribed using the SuperScript preamplification system for first-strand cDNA synthesis (Life Technologies BRL, Gaithersburg, MD) (17, 19). The AGT primers used in the present study included: sense, 5′-TTGTTGAGAGCTTGGGTCCCTTCA-3′ (exon 2, bases +638 to +661) and antisense, 5′-CAGACACTGAGGTGCTGTTGTCCA-3′ (exon 3, bases +901 to +878). For control, the GAPDH primers included sense, 5′-TCCCTCAAGATTGTCAGCAA-3′ (bases +421 to +440) and antisense, 5′-AGATCCACAACGGATACATT-3′ (bases +728 to +709). Quantitative RT-PCR was then performed with each RT-PCR reaction containing 0.25–4 μl of the RT mixture, 2.5 μl of 10× buffer, 37.5 nmol of MgCl2, 5 nmol of each dNTP, 5 pmol of each sense and antisense primer for AGT or GAPDH, and 0.625 U of Taq DNA polymerase (Life Technologies BRL), in a final volume of 25 μl, respectively (17, 19). PCR was carried out in a DNA thermal cycler 480 (PerkinElmer, Norwalk, CT) for 15–40 cycles, with a 45-s denaturation at 94°C, a 30-s annealing at 52°C, and a 90-s extension at 72°C, with a final 10-min extension at 72°C (17, 19). AGT mRNA expression was normalized as the AGT mRNA-to-GAPDH mRNA ratio for comparisons.
Measurement of AGT protein expression in the renal cortex by Western blot analysis.
AGT protein expression in the renal cortex was determined by Western blot analysis as previously described (17, 19). Briefly, proteins samples from the cortex were extracted from each kidney in the buffer containing a protease inhibitor cocktail (100 μg/ml PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 2 mM EDTA, final concentrations in lysis buffer) and quantified accordingly. Protein samples were then electrophoretically separated on 3–8% Tris-glycine stacking gels at 125 V for 2.5 h and transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA) for 90 min at 25 V (XCell II Mini-Cell; Novex, San Diego, CA). The membranes were incubated with the primary AGT antibody for 3 h, washed, incubated with horseradish peroxidase-conjugated secondary antibody (donkey anti-sheep IgG, 1:30,000; Sigma) for 1 h, and washed again. This was followed by detection using enhanced chemiluminescence Western blotting (ECL system; Amersham Pharmacia Biotech) and exposed to X-ray film (Hyperfilm-ECL; Amersham Pharmacia Biotech). The integrated densitometric values (IDV) were normalized using the average values for the control group as previously described (17, 19).
Measurement of plasma AGT concentration and 24-h urinary AGT excretion.
Plasma AGT levels and 24-h urinary AGT excretion were measured by a specific AGT ELISA as previously described (18, 20). Briefly, highly purified recombinant rodent AGT protein was used as the standard. One hundred microliters per well of rodent AGT (0.08–5.0 ng/ml for rat AGT diluted in ELISA buffer), plasma (1:2,500 diluted in ELISA buffer), and urine samples (1:25 diluted in ELISA buffer) were added into each well of the plates and incubated at 37°C for 1 h. After the incubation, the plates were washed with a washing buffer (PBS containing Tween 20, 0.05%, pH 7.5). This was followed by incubation with the 100 μl/well of the horseradish peroxidase-labeled C-terminal antibody (1:30 diluted in antibody solution) at 37°C for 30 min. Finally, the plates were washed with the washing buffer, and further incubated with the 100 μl/well of 3,3′,5,5′-tetramethylbenzidine solution under light-protected conditions at room temperature for 30 min. The reactions were stopped with the 100 μl/well of sulfuric acid (0.5 mol/l), and the absorbance were measured at 450 nm (18).
Measurements of plasma and renal cortical ANG II levels.
Plasma and kidney cortical samples were collected at the end of the experiment to measure plasma and proximal tubule ANG II levels as described previously using a sensitive ELISA kit (S-1133; Bachem) (25, 36). ANG II was extracted from plasma and kidney samples using a phenyl-bonded solid-phase peptide extraction column (Elut-C18, Varian), vacuum dried overnight, and reconstituted in an ANG II assay buffer.
Imaging of overexpression of ECFP/ANG II selectively in proximal tubules of rat and mouse kidneys.
To visualize whether ECFP/ANG II or its control ECFP/ANG IIc was similarly overexpressed selectively in the proximal tubules of the rat and mouse kidneys, fresh sections of the kidneys (6 μm thick) from control and experimental animals were cut on a cryostat and thaw-mounted on glass slides. Sections were briefly counterstained with the cell nuclear marker 4,6-diamidino-2-phenylindole (DAPI; 300 nM, Molecular Probes) for 5 min to stain the nuclei, or with the high-affinity F-actin probe Alexa Fluor 568 phalloidin to stain apical and basolateral membranes of the renal tubules, as well as the glomeruli with red-orange fluorescence (A12380, 1:40 from the stock solution, Molecular Probes). Sections were washed with phosphate-buffered saline and mounted on a microscopic stage. A Nikon-Eclipse TE2000-U inverted fluorescence microscope and a dual DAPI-CFP band-pass excitation filter set (excitation: 440 nm; emission: 495/50 nm) or a filter for Alexa Fluor 568 phalloidin (excitation: 578 nm; emission: 600 nm) were used for imaging as we described previously (25, 35, 36).
Overexpression of intracellular ECFP/ANG II fusion proteins in mouse proximal tubule cells.
To determine the potential signaling mechanisms of intracellular ECFP/ANG II-induced augmentation of AGT expression in the kidneys, immortalized mouse proximal tubule cells (mPCTs) derived from the S1 segment of the proximal tubule of male C57BL/6J mice were used in in vitro cell culture studies (57). mPCTs were a generous gift of Dr. Ulrich Hopfer of Case Western Reserve University. Briefly, mPCT cells were subcultured to 80% confluence in six-well plates, as appropriate, in the complete DMEM/F-12 growth medium at 37°C, supplied with 95% O2-5% CO2, 50 nM hydrocortisone, 5% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin as we described previously (27, 31, 35, 57). mPCTs were then transfected with the plasmid ECFP/ANG II (4 μg/well) for 48 h using the transfection protocol as we described previously (31, 35, 60, 62) and concurrently treated with or without the AT1 receptor blocker losartan (10 μM), the AT2 receptor blocker PD123319 (10 μM), the MEK1/MEK2 kinase inhibitor U0126 (1 μM), or the NF-κB inhibitor RO 106–9920 (1 μM), respectively (28). Protein and/or RNA samples were extracted from control and treated mPCTs, and the changes in NF-κB, p65, and AGT proteins were measured by Western blotting using a mouse monoclonal primary antibody recognizing an epitope overlapping the nuclear localization signal (NLS) of the p65 subunit of the NF-κB heterodimer (MAB3026, 1:500 dilution, Millipore) (24, 28) or a rabbit monoclonal primary anti-AGT antibody (MABC123, 1:1,000, Millipore), respectively.
Statistical analysis.
All results are presented as means ± SE. One-way ANOVA was first to compare the differences in the same parameters between groups of rats. If the P value was <0.05, a post hoc Newman-Keuls multiple comparison test or Student's unpaired t-test was performed to compare two different group means. The significance was set at P < 0.05.
RESULTS
Overexpression of ECFP/ANG II selectively in the proximal tubule of the kidney.
Adenovirus-mediated sglt2 promoter-driven overexpression of intracellular ECFP/ANG II or its scrambled control, ECFP/ANG IIc, selectively in the proximal tubule of the rat kidney is shown in Fig. 1. We previously reported in detail that the overexpression of ECFP/ANG II peaked at 7–14 days but persisted 4 wk after intrarenal gene transfer (25). The present study focused on the relatively long-term expression and its effect on AGT expression in the kidney and blood pressure 4 wk after the overexpression was initiated. In control rats, there were minimal levels of CFP visualized in the cortex (Fig. 1A) and medulla (Fig. 1D), respectively. However, high levels of ECFP/ANG II expression were observed in nearly all proximal tubules in the superficial cortex of the kidney, with blue showing the nuclei (Fig. 1B). Both apical and basolateral membranes of the proximal tubules, as well as the glomeruli, were imaged in red-orange fluorescence using the high-affinity F-actin probe Alexa Fluor 568 phalloidin. Very low levels of ECFP/ANG II expression were observed in the glomeruli and cortical collecting ducts of the cortex (Fig. 1B). Furthermore, no ECFP/ANG II expression was detected in distal tubules and collecting ducts of the renal medulla (Fig. 1E). In the rat kidneys transferred with the scrambled Ad-sglt2-ECFP/ANG IIc, similar levels of CFP expression were visualized in the proximal tubules and glomeruli in the cortex (Fig. 1C), but not in the medulla (Fig. 1F).
Fig. 1.

Expression of intracellular cyan fluorescent fusion of ANG II, cyan fluorescent fusion protein (ECFP)/ANG II, or its scrambled control, ECFP/ANG IIc, in the rat kidney 4 wk after adenovirus-mediated, sglt2-driven, ECFP/ANG II transfer in the superficial cortex. A (cortex) and D (medulla): representative control rat kidney showing only basal levels of autofluorescence in the proximal tubules of the cortex and in the renal medulla. Apical and basolateral membranes of the renal tubules as well as the glomeruli were stained by the high-affinity F-actin probe Alexa Fluor 568 phalloidin, in red-orange fluorescence. B (cortex) and E (medulla): expression of the specific ECFP/ANG II in the cytoplasm and perinuclear region of proximal tubules of a representative rat kidney, shown as blue-green, with nuclei stained by 4,6-diamidino-2-phenylindole (DAPI) in blue, and apical and basolateral membranes as well as the glomeruli in red-orange fluorescence. C (cortex) and F (medulla): expression of the scrambled ECFP/ANG IIc fusion protein in the cytoplasm and perinuclear region of proximal tubules of a representative rat kidney. Note the lack of ECFP/ANG II expression in the renal medulla of the kidney 4 wk after ECFP/ANG II transfer in the cortex of the same kidney. G, glomerulus; PT, proximal tubules; CD, collecting ducts. Bar = 20 μm.
Effects of proximal tubule-selective overexpression of ECFP/ANG II on blood pressure and urinary sodium excretion.
The time course of blood pressure and urinary electrolyte excretory responses to the proximal tubule-selective overexpression of ECFP/ANG II in the rat kidney was reported previously (25). At 4 wk after ECFP/ANG II overexpression in the proximal tubules (Fig. 2A), a moderate and significant increase in systolic blood pressure persisted in rats receiving the transfer of ECFP/ANG II (140 ± 5 mmHg), but not the scrambled control ECFP/ANG IIc (122 ± 6 mmHg, P < 0.01). No difference in basal blood pressure was found among the three groups (Fig. 2). The blood pressure response was associated with a moderate, but significant, decrease in 24-h urinary sodium excretion in rats receiving the transfer of ECFP/ANG II (1.82 ± 0.1 mmol/24 h vs. basal: 2.33 ± 0.13 mmol/24 h, P < 0.01), but not the scrambled control ECFP/ANG IIc [2.16 ± 0.06 mmol/24 h vs. basal: 2.10 ± 0.12 mmol/24 h, not significant (n.s.)] (Fig. 2B).
Fig. 2.

Effects of expression of intracellular ECFP/ANG II or its scrambled ECFP/ANG IIc selectively in the proximal tubules of the rat kidneys for 4 wk on systolic blood pressure (A) and 24-h urinary sodium excretion (UNaV; B). **P < 0.01 vs. basal in the same group of animals. ++P < 0.01 vs. the time-control group at week 4.
Effect of proximal tubule-selective overexpression of ECFP/ANG II on plasma AGT, PRA, and ANG II levels.
Unlike systemic infusion of ANG II which markedly increases circulating and tissue ANG II levels and directly stimulates AGT biosynthesis and release from the liver (45, 48), adenovirus-mediated overexpression of ECFP/ANG II selectively in the proximal tubules in rats had no significant effects on plasma AGT (Fig. 3A), PRA, or ANG II levels (Fig. 3B).
Fig. 3.

Effects of expression of intracellular ECFP/ANG II selectively in the proximal tubules of the rat kidneys for 4 wk on plasma angiotensinogen (AGT; A), renin activity (PRA; B), and ANG II levels (C). No significant difference (n.s.) was detected between the time control and ECFP/ANG II-overexpressing groups.
Effect of proximal tubule-selective overexpression of ECFP/ANG II on AGT mRNA and protein expression and ANG II levels in the renal cortex.
Figure 4 shows that the expression of AGT mRNA in the renal cortex was significantly increased by >1.5-fold in response to overexpression of ECFP/ANG II selectively in the proximal tubules (Fig. 4, left, P < 0.05). Similarly, there were corresponding increases in AGT protein expression, as estimated by Western blot analysis (Fig. 4, P < 0.01) and ANG II levels in the renal cortex (Fig. 5, P < 0.01), respectively. By comparison, overexpression of the control ECFP/ANG IIc selectively in the proximal tubules of the mouse kidney had no effects on AGT mRNA and protein expression (see Fig. 8), as well as on ANG II levels in the renal cortex (Fig. 5).
Fig. 4.

Effects of expression of intracellular ECFP/ANG II selectively in the proximal tubules of the rat kidneys for 4 wk on AGT mRNA (A) and protein expression (B) in the renal cortex. *P < 0.05 vs. the time control group.
Fig. 5.

Effects of expression of intracellular ECFP/ANG II selectively in the proximal tubules of the rat kidneys for 4 wk on ANG II levels in the renal cortex 4 wk after ECFP/ANG II transfer in the cortex. **P < 0.01 vs. the time control group.
Fig. 8.
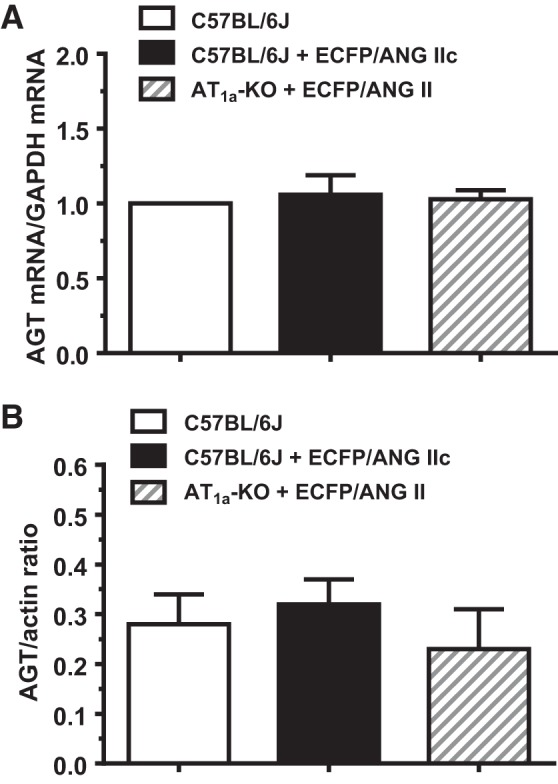
Effects of overexpression of intracellular ECFP/ANG II selectively in the proximal tubules of AT1a-KO mice or scrambled control ECFP/ANG IIc selectively in the proximal tubules of c57BL/6J mice for 4 wk on AGT mRNA (A) and protein expression (B) in the renal cortex. No differences in AGT mRNA and AGT protein levels were detected in control C57BL/6J mice, C57BL/6J mice expressing the control ECFP/ANG IIc fusion protein, and AT1a-KO mice with the expression of specific ECFP/ANG II; n = 6–8/group.
Effect of proximal tubule-selective overexpression of ECFP/ANG II on urinary protein and AGT excretion.
To determine whether AGT was secreted into the proximal tubule luminal fluid and then excreted in urine, 24-h urinary protein and AGT excretion were compared in rats receiving proximal tubule-specific overexpression of ECFP/ANG II or vehicle control (Fig. 6A). While there was no significant difference in 24-h urinary protein excretion between groups, 24-h urinary AGT excretion was significantly increased in rats overexpressing ECFP/ANG II in the proximal tubules 4 wk after the gene transfer (basal: 58.4 ± 6.3 ng/day vs. ECFP/ANG II: 123.6 ± 20.9 ng/day, P < 0.01) (Fig. 6B). Interestingly, 24-h urinary AGT excretion was also increased moderately in control rats (P < 0.05). However, the increase in 24-h urinary AGT excretion in control rats was significantly smaller than in rats overexpressing the intracellular ECFP/ANG II selectively in the proximal tubules.
Fig. 6.

Effects of expression of intracellular ECFP/ANG II selectively in the proximal tubules of the rat kidneys on urinary protein (A) and AGT excretions (B) 4 wk after ECFP/ANG II transfer in the cortex. No difference in 24-h urinary protein excretion was detected between the time control (open bars) and ECFP/ANG II-expressing groups (filled bars). However, 24-h urinary AGT excretion was significantly increased in the ECFP/ANG II-expressing group compared with their basal levels (*P < 0.05) and the time control group (+P < 0.05).
Role of AT1a receptors in mediating the effects of proximal tubule-selective overexpression of ECFP/ANG II on blood pressure and AGT expression.
In a previous study, we reported that the AT1 receptor blocker losartan largely blocked the effects of proximal tubule-selective overexpression of ECFP/ANG II on blood pressure responses in rats and mice (25). To determine whether AT1a receptors are involved in mediating the effect of ECFP/ANG II overexpression on AGT mRNA and protein expression, additional experiments were performed in AT1a receptor-KO mice. Figure 7 shows that compared with a representative control mouse (Fig. 7A), ECFP/ANG II was expressed similarly in the proximal tubules, but not the glomeruli, of the renal cortex in a male representative C57BL/6J mouse (Fig. 7B), as well as in a male representative AT1a-KO mouse (Fig. 7C). Compared with control C57BL/6J mice, overexpression of ECFP/ANG II selectively in the proximal tubules was associated with a significant increase in systolic blood pressure (Fig. 7D) and a significant decrease in 24-h urinary sodium excretion (Fig. 7E) in C57BL/6J, but not in AT1a-KO mice. Twenty-four-hour urinary potassium excretion was not significantly altered by proximal tubule-selective overexpression of ECFP/ANG II. Overexpression of ECFP/ANG II in the proximal tubules had no significant effects on AGT mRNA expression and AGT proteins in the renal cortex of AT1a-KO mice compared with control C57BL/6J mice (Fig. 8).
Fig. 7.

Overexpression of intracellular ECFP/ANG II selectively in the proximal tubule of the kidney in C57BL/6J or AT1a-knockout (KO) mice and its effects on systolic blood pressure and 24-h UNaV and potassium excretion (UKV). A: representative control C57BL/6J kidney showing a low level of autofluorescence in the cortex. B: representative control C57BL/6J kidney showing overexpression of ECFP/ANG II selectively in the proximal tubule of the superficial cortex in blue green. C: representative AT1a-KO mouse kidney also showing overexpression of ECFP/ANG II selectively in the proximal tubule. Red represents DAPI-stained nuclei in the cortex after conversion from blue. D: effect on systolic blood pressure (SBP). E: effect on UNaV. F: effect on UKV. **P < 0.01 vs. control, whereas ++P < 0.01 vs. ECFP/ANG II overexpression; n = 6–8/group.
Roles of MAP kinases and NF-кB signaling mechanisms.
To elucidate the signaling mechanism involved, mPCTs were transfected with ECFP/ANG II and treated with ANG receptor blockers, MAP kinase inhibitor U0126, or NF-кB inhibitor RO 106–9920. Overexpression of ECFP/ANG II in mPCTs increased NF-кB, p65 proteins by >3-fold (P < 0.01), and the response was significantly attenuated by the AT1 receptor blocker losartan (10 μM) and the MAP kinase MER1 and MEK2 inhibitor U0126 (1 μM, P < 0.01), but not by the AT2 receptor blocker PD123319 (10 μM, n.s.) (Fig. 9A). The overexpression of ECFP/ANG II also significantly increased AGT protein (P < 0.01), which was significantly attenuated by the MAP kinase MER1 and MEK2 inhibitor U0126 and the NF-кB inhibitor R0 106–9920 (P < 0.01), respectively (Fig. 9B).
Fig. 9.

Effects of overexpression of intracellular ECFP/ANG II in cultured mouse proximal tubule cells (mPTCs) on NF-кB (A) or AGT protein expression (B). **P < 0.01 vs. control mPCTs. ++P < 0.01 vs. mPCTs expressing ECFP/ANG II (n = 4–6 wells each).
DISCUSSION
The role of circulating, endocrine, or extracellular ANG II in the regulation of AGT expression in the kidney has been extensively investigated previously in ANG II-induced hypertensive rats and mice (11, 16, 17, 21, 22). In those studies, chronic infusion of pressor doses of exogenous ANG II for weeks was found to induce severe hypertension and kidney injury, associated with significant augmentation of renal AGT expression and urinary AGT excretion (11, 16, 17, 21, 22). These studies indicate that unlike renin expression in the JGAs of the cortex, AGT mRNA expression and AGT protein production in the kidney are directly stimulated by ANG II, which provides a powerful and sustained feed-forward mechanism to further increase intrarenal ANG II formation and therefore lead to persistent hypertension. Indeed, intrarenal ANG II levels are significantly higher than can be explained by circulating AGT and ANG II under physiological conditions as well as during ANG II-induced hypertension (44, 53, 56, 61, 63). However, the source of intrarenal AGT in physiology and renal diseases such as in ANG II-dependent hypertension remains an issue of current debate. Recently, the study of Matsusaka et al. (39) has suggested that liver-derived AGT is the primary source of intrarenal AGT and ANG II and taken up via the megalin-dependent mechanism in normal mice, but they did not address the responses in hypertensive mice. Similarly, Pohl et al. (48) reported that immunoreactive AGT that was detected in the early proximal tubule was primarily derived from the uptake of filtered liver AGT, whereas AGT mRNA expression was detected in the proximal straight tubule. Nevertheless, the latter two studies may not be adequate to determine whether liver-derived or kidney-derived AGT contributes to augmented kidney or intratubular ANG II levels in ANG II-dependent hypertension or kidney diseases (45).
The present study may provide some new insights into the potential role of kidney derived AGT and intracellular ANG II to the current debate. One important difference in the experimental approach of the present study was that pressor doses of exogenous ANG II were not infused to induced severe hypertension, as we previously reported (11, 17, 21). Systemic administration of ANG II is well recognized to stimulate AGT biosynthesis and release from the liver (45, 48). Increased liver-derived AGT by circulating ANG II is expected to be delivered into the kidney, making it difficult to separate the role of kidney AGT from that of liver-derived AGT. Instead, we used a proximal tubule-specific sglt2 promoter to overexpress an intracellular ECFP-tagged ANG II fusion protein selectively in the proximal tubules of the kidney in rats and mice (Figs. 1 and Fig. 7) (25, 28, 36). With this unique approach, the expression of ECFP/ANG II is confined within the infected cells and increases intracellular ANG II levels in the proximal tubules, but does not release or secrete ANG II into the peritubular interstitium or microcirculation to increase plasma ANG II, or into the proximal tubular lumen to act on AT1 receptors or tubular cells (25). Indeed, plasma AGT, PRA, and ANG II were not significantly elevated in rats with overexpression of intracellular ECFP/ANG II selectively in the proximal tubules in the present study (Fig. 3); therefore, the contribution of systemically ANG II-stimulated, liver-derived AGT biosynthesis and secretion may most likely be excluded. The present study demonstrates that overexpression of intracellular ECFP/ANG II selectively in the proximal tubules of rats and mice significantly increased AGT mRNA expression and AGT proteins in the renal cortex (Fig. 4), ANG II levels in the cortex (Fig. 5), as well as augmented urinary AGT excretion (Fig. 6). Our results may be best interpreted as a direct effect of intracellular ANG II, independently of the circulating RAS, to enhance proximal tubule-specific AGT formation. As reported previously (25, 28, 36), we again found that ECFP/ANG II was expressed selectively in the proximal tubules of the renal cortex, with very low levels of ECFP/ANG II expressed in the glomeruli, the cortical collecting tubules, or the entire medulla (Figs. 1 and Fig. 7). Furthermore, we expressed the Ad-sglt2-ECFP/ANG IIc, a scrambled ECFP fusion protein, in different groups of rats or mice as a vehicle control to determine whether the effect of ECFP/ANG II is specific. Indeed, overexpression of the Ad-sglt2-ECFP lacking ANG II fusion protein in the proximal tubules had no effects on blood pressure, urinary sodium excretion in rats (Fig. 2), or AGT mRNA and AGT protein in the renal cortex of mice (Fig. 8). Thus our approaches effectively exclude the potential nonspecific, or off-target, effects of intracellular ECFP on blood pressure and intrarenal AGT expression in the present study.
We and others have previously shown that ANG II stimulates AGT mRNA and AGT protein expression in the kidney or cultured rat or human proximal tubule cells via AT1 receptors (22, 51). However, these effects were induced by extracellular ANG II acting via cell surface AT1 receptors. No study has been reported for a role of intracellular ANG II on AGT expression in cultured proximal tubule cells or the proximal tubules of the rat and mouse kidney. One exception is a recent study in which overexpression of this intracellular ECFP/ANG II did not alter AGT accumulation in major tissues, such as kidney, liver, and brain (54). However, this study was different from the current study in a critical way. The overexpression of ECFP/ANG II in that study was driven by the mouse metallothionein promoter in all tissues, whereas in the present study the proximal tubule-specific sglt2 promoter was used to drive the expression of this intracellular protein selectively in the proximal tubules of the kidneys. The results of the present study suggest that intracellular ANG II significantly augments AGT mRNA expression and AGT protein production in the proximal tubules of the cortex, and the effects are also mediated by AT1a receptors (Figs. 7–9). This conclusion is supported by experiments using mutant mice with a deficiency of AT1a receptors, in which the effects of ECFP/ANG II on AGT mRNA and AGT protein expression in the renal cortex, blood pressure, and 24-h urinary sodium excretion were largely blocked (Figs. 8 and 9). This AT1a receptor-dependent mechanism is consistent with previous studies showing that the effects of exogenous ANG II to augment AGT mRNA and protein expression in rats or mice or in cultured proximal tubule cells were blocked by AT1 receptor blockers (22, 51). Whether AT1b receptors, which are also expressed in rodents (8), play a role in ECFP/ANG II-induced AGT mRNA or AGT protein expression in the kidney was not addressed in this study. The present study was not designed to determine the role of the AT1b receptor due to the lack of AT1b receptor-specific inhibitors or AT1b-KO animal models. However, the responses were completely prevented in the AT1a-KO mice probably due to the fact that AT1a receptors account for >90% of the total AT1 receptor abundance (8). Thus the role of the AT1b receptor may be minor and difficult to demonstrate in the presence of intact AT1a receptors.
The signaling mechanisms underlying the effect of ECFP/ANG II on intrarenal AGT mRNA and AGT protein expression in the proximal tubules have not been determined previously. We and other groups have previously shown that extracellular ANG II stimulated AGT mRNA and AGT protein expression in proximal tubule cells via the IL-6 (51), NF-кB (2, 41), IFN-γ/JAK-STAT/SOCS1 (52), or ROCK/NF-кB signaling pathways (41). The results of the present study showed that the AT1 blocker losartan, the MAP kinases MEK1 and MEK2 inhibitor U0126, and the NF-кB inhibitor RO 106–9920, but not the AT2 blocker PD123319, significantly attenuated the effects of ECFP/ANG II on AGT expression in cultured mPCTs (Fig. 9). This suggests that intracellular ANG II may stimulate cytoplasmic or nuclear AT1 (AT1a) receptors to activate the MAP kinase pathways, which alone and/or in turn activate NF-кB signaling to augment AGT expression in the proximal tubules (Figs. 9 and Fig. 10). This explanation is further supported by the reports that AT1 (AT1a) receptors are localized in the endosomal (15, 61), mitochondrial (1), and nuclear compartments (13, 33, 38, 47). However, it should be pointed out that, not confined to AGT expression, the AT1a/MAP kinase signaling pathways also mediate the effects of ECFP/ANG II on transcriptional responses of transforming growth factor-β, MCP-1, and NHE3 expression (28, 33).
Fig. 10.

Schematic summary showing that expression of an intracellular ANG II fusion protein selectively in the proximal tubules of the kidney stimulates AT1 (AT1a) receptors in the cytoplasm and nuclei, leading activation of MAP kinases and NF-кB signaling and subsequent AGT mRNA and protein expression. Some of increased AGT proteins may be secreted from the proximal tubules into the tubular lumen and appear in urine.
Although the present study clearly shows that overexpression of an intracellular ANG II fusion protein selectively in the proximal tubule of the rat and mouse kidney significantly augmented AGT mRNA and AGT protein expression, a number of limitations should be recognized. First, the present study did not systematically determine the time- and dose-dependent effects of ECFP/ANG II on AGT expression in the kidney. Second, the present results do not allow us to delineate the separate roles of cell surface AT1 receptors vs. that of cytoplasmic or nuclear AT1 receptors. Third, although we found that the ANG II levels were significantly increased in the cortex in response to overexpression of ECFP/ANG II selectively in the proximal tubules, it remains unknown whether this was due to the persistent overexpression of ECFP/ANG II or to local ANG II production as a result of augmented AGT expression by ECFP/ANG II. Finally, since the present study was not designed to directly determine the functional significance of the effect of intracellular ANG II on AGT expression in the proximal tubules, it is unlikely that increased AGT mRNA and protein expression in the renal cortex alone contribute to the moderate antinatriuretic response and elevated blood pressure in rats and mice overexpressing ECFP/ANG II selectively in the proximal tubules (Figs. 2 and 7). It is likely, however, that the effect of ECFP/ANG II on AGT expression partly contributes to increased intracellular ANG II levels in the proximal tubules, which in turn stimulate intracellular/nuclear AT1 (AT1a) receptors to activate major downstream signaling cascades to induce long-term transcriptional responses. The combined stimulatory and transcriptional effects of ECFP/ANG II and/or local ANG II on the expression of various sodium transporters, solute transporters, and cytokines or proinflammatory factors ultimately play important roles. Consistent with this interpretation, overexpression of human AGT and renin genes selectively in the proximal tubules of mice driven by the kidney androgen-regulated protein promoter (KAP) resulted in significantly elevated blood pressure and renal injury (9, 23, 50, 58).
In summary, the present study demonstrates that overexpression of an intracellular ANG II fusion protein, but not its scrambled control, selectively in the proximal tubules of the rat and mouse kidney significantly augments AGT mRNA and AGT protein expression, as well as increases urinary AGT excretion. This effect is associated with a moderately antinatriuretic response and elevated blood pressure and appears to involve the AT1 (AT1a) receptors/MAP kinase/NF-кB signaling pathways. Our results suggest that the effect of ECFP/ANG II on AGT expression may at least in part contribute to increased intracellular ANG II levels in the proximal tubules, which in turn stimulate intracellular/nuclear AT1 (AT1a) receptors to activate major downstream signaling cascades to induce long-term transcriptional effects in the kidney.
GRANTS
This work was supported by grants from National Institute of Diabetes and Digestive and Kidney Diseases to J. Zhuo (2RO1DK067299-06A2, 1R01DK102429-01) and the National Institute of General Medical Sciences IDeA Program to L. G. Navar (COBRE, P30GM103337).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.L.Z., H.K., X.C.L., and L.G.N. provided conception and design of research; J.L.Z., H.K., X.C.L., R.S., and A.K. performed experiments; J.L.Z., H.K., X.C.L., R.S., A.K., and L.G.N. analyzed data; J.L.Z., H.K., X.C.L., and L.G.N. interpreted results of experiments; J.L.Z., H.K., X.C.L., R.S., and A.K. prepared figures; J.L.Z., H.K., X.C.L., and L.G.N. drafted manuscript; J.L.Z., H.K., X.C.L., and L.G.N. edited and revised manuscript; J.L.Z., H.K., X.C.L., R.S., A.K., and L.G.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Julia L. Cook of the Ochsner Health System for providing us with the ECFP/ANG II and ECFP/ANG IIc constructs for the present study and Dr. Ulrich Hopfer of Case Western Reserve University for providing immortalized mouse proximal tubule cells (mPCTs).
REFERENCES
- 1.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O'Rourke B, Walston JD. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci USA 108: 14849–14854, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acres OW, Satou R, Navar LG, Kobori H. Contribution of a nuclear factor-kappaB binding site to human angiotensinogen promoter activity in renal proximal tubular cells. Hypertension 57: 608–613, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braam B, Mitchell KD, Fox J, Navar LG. Proximal tubular secretion of angiotensin II in rats. Am J Physiol Renal Fluid Electrolyte Physiol 264: F891–F898, 1993. [DOI] [PubMed] [Google Scholar]
- 4.Brechue WF, Kinne-Saffran E, Kinne RK, Maren TH. Localization and activity of renal carbonic anhydrase (CA) in CA-II deficient mice. Biochim Biophys Acta 1066: 201–207, 1991. [DOI] [PubMed] [Google Scholar]
- 5.Cervenka L, Horacek V, Vaneckova I, Hubacek JA, Oliverio MI, Coffman TM, Navar LG. Essential role of AT1A receptor in the development of 2K1C hypertension. Hypertension 40: 735–741, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Cook JL, Re R, Alam J, Hart M, Zhang Z. Intracellular angiotensin II fusion protein alters AT1 receptor fusion protein distribution and activates CREB. J Mol Cell Cardiol 36: 75–90, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 103: 17985–17990, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52: 415–472, 2000. [PubMed] [Google Scholar]
- 9.Ding Y, Sigmund CD. Androgen-dependent regulation of human angiotensinogen expression in KAP-hAGT transgenic mice. Am J Physiol Renal Physiol 280: F54–F60, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Villalobos Klassen R, Allen RB, Navar LG, Hammond TG. Megalin binds and internalizes angiotensin II. Am J Physiol Renal Physiol 288: F420–F427, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Villalobos RA, Seth DM, Satou R, Horton H, Ohashi N, Miyata K, Katsurada A, Tran DV, Kobori H, Navar LG. Intrarenal angiotensin II and angiotensinogen augmentation in chronic angiotensin II-infused mice. Am J Physiol Renal Physiol 295: F772–F779, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan S, Fox J, Mitchell KD, Navar LG. Angiotensin and angiotensin converting enzyme tissue levels in two-kidney, one-clip hypertensive rats. Hypertension 20, 763–767. 1992. [DOI] [PubMed] [Google Scholar]
- 13.Gwathmey TM, Alzayadneh EM, Pendergrass KD, Chappell MC. Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am J Physiol Regul Integr Comp Physiol 302: R518–R530, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison-Bernard LM, Zhuo J, Kobori H, Ohishi M, Navar LG. Intrarenal AT1 receptor and ACE binding in ANG II-induced hypertensive rats. Am J Physiol Renal Physiol 282: F19–F25, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imig JD, Navar GL, Zou LX, O'Reilly KC, Allen PL, Kaysen JH, Hammond TG, Navar LG. Renal endosomes contain angiotensin peptides, converting enzyme, and AT1A receptors. Am J Physiol Renal Physiol 277: F303–F311, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Kobori H, Harrison-Bernard LM, Navar LG. Enhancement of angiotensinogen expression in angiotensin II-dependent hypertension. Hypertension 37: 1329–1335, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNA and protein in angiotensin II- dependent hypertension. J Am Soc Nephrol 12: 431–439, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobori H, Katsurada A, Miyata K, Ohashi N, Satou R, Saito T, Hagiwara Y, Miyashita K, Navar LG. Determination of plasma and urinary angiotensinogen levels in rodents by newly developed ELISA. Am J Physiol Renal Physiol 294: F1257–F1263, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobori H, Nishiyama A, Abe Y, Navar LG. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension 41: 592–597, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG. Urinary angiotensinogen as an indicator of intrarenal angiotensin status in hypertension. Hypertension 41: 42–49, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobori H, Ozawa Y, Satou R, Katsurada A, Miyata K, Ohashi N, Hase N, Suzaki Y, Sigmund CD, Navar LG. Kidney-specific enhancement of ANG II stimulates endogenous intrarenal angiotensinogen in gene-targeted mice. Am J Physiol Renal Physiol 293: F938–F945, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lara LS, McCormack M, Semprum-Prieto LC, Shenouda S, Majid DS, Kobori H, Navar LG, Prieto MC. AT1 receptor-mediated augmentation of angiotensinogen, oxidative stress, and inflammation in ANG II-salt hypertension. Am J Physiol Renal Physiol 302: F85–F94, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavoie JL, Lake-Bruse KD, Sigmund CD. Increased blood pressure in transgenic mice expressing both human renin and angiotensinogen in the renal proximal tubule. Am J Physiol Renal Physiol 286: F965–F971, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Brasier AR. Angiotensinogen gene activation by angiotensin II is mediated by the rel A (nuclear factor-kappaB p65) transcription factor: one mechanism for the renin angiotensin system positive feedback loop in hepatocytes. Mol Endocrinol 10: 252–264, 1996. [DOI] [PubMed] [Google Scholar]
- 25.Li XC, Cook JL, Rubera I, Tauc M, Zhang F, Zhuo JL. Intrarenal transfer of an intracellular cyan fluorescent fusion of angiotensin II selectively in proximal tubules increases blood pressure in rats and mice. Am J Physiol Renal Physiol 300: F1076–F1088, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li XC, Gu V, Miguel-Qin E, Zhuo JL. Role of caveolin 1 in AT1a receptor-mediated uptake of angiotensin II in the proximal tubule of the kidney. Am J Physiol Renal Physiol 307: F949–F961, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XC, Hopfer U, Zhuo JL. AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through the microtubule-dependent endocytic pathway. Am J Physiol Renal Physiol 297: F1342–F1352, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li XC, Hopfer U, Zhuo JL. Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am J Physiol Renal Physiol 303: F1617–F1628, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li XC, Navar LG, Shao Y, Zhuo JL. Genetic deletion of AT1a receptors attenuates intracellular accumulation of angiotensin II in the kidney of AT1a receptor-deficient mice. Am J Physiol Renal Physiol 293: F586–F593, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li XC, Widdop RE. Regional hemodynamic effects of the AT1 receptor antagonist CV-11974 in conscious renal hypertensive rats. Hypertension 26: 989–997, 1995. [DOI] [PubMed] [Google Scholar]
- 31.Li XC, Zhuo JL. Selective knockdown of AT1 receptors by RNA interference inhibits Val5-ANG II endocytosis and NHE-3 expression in immortalized rabbit proximal tubule cells. Am J Physiol Cell Physiol 293: C367–C378, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li XC, Zhuo JL. In vivo regulation of AT1a receptor-mediated intracellular uptake of [125I]-Val5-angiotensin II in the kidneys and adrenal glands of AT1a receptor-deficient mice. Am J Physiol Renal Physiol 294: F293–F302, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li XC, Zhuo JL. Intracellular ANG II directly induces in vitro transcription of TGF-β1, MCP-1, and NHE-3 mRNAs in isolated rat renal cortical nuclei via activation of nuclear AT1a receptors. Am J Physiol Cell Physiol 294: C1034–C1045, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li XC, Zhuo JL. Nuclear factor-kappaB as a hormonal intracellular signaling molecule: focus on angiotensin II-induced cardiovascular and renal injury. Curr Opin Nephrol Hypertens 17: 37–43, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li XC, Zhuo JL. Phosphoproteomic analysis of AT1 receptor-mediated signaling responses in proximal tubules of angiotensin II-induced hypertensive rats. Kidney Int 80: 620–632, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li XC, Zhuo JL. Proximal tubule-dominant transfer of AT1a receptors induces blood pressure responses to intracellular angiotensin II in AT1a receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol 304: R588–R598, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li XC, Zhuo JL. Mechanisms of AT1a receptor-mediated uptake of angiotensin II by proximal tubule cells: a novel role of the multiligand endocytic receptor megalin. Am J Physiol Renal Physiol 307: F222–F233, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Licea H, Walters MR, Navar LG. Renal nuclear angiotensin II receptors in normal and hypertensive rats. Acta Physiol Hung 89: 427–438, 2002. [DOI] [PubMed] [Google Scholar]
- 39.Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, Nishiyama A, Ichikawa I. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol 23: 1181–1189, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitchell KD, Jacinto SM, Mullins JJ. Proximal tubular fluid, kidney, and plasma levels of angiotensin II in hypertensive ren-2 transgenic rats. Am J Physiol Renal Physiol 273: F246–F253, 1997. [DOI] [PubMed] [Google Scholar]
- 41.Miyata K, Satou R, Shao W, Prieto MC, Urushihara M, Kobori H, Navar LG. ROCK/NF-κB axis-dependent augmentation of angiotensinogen by angiotensin II in primary-cultured preglomerular vascular smooth muscle cells. Am J Physiol Renal Physiol 306: F608–F618, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullins JJ, Peters J, Ganten D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature 344: 541–544, 1990. [DOI] [PubMed] [Google Scholar]
- 43.Navar LG, Imig JD, Zou L, Wang CT. Intrarenal production of angiotensin II. Semin Nephrol 17: 412–422, 1997. [PubMed] [Google Scholar]
- 44.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension 57: 355–362, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navar LG, Satou R, Gonzalez-Villalobos RA. The increasing complexity of the intratubular renin-angiotensin system. J Am Soc Nephrol 23: 1130–1132, 2012. [DOI] [PubMed] [Google Scholar]
- 46.Navar LG, Zou L, Von Thun A, Tarng WC, Imig JD, Mitchell KD. Unraveling the mystery of Goldblatt hypertension. News Physiol Sci 13: 170–176, 1998. [DOI] [PubMed] [Google Scholar]
- 47.Pendergrass KD, Averill DB, Ferrario CM, Diz DI, Chappell MC. Differential expression of nuclear AT1 receptors and angiotensin II within the kidney of the male congenic mRen2. Lewis rat. Am J Physiol Renal Physiol 290: F1497–F1506, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Pohl M, Kaminski H, Castrop H, Bader M, Himmerkus N, Bleich M, Bachmann S, Theilig F. Intrarenal renin angiotensin system revisited: role of megalin-dependent endocytosis along the proximal nephron. J Biol Chem 285: 41935–41946, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubera I, Poujeol C, Bertin G, Hasseine L, Counillon L, Poujeol P, Tauc M. Specific Cre/Lox recombination in the mouse proximal tubule. J Am Soc Nephrol 15: 2050–2056, 2004. [DOI] [PubMed] [Google Scholar]
- 50.Sachetelli S, Liu Q, Zhang SL, Liu F, Hsieh TJ, Brezniceanu ML, Guo DF, Filep JG, Ingelfinger JR, Sigmund CD, Hamet P, Chan JS. RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney Int 69: 1016–1023, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Satou R, Gonzalez-Villalobos RA, Miyata K, Ohashi N, Katsurada A, Navar LG, Kobori H. Costimulation with angiotensin II and interleukin 6 augments angiotensinogen expression in cultured human renal proximal tubular cells. Am J Physiol Renal Physiol 295: F283–F289, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Satou R, Miyata K, Gonzalez-Villalobos RA, Ingelfinger JR, Navar LG, Kobori H. Interferon-gamma biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J 26: 1821–1830, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shao W, Seth DM, Navar LG. Augmentation of endogenous intrarenal angiotensin II levels in Val5-ANG II infused rats. Am J Physiol Renal Physiol 296: F1067–F1071, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh A, Vitko J, Re RN, Cook JL. Intracellular enhanced cyan fluorescent protein/angiotensin II does not modify angiotensinogen accumulation in transgenic mice. Ochsner J 13: 37–41, 2013. [PMC free article] [PubMed] [Google Scholar]
- 55.van Kats JP, Schalekamp MA, Verdouw PD, Duncker DJ, Danser AH. Intrarenal angiotensin II: interstitial and cellular levels and site of production. Kidney Int 60: 2311–2317, 2001. [DOI] [PubMed] [Google Scholar]
- 56.von Thun AM, Vari RC, El Dahr SS, Navar LG. Augmentation of intrarenal angiotensin II levels by chronic angiotensin II infusion. Am J Physiol Renal Fluid Electrolyte Physiol 266: F120–F128, 1994. [DOI] [PubMed] [Google Scholar]
- 57.Woost PG, Kolb RJ, Finesilver M, Mackraj I, Imboden H, Coffman TM, Hopfer U. Strategy for the development of a matched set of transport-competent, angiotensin receptor-deficient proximal tubule cell lines. In Vitro Cell Dev Biol Anim 42: 189–200, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Ying J, Stuart D, Hillas E, Gociman BR, Ramkumar N, Lalouel JM, Kohan DE. Overexpression of mouse angiotensinogen in renal proximal tubule causes salt-sensitive hypertension in mice. Am J Hypertens 25: 684–689, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhuo JL, Ohishi M, Mendelsohn FA. Roles of AT1 and AT2 receptors in the hypertensive Ren-2 gene transgenic rat kidney. Hypertension 33: 347–353, 1999. [DOI] [PubMed] [Google Scholar]
- 60.Zhuo JL, Carretero OA, Li XC. Effects of AT1 receptor-mediated endocytosis of extracellular Ang II on activation of nuclear factor-kappa B in proximal tubule cells. Ann NY Acad Sci 1091: 336–345, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhuo JL, Imig JD, Hammond TG, Orengo S, Benes E, Navar LG. Ang II accumulation in rat renal endosomes during Ang II-induced hypertension: role of AT1 receptor. Hypertension 39: 116–121, 2002. [DOI] [PubMed] [Google Scholar]
- 62.Zhuo JL, Li XC, Garvin JL, Navar LG, Carretero OA. Intracellular angiotensin II induces cytosolic Ca2+ mobilization by stimulating intracellular AT1 receptors in proximal tubule cells. Am J Physiol Renal Physiol 290: F1382–F1390, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zou LX, Hymel A, Imig JD, Navar LG. Renal accumulation of circulating angiotensin II in angiotensin II-infused rats. Hypertension 27: 658–662, 1996. [DOI] [PubMed] [Google Scholar]
- 64.Zou LX, Imig JD, Hymel A, Navar LG. Renal uptake of circulating angiotensin II in Val5-angiotensin II infused rats is mediated by AT1 receptor. Am J Hypertens 11: 570–578, 1998. [DOI] [PubMed] [Google Scholar]