

Abstract
Lysine acetylation has emerged as one of the major post-translational modifications, as indicated by its roles in chromatin remodeling, activation of transcription factors and, most recently, regulation of metabolic enzymes. Identification of acetylation sites in a protein is the first essential step for functional characterization of acetylation in physiological regulation. However, the study of the acetylome is hindered by the lack of suitable physical and biochemical properties of the acetyl group and existence of high-abundance acetylated histones in the cell, and needs a robust method to overcome these problems. Here we present protocols for (i) using chemically acetylated ovalbumin and synthetic acetylated peptide to generate a pan-acetyllysine antibody and a site-specific antibody to Lys288-acetylated argininosuccinate lyase, respectively; (ii) using subcellular fractionation to reduce highly abundant acetylated histones; and (iii) using acetyllysine antibody affinity purification and mass spectrometry to characterize acetylome of human liver tissue. The entire characterization procedure takes ~2–3 d to complete.
INTRODUCTION
Protein lysine acetylation refers to post-translational addition of an acetyl group to the ε-amino group of the side chain of a lysine residue. This modification is different from N-terminal α-amino group acetylation with respect to the nature of modifying enzymes and physiological functions1. Multiple acetyltransferases and deacetylases are responsible for lysine acetylation, which has major roles in regulating various biological functions, whereas N-terminal acetylation often functions to stabilize proteins1,2. Internal lysine acetylation was discovered in histones in the early 1960s (refs. 3,4), and rapid progress has been made in the field in the past one-and-a-half decades. Imbalance in histone acetylation has been found to change chromatin structure and to be associated with transcriptional dysregulation of genes that are involved in the control of proliferation, cell-cycle progression, differentiation and/or apoptosis5,6. Nonhistone proteins, mainly nuclear transcription regulators such as p53 and E2F, have also been found to be acetylated, and their activities are regulated by acetylation7–9. Therefore, extensive studies have revealed that acetylation has a fundamental role in transcription regulation, by either altering chromatin structure through histone modification or modulating individual transcription factors or coregulators.
With more biological functions of acetylation being revealed, an easy and robust protocol for identifying acetylation will greatly benefit the field of protein acetylation. Detection of protein acetylation is challenging mainly because of the lack of usable physical and biochemical properties of the acetyl group. It is, however, possible to generate antibodies to acetylated lysine residues (anti-acetyllysine), and antibody affinity purification offers the possibility of enriching acetylated proteins. The development of such antibodies provides a valuable tool for detecting acetylation, but this approach is also complicated by the extremely high abundance of some acetylated proteins, such as histones and tubulin, in the cell. Thus, the abundant acetylation of histone and tubulin hinders the detection of acetylation of low-abundance proteins.
Kim et al.10 developed a method of coupling anti-acetyllysine affinity purification to mass spectrometry (MS). By enriching acetylated peptides through affinity purification and reducing interference from high-abundance acetylated histone peptides through subcellular fractionation, Kim et al.10 successfully detected many low-abundance acetylated peptides. Further improvement of this approach resulted in two recent proteomic studies, one from our group11 and the other from Choudhary et al.12. Whereas Kim et al.10 identified close to 200 acetylated proteins from mouse liver, our improved approach has identified more than 1,000 acetylated proteins from human liver11. The substantially higher number of acetylated proteins identified in our study may be attributed to the high affinity of our homemade anti-acetyllysine, as well as to the fact that we have carried out multiple rounds of identification procedures to ensure more comprehensive coverage. Our number is similar to that of Choudhary et al.12, who identified more than 1,000 acetylated proteins from a human leukemia cell line. These studies, taking similar approaches, have substantially expanded our knowledge of the acetylome and functions of protein acetylation.
The protocols presented here provide detailed information about the generation of pan-acetyllysine and site-specific acetyllysine antibodies, as well as about procedures for subcellular fractionation and affinity enrichment of acetylated peptides from human liver tissue (Fig. 1). This approach should be applicable to protein acetylation studies in other tissues and organisms.
Figure 1.
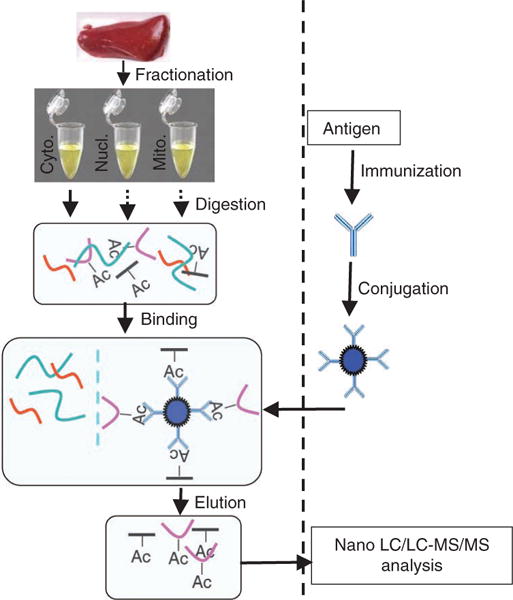
Schematic diagram of enrichment of acetylated peptides from human liver. Major steps of antibody generation, sample preparation and enrichment of acetylated peptides are illustrated.
Experimental design and considerations
The protocol described in this article is based on subcellular fractionation and anti-acetyllysine affinity purification of digested total peptide mixtures, followed by MS. Key factors for the success of this approach include the specificity and affinity of anti-acetyllysine, the accessibility of acetylated lysine residues, the separation of different cellular fractions and the quality of MS analyses. The low affinity of most antibodies to acetyllysine means that they are ineffective in precipitating whole protein even if the protein is acetylated. Moreover, the acetylated lysine residue may be buried inside a protein and not accessible to antibody binding. These problems can be solved by trypsin digestion. The acetylated peptides generated by protease digestion are much smaller than a whole protein and can be immunoprecipitated by anti-acetyllysine even if the antibody has low affinity. Furthermore, acetylated lysine residues will be readily accessible to the antibody because digested peptides lack secondary structure. To avoid interference by highly abundant acetylated proteins such as histone, cell fractionation can be used to separate the nucleus from other organelles or cytosol.
The lack of polarity and the unique steric structure of the acetyl group lead to it offering very few useful chemical and physical properties that can be used to enrich acetylated proteins. Therefore, antibody-based affinity purification offers an attractive approach to identifying acetylated proteins. However, the small acetyl group (42 Da) also has low antigenicity, and it is thus challenging to generate a high-affinity antibody. Antigens used to generate antibodies to acetyllysine are usually either acetyllysine-containing synthetic peptides or chemically acetylated carrier proteins13. The benefits of using chemically acetylated carrier protein as antigen are twofold: first, there are many acetyllysine residues in one carrier protein, which is likely to increase the chance of obtaining anti-acetyllysine antibody and, second, the flanking sequences of each acetyllysine residue vary greatly, according to the carrier protein’s sequence, making it possible to generate an anti-acetyllysine pool that can recognize acetyllysine independent of flanking sequences. This is particularly important for isolation of total cellular acetylated peptides, given that lysine acetylation sites have minimal consensus sequence. Lysine ε-amino group acetylation can be easily carried out by a single-step chemical reaction using acetic anhydride13, making it simple in practice to obtain sufficient antigen.
Interference caused by the high abundance of some acetylated proteins during enrichment can be resolved, at least in part, by subcellular fractionation. For example, the highly abundant histones are mainly in the nuclear fraction and can be easily separated from other subcellular compartments by centrifugation. Mitochondrial proteins are now known to be extensively acetylated14. Because intact mitochondria can be readily separated from the cytosolic fraction by centrifugation, mitochondrial proteins can be enriched and analyzed separately without the interference of cytoplasmic proteins, such as the highly abundant and heavily acetylated tubulin15,16. MS analyses of separate subcellular fractions also greatly reduce the complexity of each fraction, thereby increasing the coverage of acetylated peptide identification.
By these manipulations, we expect to detect more acetylated proteins, especially the low-abundance ones. Compared with directly analyzed acetylated peptides in samples, which usually yield 1%–2% positive hits among the total peptides detected, affinity-enriched samples should yield a much higher percentage of positive hits. In addition, as a single round of manipulation usually results in insufficient coverage, experiments will need to be repeated 3–4 times to ensure the identification of a large number of acetylated peptides and thus to define a comprehensive acetylome10–12.
Comments regarding the procedure
Acetylated BSA was used to analyze pan-anti-acetyllysine specificity. It was generated using the same protocol from Steps 1 to 10, except that ovalbumin (OVA) was replaced by BSA.
MATERIALS
REAGENTS
BSA (Sangon, cat. no. 0332)
OVA (Sangon, cat. no. A5503)
Na2CO3 (Sangon, cat. no. ST0840)
Pyridine (Sinopharm Chemical Reagent Co. (SCRC), cat. no. 10018118)
Acetic anhydride (SCRC, cat. no. 10000318)
Tris base (Sangon, cat. no. TB0194)
Glycerol (Bio Basic, Inc. (BBI), cat. no. GB0232)
β-Mercaptoethanol (AMRESCO, cat. no.0482)
SDS (Sangon, cat. no. 0227)
Bromophenol blue (Sangon, cat. no. BT0922)
Acrylamide (Sangon, cat. no. 0341)
Bisacrylamide (Sangon, cat. no. 0172)
Ammonium persulfate (Sigma, cat. no. A3678)
TEMED (BBI, cat. no. TB0508)
DMEM (GIBCO, cat. no. 12100-061)
Triclostatin A (Cell Signaling, cat. no. 9950)
Nicotinamide (Sigma, cat. no. N0636)
NaCl (Sangon, cat. no. SB0476)
Nonidet-P40 (NP-40; BBI, cat. no. NDB0385)
PMSF (Fluka, cat. no. P0754)
Aprotinin (BBI, cat. no. AD0153)
Leupeptin (Sangon, cat. no. LJ580)
Pepstatin (Sangon, cat. no. J583)
Peptone (Oxoid, cat. no. LP0037)
Tween-20 (Sangon, cat. no. 0777)
KCl (BBI, cat. no. PB0440)
MgCl2 (Fluka, cat. no. 63063)
DTT (Merck, cat. no. 233156)
Iodoacetamide (AMRESCO, cat. no. M216)
Cysteine (BBI, cat. no. CB0088)
NH4HCO3 (BBI, cat. no. AB0032)
Trypsin (Promega, cat. no. V5113)
Trifluoroacetic acid (TFA; Sigma, cat. no. T6508) ! CAUTION TFA is corrosive and causes severe burns. Inhalation may cause lung damage.
Acetonitrile (Sigma, cat. no. 34851)
Sulfolink coupling gel (Pierce, cat. no. 20401)
Formic acid (Fluka, cat. no. 06440) ! CAUTION Formic acid is corrosive and causes severe burns. Inhalation may cause lung damage.
Na2HPO4 (Sangon, cat. no. ST1288)
NaH2PO4 (Sangon, cat. no. SB0879)
EDTA (Sangon, cat. no. 0105)
Keyhole limpet hemocyanin (KLH; Pierce, cat. no. 0077605)
Complete Freund’s adjuvant (Sigma, cat. no. F5881)
Acetone (100%; Sigma, cat. no. 270725) ! CAUTION Acetone can damage the mucosa of the mouth and can irritate and damage skin. Acetone should be used with appropriate safety measures, such as protective gloves, glasses and clothing, and adequate ventilation. Store acetone in an explosion- or flame-proof freezer.
N,N,N′,N′-Tetramethylethylenediamine (TEMED; BBI, cat. no. TB0508)
Glacial acetic acid (SCRC, cat. no. 10000318)
Coomassie brilliant blue G-250 (Serva, cat. no. 17524)
Secondary antibody (Genscript, cat. no. A00098)
ECL Plus (chemiluminescent detection reagent)(GE, cat. no. RPN2132)
Mitochondrial lysis buffer (Pierce, cat. no. 89874)
Tris-HCl (BBI, cat. no. 77-86-1)
DMSO (Sigma, cat. no. D8418)
HEK293T cells (ATCC, cat. no. CRL-11268)
Bradford reagent (Sigma, cat. no. B6916)
ELISA plate (Bio-Rad, cat. no. 224-0096)
Horseradish peroxidase color development solution (Beyotime, cat. no. P0202)
Deionized water (from Milli-Q Elix, Millipore)
Nonfat milk (BD, cat. no. 232100)
Purify buffer salts (Pierce, cat. no. 77159)
EQUIPMENT
50-ml conical tubes (for gels)
15-ml conical tubes (for gels)
1.5-ml tubes (for RP-HPLC and gels)
Dounce homogenizer (for tissue homogenization)
Concentrator 5301 (for vacuum drying; Eppendorf, cat. no. 5301)
Glass Hamilton syringe with metal needle (1 ml, for reverse phase (RP)-HPLC; Hamilton, cat. no. 1001)
Spacers (SDS gel; 0.75 mm)
Gel electrophoresis apparatus (SDS gels; Bio-Rad
Rocking platform (for western blotting)
Polyvinylidene difluoride membrane (for western blotting) or nitrocellulose membrane (Whatman, cat. no. 9053815)
3 MM paper (Whatman, for western blotting)
Wet-transfer chamber (Bio-Rad; for western blotting)
Power supply (500 V, 500 mA, SDS-PAGE; Bio-Rad)
Fast protein liquid chromatography (FPLC) system with pumps, UV detector and fraction collector, and 2-ml sample loop (AKTA-FPLC, Amersham Biosciences)
Sephadex G25 column (10 ml; GE, cat. no. 17-1408-01)
Typhoon Trio imaging system (GE)
REAGENT SETUP
New Zealand rabbit (antibody preparation)
Rabbits (~2.5 kg each in weight at the time of starting experiments) were accustomed to an animal facility for 2–4 d before starting the immunization process. ! CAUTION Rabbits must be maintained according to relevant national regulations using protocols and conditions approved by the institutional animal care and use committee.
Biological samples
Human liver tissue samples were obtained by collecting the surrounding liver tissues of liver cancer samples. ! CAUTION A physician or a nurse practitioner must obtain informed consent from donors.
High-salt extraction buffer (high-salt extraction of nuclear proteins)
Prepare 100 ml of a solution containing 20 mM Tris-Cl (pH 7.4), 25% (vol/vol) glycerol, 1.5 mM MgCl2, 0.2 mM EDTA and 1.2 M KCl. Add protease and phosphatase inhibitors described below to a 1× concentration just before use in lysis of cells.
Proteinase inhibitor cocktail (50×)
Prepare a solution containing 50 mM PMSF, 0.05 mg ml−1 aprotinin, 0.05 mg ml−1 leupeptin and 0.05 mg ml−1 pepstatin in DMSO (can be stored for up to 12 weeks at −20 °C).
▲ CRITICAL Add DTT, PMSF (final concentration of 1 mM) and protease inhibitors just before use.
Acrylamide: bisacrylamide solution (SDS gels)
This solution consists of 250 g acrylamide in 417 ml ddH2O. Stir overnight and add 1.67 g bisacrylamide. Filter the solution and store it at room temperature (20–25 °C) in a dark container protected from light. ! CAUTION Acrylamide and bisacrylamide are potent neurotoxins. Use appropriate safety measures, such as protective gloves and safety goggles, and handle under adequate ventilation.
Running buffer (SDS gels)
The buffer is prepared by dissolving 3.02 g Tris base, 18.8 g glycine and 1 g SDS in 1 liter deionized water. This buffer can be stored at room temperature until it is used.
Coomassie brilliant blue
This solution is prepared with 0.25% (wt/vol) Coomassie brilliant blue G-250, 45% (vol/vol) methanol, 10% (vol/vol) acetic acid. Solubilize brilliant blue G-250 powder in methanol first, then add acetic acid and finally ddH2O. Filter the solution and store at room temperature.
NETN buffer
This solution contains final concentrations of 50 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA and 0.5% (wt/vol) NP-40; pH is adjusted to 8.0 before use.
Transfer buffer (western blotting)
This buffer is prepared by dissolving 4.55 g Tris base and 21.6 g glycine in 500 ml deionized water; add methanol and water to obtain a final concentration of 20% (vol/vol) methanol and a total volume of 1 liter. This buffer can be stored at room temperature until it is used.
Purification buffer salts (Pierce)
See Box 1 for preparation instructions.
BOX 1. COUPLING ASL Ack288 ANTIGEN TO KLH ● TIMING ~1.5 D.
▲ CRITICAL STEP Synthesize the corresponding peptide of Lys288-acetylated argininosuccinate lyase (ASL AcK288: CSTGSSLMPQKK(Ac)N) (20 mg); a cysteine residue (C) is added to the C-terminus of the peptide to facilitate conjugation in the following steps.
Weigh 2 mg of peptide and dissolve it in 200 μl of PBS-EDTA (pH 7.5; PBS with 1 mM EDTA); EDTA is included to chelate metal ions that could interfere with cross-linking.
Weigh 10 mg of KLH and dissolve it in 1 ml of water.
-
Mix 200 μl of peptide solution with 200 μl of KLH solution.
▲ CRITICAL STEP Keep peptide/KLH ratio > 10 (mol/mol).
Incubate the reaction mixture at 27 °C for 2 h.
Dissolve purification buffer salts (Pierce) in 60 ml water.
Dialyze reaction mixture in purification buffer salts solution overnight.
Determine conjugated KLH concentration by the Bradford method.
Aliquot conjugated KLH (200 μg per vial) and freeze at −40 °C for further use.
Coupling buffer
See Box 2 for preparation instructions.
BOX 2. PREPARATION OF AFFINITY COLUMN AND AFFINITY ANTIBODY PURIFICATION ● TIMING 2 D.
Preparation of affinity column
▲ CRITICAL STEP Prepare chemical acetylated BSA or synthesized ASL AcK288 corresponding peptide [CSTGSSLMPQKK(Ac)N]; a cysteine residue is added to the C-terminus of the peptide.
-
1
Add 2 ml of Sulfolink coupling gel slurry to a 15-ml conical tube.
-
2
Add 5 ml of coupling buffer to the tube, close the cap and wash the gel by inversion.
Coupling buffer
|
| |
| Components | |
|
| |
| Tris | 50 mM |
| EDTA-Na (pH 8.5) | 5 mM |
|
| |
-
3
Centrifuge at 1,000g for 3 min.
-
4
Repeat Steps 2 and 3 two or three times.
-
5
Dissolve Ac-BSA or peptide in coupling buffer to a final concentration of 5 mg ml−1.
-
6
Measure OD280 of the solution.
▲ CRITICAL STEP If OD280 > 2.0, dilute Ac-BSA or peptide solution until OD280 < 2.0.
-
7
Add 3–5 ml of peptide solution to the tube and mix with gel by inversion.
-
8
Shake the solution at room temperature for 15 min.
-
9
Keep the tubes aside at room temperature for 30 min.
-
10
Centrifuge at 1,000g for 3 min, and then separate supernatant and gel slurry.
▲ CRITICAL STEP Do not discard either the supernatant or the gel slurry.
-
11
Measure OD280 of the supernatant.
Note: If the OD280 of the supernatant is significantly less than the OD280 measured in Step 7, this indicates a successful cross-linking.
-
12
Add 5 ml of coupling buffer to the tube.
-
13
Wash the gel slurry by inverting gently a few times.
-
14
Centrifuge at 1,000g for 3 min; remove and discard the supernatant.
-
15
Repeat Steps 13–15 two times.
-
16
Add 3 ml of 50 mM L-cysteine HCl solution to the gel slurry and mix by inversion.
-
17
Incubate at room temperature for 30 min.
-
18
Centrifuge at 1,000g for 3 min; remove and discard the supernatant.
-
19
Add 5 ml of 1M NaCl to the tube and mix by inversion.
-
20
Centrifuge at 1,000g for 3 min; remove and discard the supernatant.
-
21
Repeat Steps 20 and 21 three times.
-
22
Add 5 ml of PBS to the tube and mix by inversion.
-
23
Centrifuge at 1,000g for 3 min; remove and discard the supernatant.
-
24
Repeat Steps 23 and 24 three times.
-
25
Add 2 ml of PBS and store at 4 °C.
Affinity purification
-
26
Prepare an affinity column by adding 1 ml of prepared affinity gel into a 2-ml empty column.
-
27
Equilibrate the column with 20 ml of PBS.
-
28
Bind the antibody to the column by passing 20 ml of antiserum through the column at a flow rate of 0.5 ml min−1. Collect the pass-through serum.
-
29
Reload the pass-through serum from Step 29 into the column one more time.
-
30
Wash the resin with PBS until OD280 < 0.05.
-
31
Elute antibody using 0.2 M glycine (pH 2.8).
-
32
Collect fractions into tubes containing 1 M Tris (pH 8.0) (0.5 ml of eluent into each tube containing 0.15 ml of Tris).
PROCEDURE
Generation of pan-anti-acetyllysine antibody: preparation of antigen ● TIMING 2 d
1| Weigh 20 mg of OVA and dissolve it in 20 ml of 0.1 M Na2CO3 in a 50-ml Falcon tube.
-
2| Add 100 μl of acetic anhydride slowly while shaking gently.
▲ CRITICAL STEP Acetic anhydride needs to be added dropwise with constant shaking to avoid precipitation of OVA. It takes ~10 min to complete this step.
-
3| Add 400 μl of pyridine slowly while shaking gently.
▲ CRITICAL STEP Pyridine is added dropwise with constant shaking to avoid precipitation of OVA. It takes ~30 min to complete this step.
? TROUBLESHOOTING
4| Incubate the reaction at room temperature (30 °C) for 4–5 h.
5| Add 400 μl of 1 M Tris base into the reaction mixture to stop the reaction.
-
6| Load 2 ml of reaction mixture to a 10-ml Sephadex G-25 gel filtration column and remove organic reagents by running the reaction mixture through the column with 50 mM Tris buffer in an AKTA-FPLC system; monitor optical density at 280 nm (OD280) to collect the eluted protein.
▲ CRITICAL STEP Pyridine has absorbance at 280 nm and it runs out of the column right after the protein peak.
? TROUBLESHOOTING
7| Repeat Step 6 until the entire reaction mixture is separated and acetylated OVA is collected.
-
8| Vacuum-dry acetylated OVA overnight in a concentrator (Eppendorf 5301).
■ PAUSE POINT Dried acetylated protein can be stored at 4 °C until it is needed for the next step.
-
9| Prepare SDS-PAGE samples by dissolving both acetylated and unacetylated OVA in 1 ml of 5× SDS loading buffer (prepared as described in the table below); heat at 99 °C for 5 min.
Component Final concentration
Tris-HCl (pH 6.8) 0.25 M Glycerol 50% β-Mercaptoethanol 20% SDS 10% Bromophenol blue 0.0012%
-
10| Run unacetylated and acetylated OVA in 10% (wt/vol) SDS gel (prepared as described in the table below) and check molecular weight gain of acetylated OVA to verify sufficient acetylation of OVA (Fig. 2).
Component ml
H2O 4.0 Acrylamide (30%) 3.3 Tris (pH 8.8; 1.5 M) 2.5 SDS (10%) 0.1 Ammonium persulfate (10%) 0.1 TEMED 0.004
Figure 2.
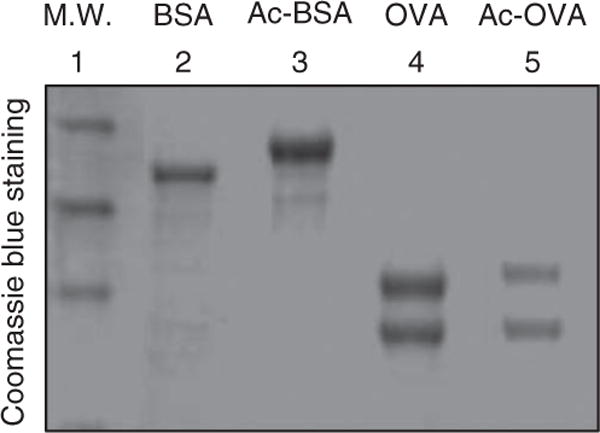
Acetylated proteins have higher molecular weight. Unacetylated and acetylated BSA and OVA proteins are resolved in 10% SDS-PAGE. M.W. indicates molecular weight marker.
Generation of antibody: day 0
-
11| Dissolve antigen in saline (0.9% (wt/vol) sodium chloride) to a final concentration of 0.5 mg ml−1.
Note: For generation of site-specific acetyllysine antibody, use the same protocol from Steps 11–24; see Box 1 for preparation of antigen to Lys288-acetylated argininosuccinate lyase11.
12| Mix the antigen solution and complete Freund’s adjuvant (1:1, vol/vol).
13| Inject 1 ml of mixed antigen into a New Zealand rabbit subcutaneously (s.c.) in multiple places; antigen dosage should be 200–400 μg per rabbit.
Days 28–30 (perform these steps on any day during this time period)
14| Prepare boost antigen by mixing antigen solution with incomplete Freund’s adjuvant (1:1, vol/vol).
15| Inject 100–200 μg per rabbit s.c.
Days 58–60 (perform these steps on any day during this time period)
16| Boost immunization again by injecting rabbits with boost antigen at 100–200 μg per rabbit s.c.
17| Obtain the blood sample (1 ml) from rabbit ear and determine antibody titer (Box 3).
BOX 3. INDIRECT ELISA PROTOCOL ● TIMING 1.5 D.
Dilute antigen with 0.1 M sodium carbonate solution (pH 9.6) to a final concentration of 2–5 μg ml−1.
Add 50 μl of antigen solution to each ELISA plate well.
Incubate the ELISA plate at 4 °C overnight to allow antigen coating.
Wash the wells with TBST five times.
Remove TBST completely.
Add 200 μl of TBST with 5% nonfat milk to each well.
Block wells at 37 °C in an incubator for at least 2 h.
Remove TBST completely.
Prepare a serial dilution of antiserum (1/1,000 to 1/500,000).
Add 50 μl each of diluted antiserum to wells (3 wells of each dilution; 4–6 dilutions).
Incubate at 37 °C for 1 h.
Wash wells with TBST five times.
Remove TBST completely.
Add 50 μl 3-3′,5,5′-tetramethylbenzidine (TMB) horseradish peroxidase color development solution to each well.
Leave the ELISA plate at room temperature for 3–5 min.
Add 20 μl of 2 M sulfuric acid to each well to stop the reaction.
Measure color intensity at 450 and 630 nm.
Days 88–90 (perform these steps on any day during this time period)
-
18| Boost immunization again by injecting rabbits with boost antigen at 100–200 μg per rabbit s.c.
■ PAUSE POINT Wait 10–15 days after final immunization to kill the rabbit and continue the procedure.
19| 10–15 days after final immunization, kill the rabbit and collect blood (~50 ml per rabbit) from the carotid arteries.
20| Place collected blood in a 37 °C incubator for 2 h.
-
21| Transfer blood from 37 °C to 4 °C and maintain at 4 °C overnight.
■ PAUSE POINT Store blood at 4 °C overnight before continuing to the next step.
22| The next day, centrifuge blood at 6,000g at 4 °C for 20 min.
23| Carefully collect antiserum and discard pellets.
24| Determine antibody titer by indirect ELISA (for protocol, see Box 3). Note that antiserum can be used directly for the following manipulations or further purification can be carried out. For antibody purification, see Box 2.
Prepare cells for pan-anti-acetyllysine antibody verification ● TIMING 2–3 d
25| Grow HEK293T cells in DMEM to ~80% confluence.
26| Split cells equally into two culture plates, one for treatment and one for control.
-
27| 18 h before harvesting cells, add Triclostatin A to the treatment plate to a final concentration of 5 μM.
? TROUBLESHOOTING
-
28| 6 h before harvesting cells, add nicotinamide to the treatment plate to a final concentration of 5 mM.
? TROUBLESHOOTING
29| Centrifuge plates at 1,000g for 5 min to harvest cells.
Verification of pan-anti-acetyllysine antibody ● TIMING 1 d
-
30| Lyse cells using Nonidet-P40 buffer.
Component
NaCl 150 mM Tris-HCl (pH 7.5) 50 mM NP-40 0.1–0.5% Proteases inhibitors Indicated in reagents setting
31| Centrifuge lysates at 12,000g at 4 °C for 15 min.
32| Retain the supernatant and discard the pellet.
33| Quantify total protein in the supernatant using Bradford reagent.
-
34| Run treated and control samples with an equal amount of total proteins on a 12% (wt/vol) SDS gel.
SDS gel (12%) component ml
H2O 3.3 Acrylamide (30%) 4.0 Tris (pH 8.8; 1.5 M) 2.5 SDS (10%) 0.1 Ammonium persulfate (10%) 0.1 TEMED 0.004
35| Transfer proteins from the gel to a nitrocellulose membrane.
-
36| Block the membrane with peptone buffer.
Peptone blocking buffer component Concentration
Tris-HCl (pH 7.5) 0.1 M NaCl 100 mM Peptone 1% Tween-20 10%
-
37| Wash the membrane twice with Tris-buffered saline and Tween 20 (TBST) buffer.
TBST buffer component Concentration
Tris-HCl (pH 7.5) 0.1 M NaCl 100 mM Tween-20 0.5%
38| Incubate the membrane with pan-anti-acetyllysine antibody (titer 1:100,000) diluted at 1:500 (vol/vol) in 10 ml of peptone-antibody buffer (prepared according to the in-text table below), shaking at room temperature for 4–6 h. The dilution factor varies among different batches of antiserum.
39| Wash the membrane three times with TBST buffer.
-
40| Incubate the membrane with secondary antibody diluted at 1:1,000 (vol/vol) in 10 ml peptone-antibody buffer, along with shaking at room temperature for 2–3 h.
Peptone-antibody buffer component Concentration
Tris-HCl (pH 7.5) 0.1 M NaCl 100 mM Peptone 0.1% Tween-20 0.5%
41| Wash the membrane three times with TBST buffer.
42| Apply ECL Plus to the membrane, keep it in the dark for 5 min and detect signal with Typhoon Trio. Alternatively, ECL can be developed by exposure to films. See examples in Supplementary Figure 1 of reference 11 (http://www.sciencemag.org/cgi/data/327/5968/1000/DC1/1).
Subcellular fractionation of tissue sample ● TIMING 1 d
43| Weigh 1 g of human liver tissue.
-
44| Mince the tissue into small pieces with a razor blade and scissors.
▲ CRITICAL STEP This and the following steps (Steps 44–51) must be performed on ice.
-
45| Transfer minced tissue to a 10 ml glass Dounce homogenizer (on ice) and add 5 ml of ice-cold homogenization buffer.
Homogenization buffer component Concentration
KCl 10 mM MgCl2 1.5 mM Tris-Cl (pH 7.4) 10 mM Triclostatin A 5 μM Nicotinamide 50 mM
46| Homogenize the tissue by applying 30–40 strokes.
47| Pass homogenized mixture through a 100-mm-diameter cell filter (pore size 125 μm) to remove debris.
48| Centrifuge the filtered mixture at 1,000g at 4 °C for 10 min.
49| Carefully transfer the supernatant to another centrifuge tube and retain the pellet. Note that pellets contain nuclei; see Box 4 for extraction of nuclear proteins.
50| Centrifuge at 10,000g at 4 °C for 30 min.
51| Separate the supernatant (contains cytosolic proteins) and the pellet (contains mitochondria); retain both fractions. Note that this pellet contains mitochondrial proteins; see Box 5 for extraction of mitochondrial proteins.
BOX 4. EXTRACTION OF NUCLEIC PROTEINS ● TIMING ~1.5 H.
Add 1 ml high-salt buffer to the nuclear pellet.
Shake at 4 °C on a rotary shaker for 30 min.
Centrifuge at 12,000g at 4 °C for 10 min.
Collect the supernatant.
Repeat Steps 1–4 and combine the supernatants from both; discard the pellet.
BOX 5. EXTRACTION OF MITOCHONDRIAL PROTEINS ● TIMING ~1 H.
Add 1 ml of mitochondrial lysis buffer to mitochondrial pellets.
Mix gently at 4 °C on a rotary shaker for 30 min.
Centrifuge at 12,000g for 15 min at 4 °C.
Retain the supernatant and discard the pellet.
Enrichment of acetylated peptides ● TIMING 2 d
52| Add DTT to the protein solution to a final concentration of 5 mM to break disulfide bonds within proteins; allow the reaction to proceed for 10 min. Manipulations from this point on are identical for cytosolic, mitochondrial and nucleic proteins.
53| Add freshly prepared iodoacetamide to the protein solution to a final concentration of 15 mM to modify SH groups; allow the reaction to proceed in the dark at room temperature for 30 min.
54| Add cysteine to a final concentration of 15 mM to stop the reaction; allow the reaction to proceed at room temperature for 30 min.
55| Add chilled acetone to the solution to reach a final concentration of 85% (vol/vol).
56| Store the solution at −20 °C for 1 h to allow complete precipitation of proteins.
-
57| Centrifuge at 800g at 4 °C for 15 min and discard the supernatant.
▲ CRITICAL STEP In Steps 57 and 58, do not exceed 800g each time in order to avoid solubility problems caused by tight pellet formation.
? TROUBLESHOOTING
-
58| Wash the pellet three times with 5 ml of chilled acetone by thoroughly mixing and then centrifuge it at 800g at 4 °C for 5 min.
? TROUBLESHOOTING
59| Vacuum-dry protein pellets with a vacuum concentrator (Eppendorf 5301) for ~30 min.
-
60| Dissolve the protein pellet in 5 ml of 50 mM NH4HCO3 (pH 8.0) and sonicate for 5 min at room temperature to increase solubility.
▲ CRITICAL STEP Turbidity in the solution may be observed at this point. Do not centrifuge to remove it; use it directly for the following steps.
61| Determine the protein concentration of the suspension by the Bradford method.
-
62| In a fresh tube, take 3–10 mg protein, add sequencing-grade trypsin at a 1:50 (trypsin:protein) ratio, and hydrolyze at 37 °C overnight.
▲ CRITICAL STEP Although the reaction solution becomes clear after ~1 h, continue the digestion process overnight.
63| Add additional trypsin to the tube at a ratio of 1:100 (trypsin:protein) and incubate for 3 h at 37 °C.
64| Heat the sample at 99 °C for 5 min to inactivate trypsin.
65| Centrifuge at 12,000g for 10 min; retain the supernatant and discard the pellet (mainly contains insoluble proteins).
66| Vacuum-dry the supernatant and redissolve the pellet in 1 ml of deionized water.
-
67| Repeat Step 66 three to four times.
▲ CRITICAL STEP Steps 66 and 67 remove NH4CO3; repeat several more times if a large amount of white powder is detected in the tube.
-
68| Redissolve the pellet in 1.5 ml of NETNA binding buffer.
NETNA binding buffer component Concentration
Tris-HCl 50 mM (pH 8.0) NaCl 100 mM EDTA 1 mM NP-40 0.5% Acetonitrile 10%
? TROUBLESHOOTING
69| Conjugate the pan-anti-acetyllysine antibody to protein A beads (for protocol, see Box 6).
70| Add antibody-conjugated beads (200 μl) to the peptide solution prepared in Step 66; allow binding to continue for 4–8 h at 4 °C with rotary shaking.
71| Centrifuge at 1,000g briefly (< 10 s) and carefully remove the supernatant.
72| Wash beads three times with 1 ml of PBS buffer.
-
73| Remove PBS buffer completely and add 150 μl of elution buffer.
Elution buffer component Concentration (%)
TFA 0.1 Acetonitrile 5
74| Repeat the elution procedure three times and combine the supernatant from each elution.
75| Vacuum-dry the supernatant; enriched acetylated peptides should be in the pellet.
-
76| Dissolve the pellet in MS sample buffer and subject the sample to liquid chromatography–MS analysis.
MS sample buffer component Concentration (%)
Acetonitrile 10 (vol/vol) Formic acid solution 0.8 (wt/wt)
BOX 6. GENERATE CONJUGATED PAN-ANTI-ACETYLLYSINE ANTIBODY ● TIMING ~5 H.
-
Add 200 μl rabbit anti-acetylated-lysine serum and 100 μl protein A beads into a 1.5-ml Eppendorf tube.
Note: If purified antibody is used, add ~200 μg of antibody.
Add NETN buffer (see REAGENT SETUP) and adjust the final volume to 1 ml.
Shake gently at 4 °C on a rotary shaker for 4 h.
Wash the beads with 1 ml of NETN buffer three times.
-
Wash with ETN buffer twice.
Note: This preparation is sufficient for enriching acetylated peptides from 3–10 mg of protein.
ETN buffer
Component
EDTA 1 mM Tris-HCl 50 mM (pH 8.0) NaCl 100 mM
● TIMING
Steps 1–10, Generation of pan-anti-acetyllysine antibody: preparation of antigen: 2 d
Steps 11–24, Generation of antibody: ~100 d
Steps 25–29, Prepare cells for pan-anti-acetyllysine antibody verification: 2–3 d
Steps 30–42, Verification of pan-anti-acetyllysine antibody: 1 d
Steps 43–51, Subcellular fractionation of tissue sample: 1 d
Steps 52–76, Enrichment of acetylated peptides: 2 d
? TROUBLESHOOTING
Generation of chemical acetylated antigen
Step 3
Precipitation usually causes low efficiency of acetylation of carrier proteins and subsequently leads to inability to generate high-quality anti-acetyllysine antibody. Although acetic anhydride and pyridine are both used in chemical acetylation processes, excess amounts of both reagents or inappropriate addition of either reagent to the reaction solution can cause protein precipitation. Each component should be added to the reaction solution according to the specified order. If precipitation is still observed during the reaction, reduce the initial OVA concentration to half of that indicated in the protocol.
Step 6
It is essential to separate pyridine and acetic acid from acetylated OVA to ensure purity of the antigen. We used a 10-ml Sephadex G-25 column loaded with 2 ml of reaction mixture to separate acetylated OVA from pyridine and acetic acid on an AKTA-FPLC system. A longer column may be needed if the separation is incomplete.
Treatment of cells by deacetylase inhibitors
Steps 27 and 28
When it comes to prepare cells for anti-acetyllysine antibody verification, successful treatment by deacetylase inhibitors is critical to the preparation of cells for anti-acetyllysine antibody verification, as unsuccessful treatment could lead to incorrect conclusions. Of the three classes of deacetylases identified in cells so far, class I and II deacetylases are successfully inhibited by 0.5 μM trichostatin A treatment and NAD +-dependent sirtuins are inhibited by 5 mM nicotinamide (NAM) treatment. In our system, the effect of trichostatin A treatment is obvious after treating cells for 18 h; NAM treatment, however, exerts the strongest effect when cells are treated for 4–6 h. A time-course experiment may be needed to determine appropriate timing for optimal inhibition results.
Avoid formation of insoluble protein pellet
Steps 57 and 58
Acetone precipitated proteins have the advantage of being easily re-dissolved in a water-based solution. However, if the precipitated proteins are in a tightly packed pellet, it is hard for all proteins to redissolve, thus affecting the completion of trypsin digestion. Once a tight pellet is formed, it is helpful to add 1 ml of acetone to the pellet and to follow this with sonication for 5–10 min to break up the pellet; thereafter, vacuum-dry to remove acetone. The resulting protein pellet powder is easy to dissolve in NH4CO3 solution.
Increase solubility of acetylated peptides
Step 68
After proteins have been digested into peptides, hydrophobic ones cannot be easily dissolved in aqueous buffer. This is particularly important for acetylated peptides because acetylation changes a positively charged lysine residue to a neutral acetyllysine, thus inevitably decreasing the solubility of acetylated peptides. Addition of acetonitrile to the buffer can increase the solubility of hydrophobic peptides (Step 68) because acetonitrile is a good solution for hydrophobic peptides but has little effect on antibody stability17, thus facilitating enrichment of acetylated peptides. However, the concentration of acetonitrile in the buffer should not exceed 20% because it will denature the antibody at a high concentration.
ANTICIPATED RESULTS
Chemical acetylation of proteins typically results in addition of acetyl groups to lysine residues stoichiometrically. Acetylated proteins thus have higher molecular weight compared with unacetylated protein when resolved in SDS-PAGE (Fig. 2). After antibody is generated, the quality of the antibody can be tested with cell lysates treated with or without deacetylase inhibitors. Typically, acetylation of prominent bands, such as histone or tubulin, should be elevated on inhibitor treatment11. As the antigen is acetylated OVA, antibodies against OVA backbone can also be generated. The specificity of the antibody can be tested using acetylated BSA as competitor11. The process of acetylated enrichment is hard to follow; however, the amount of enriched peptides can be monitored when samples are run through liquid chromatography (Fig. 3). The successful enrichment of acetylated peptides can be judged by assessing whether acetylated histone peptides are identified by tandem MS analysis. Typically, identification of various acetylated histone peptides indicates a successful enrichment (Fig. 4). A more meaningful measurement is the percentage of acetylated peptide over total peptides identified. Without antibody purification, acetylated peptides normally represent < 2% of the total liver peptides. In our studies, acetylated peptides usually account for 10–20% of the total enriched peptides. Finally, clean MS/MS spectra for acetylated peptides of non-nuclear proteins indicate a successful enrichment of such proteins (Fig. 5).
Figure 3.
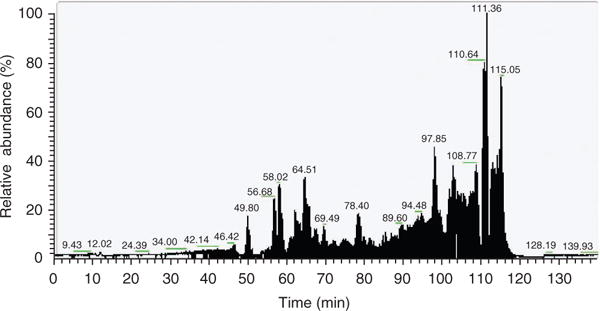
Monitoring of enriched peptides by RP-LC. An example of RP-LC (C-18 column) separation of enriched peptides is shown.
Figure 4.

Identification of multiacetylated forms of acetylated histone peptides. Multiacetylated forms of histone H4 peptides of human liver tissue identified by MS/MS are shown; red Ks indicate acetylated lysine in the peptides.
Figure 5.
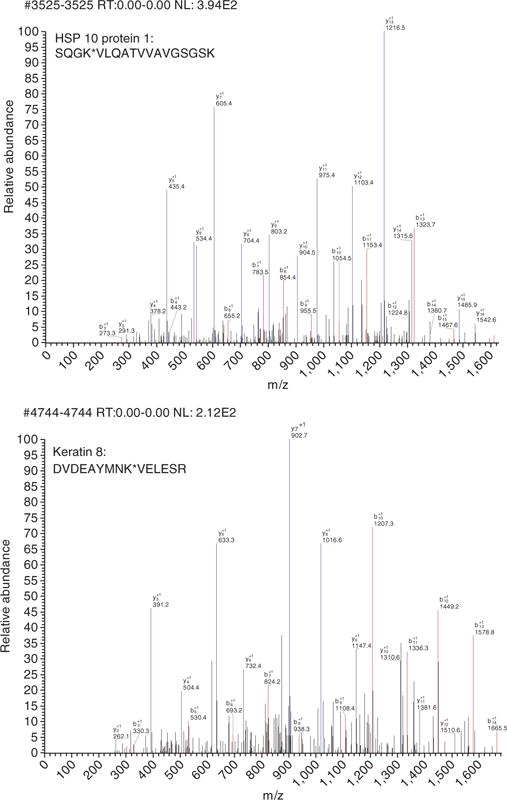
MS spectra of enriched acetylated peptides. Two MS spectra of identified acetylated peptides are shown.
Acknowledgments
We are grateful to members of the Fudan MCB lab, especially those who contributed to the development of these techniques. We thank Y. Zhen of Shanghai Genomics Inc for generating antibodies and providing detailed information. This work is supported by the 985 Program of the Chinese Ministry of Education, by the State Key Development Program of Basic Research of China (2009CB918401, 2009CB918600 and 2006CB806700), by the National High Technology Research and Development Program of China (2006AA02A308), and by grants from the Chinese National Science Foundation (30971485/C0706, 30600112 and 30871255), the Shanghai Key Project for Basic Research, China (09JC1402300, 07PJ14011 and 08JC1400900).
Footnotes
Note: Supplementary information is available via the HTML version of this article.
AUTHOR CONTRIBUTIONS K.-L.G., Y. X. and S.Z. designed the protocols; Y.L., W.Y. and S.Z. carried out the experiments; K.-L.G. and S. Z. wrote the paper.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Driessen HP, de Jong WW, Tesser GI, Bloemendal H. The mechanism of N-terminal acetylation of proteins. CRC Crit Rev Biochem. 1985;18:281–325. doi: 10.3109/10409238509086784. [DOI] [PubMed] [Google Scholar]
- 2.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 3.Phillips DM. The presence of acetyl groups of histones. Biochem J. 1963;87:258–263. doi: 10.1042/bj0870258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 6.Paolinelli R, Mendoza-Maldonado R, Cereseto A, Giacca M. Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat Struct Mol Biol. 2009;16:412–420. doi: 10.1038/nsmb.1583. [DOI] [PubMed] [Google Scholar]
- 7.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 8.Marzio G, et al. E2F family members are differentially regulated by reversible acetylation. J Biol Chem. 2000;275:10887–10892. doi: 10.1074/jbc.275.15.10887. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, et al. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11:313–324. doi: 10.1593/neo.81358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SC, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 11.Zhao S, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2006;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 13.Qiang L, Xiao H, Campos EI, Ho VC, Li G. Development of a PAN-specific, affinity-purified anti-acetylated lysine antibody for detection, identification, isolation, and intracellular localization of acetylated protein. J Immunoassay Immunochem. 2005;26:13–23. doi: 10.1081/ias-200041153. [DOI] [PubMed] [Google Scholar]
- 14.Hirschey MD, Shimazu T, Huang JY, Verdin E. Acetylation of mitochondrial proteins. Methods Enzymol. 2009;457:137–147. doi: 10.1016/S0076-6879(09)05008-3. [DOI] [PubMed] [Google Scholar]
- 15.Takemura R, et al. Increased microtubule stability and alpha tubulin acetylation in cells transfected with microtubule-associated proteins MAP1B, MAP2 or tau. J Cell Sci. 1992;103(Pt 4):953–964. doi: 10.1242/jcs.103.4.953. [DOI] [PubMed] [Google Scholar]
- 16.Gaertig J, et al. Acetylation of lysine 40 in alpha-tubulin is not essential in Tetrahymena thermophila. J Cell Biol. 1995;129:1301–1310. doi: 10.1083/jcb.129.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu B, Iwuoha EI, Smyth MR, O’Kennedy R. Effects of acetonitrile on horseradish peroxidase (HRP)-anti HRP antibody interaction. Biosens Bioelectron. 1997;12:619–625. doi: 10.1016/s0956-5663(97)00015-8. [DOI] [PubMed] [Google Scholar]