

I. Introduction
Interest in modifying the genome of animals has probably existed in some form throughout modern history, but it is only recently that techniques to achieve this experimentally have been developed. Fundamental to these techniques is the ability to culture early embryos in vitro (Reviewed by Brinster, 1972) which allowed a variety of manipulations to be performed. One of the earliest genetic manipulations of the embryo was the production of chimeric mice achieved by mixing cleavage stage embryos (Reviewed by McLaren, 1976). This technique led to the development of a method for transferring cells of the inner cell mass directly from one blastocyst to another, thereby mixing the earliest totipotent cells of the embryo proper (Gardner, 1968). Since it was known that teratocarcinoma cells arose from germ cells or the early embryo (Stevens, 1964; Sherman and Solter, 1975) and that each carcinoma cell was capable of giving rise to an individual tumor which could differentiate into many tissue types (Kleinsmith and Pierce, 1964), it seemed possible that this primitive cell might colonize the mouse blastocyst and its descendants appear in the adult. That this actually would occur was first reported in 1974, and it was shown the following year that the cells would colonize many tissues including germ cells (Brinster, 1974; Mintz and Illmensee, 1975). Employing this system it was thought possible that mutations could be made in teratocarcinoma cell lines and that these mutations would appear in progeny from the colonized mouse. Following the development of recombinant DNA techniques it was also suggested that transfected teratocarcinoma cells might carry new genes into the germ line of mice (Mintz and Cronmiller, 1981). Although these proposals are reasonable, technical difficulties have prevented their accomplishment.
A technique for modifying an animal genome that has always been conceptually popular involves nuclear transfer into the fertilized egg. The transfer of similar nuclei into many eggs would result in cloning. While this has been successful in the frog (Reviewed by Gurdon, 1977), the successful development of adult mice following transfer of nuclei into fertilized eggs has only recently been achieved (Illmensee and Hoppe, 1981; McGrath and Solter, 1983). The use of this technique to modify the genome depends on being able to produce changes in a totipotent nucleus before substituting it for the pronuclei of the fertilized egg. Undifferentiated cultured teratocarcinoma cells that have been mutated or transfected with cloned genes (as discussed above) are generally considered the best source of experimentally altered nuclei for this system. It has been proposed that teratocarcinoma cells could be grown in vitro, their genome altered, and the desired genetic modification selected for transfer into a blastocyst as a cell, or into a fertilized egg as a nucleus. In this manner, one could determine the gene expression characteristics before the nucleus or cell was transferred. However, there is no assurance that the gene will not be modified during development in an unpredictable way as sometimes occurs with genes injected directly into fertilized eggs (see later). While the above two techniques have received considerable attention, they have not been used successfully to introduce new genes into the germ line of animals. However, the availability of new embryonic cell lines that do not develop abnormal chromosomes when grown in vitro and can efficiently colonize blastocysts may facilitate the use of these two techniques (Bradley et al., 1984).
The system that has been extremely successful in introducing new genes into animals involves microinjection of a gene into the pronucleus of the fertilized egg (Fig. 1). This system arose from the complex and varied background of mammalian embryo manipulation, briefly described above, but several other experimental findings contributed to its development. First, the studies of Gurdon (1977) in which messenger RNA and DNA were shown to function in the Xenopus egg prompted similar experiments with mouse eggs (Brinster et al., 1980, 1981a). Second, experiments by McBride and Ozer (1973) indicated that genetic information could be transferred to cells by purified metaphase chromosomes. In addition, it was found that viral DNA injected into the blastocyst cavity was present in cells of the adult animal (Jaenisch and Mintz, 1974). These experiments suggested that chromosomal fragments or viral DNA might be expressed following transfer to eggs. Third, and perhaps most important, the emerging field of recombinant DNA made available reasonable quantities of purified genes that could be injected into eggs. Furthermore, it was demonstrated that these purified genes were expressed following transfection into cultured cells (Graham and van der Eb, 1973; Wigler et al., 1977).
Fig. 1.

Microinjection of new genes into a fertilized mouse egg. The upper picture shows a normal egg secured by a large holding pipette. The tip of the smaller pipette is in the male pronucleus. In the lower picture the pronucleus has been swollen by the introduction of a solution containing new genes through the small injection pipette. Egg is approximately 70 μm in diameter. The tip of the injector pipette is 1.5 μm.
II. The Metallothionein Breakthrough and Giant Mice
Between December 1980 and November 1981 six papers were published describing the results of experiments in which DNA was microinjected into fertilized mouse eggs. In five of these studies, injections were into the pronucleus, and the DNA became integrated into chromosomes (Gordon et al., 1980; E. Wagner et al., 1981; T. Wagner et al., 1981; Costantini and Lacy, 1981; Brinster et al., 1981b). In the sixth study, a lambda clone containing a retroviral sequence was injected into the cytoplasm, and it was suggested that transcription of the DNA occurred after microinjection, and a DNA copy of these transcripts integrated de novo into the mouse genome (Harbers et al., 1981). The level of expression of the introduced gene varied among the studies. In two studies, no gene expression was reported (Gordon et al., 1980; Costantini and Lacy, 1981), and in another study, low level expression was seen in one animal (E. Wagner et al., 1981). T. Wagner et al. (1981) reported that rabbit globin was found in the blood of adult animals when the rabbit β-globin gene was introduced into mice. In the experiments from our laboratories (Brinster et al., 1981b) high levels of viral thymidine kinase were found in the liver of one mouse that contained the injected gene. Following de novo viral DNA integration, Harbers et al. (1981) detected viral messenger RNA in several adult tissues. Animals into which foreign DNA has integrated are called transgenic (Gordon and Ruddle, 1981).
In the experiments described by Brinster and Palmiter, a gene composed of the mouse metallothionein I (MT) promoter/regulator fused to the herpes simplex virus (HSV) thymidine kinase (TK) structural gene was injected into mouse eggs (Fig. 2). The MT promoter was selected for construction of the fusion gene because: (1) it was a mouse DNA sequence and homology with mouse sequences might facilitate integration and expression; (2) MT was induced to high levels of expression by heavy metals and glucocorticoids in several types of cells (Durnam and Palmiter, 1981; Mayo and Palmiter, 1981), and by cadmium in mouse eggs (Brinster et al., 1982); (3) the 5′ flanking region of MT had been sequenced, and this would facilitate subsequent promoter modifications (Glanville et al., 1981). The viral thymidine kinase structural gene was selected because it provided a unique protein not present in animal cells, and a sensitive enzyme activity assay was available. The metallothionein-thymidine kinase fusion gene was designated MK. The choice of this construction was fortunate. In one of the transgenic mice from the first group of animals, thymidine kinase activity was 200 times greater in the liver of the animal following cadmium (Cd) injection than in control animals without the gene, and more than 95 % of this activity was blocked by antibody specific for the herpes enzyme. Subsequent studies indicated that the fusion gene was expressed in 60–70% of the mice carrying it (Palmiter et al., 1982a). Although MK gene expression was induced by cadmium, dexamethasone (a potent glucocorticoid) was without effect. The absence of induction by dexamethasone was also found when the gene was transfected into tissue culture cells (Mayo et al., 1982). Several studies demonstrated that integrated genes were transmitted through the germ line (Costantini and Lacy, 1981; T. Wagner et al., 1981; Stewart et al., 1982) and that progeny also expressed genes that were transmitted (Palmiter et al., 1982a). Success with the MK fusion gene clearly indicated the feasibility of a variety of experiments designed to study the expression of foreign genes in mice. The technique provided animals containing and expressing new genes that could be analyzed for changes in many biological functions. The basic strategy for making and using transgenic mice is illustrated in Figure 3.
Fig. 2.

Structure of plasmid pMK. (A) Plasmid pMT-TK contains a 3.8 kb genomic Eco R1 fragment that includes the MT-1 gene inserted into the Eco R1 site of pBR322 and the 3.5 kb Barn H1 fragment of the herpes simplex virus type I containing the thymidine kinase (TK) gene inserted into the Barn H1 site of pBR322. The two genes are in the same transcriptional orientation, as shown by the arrows. (B) The fusion plasmid, pMK, was created by digestion of pMT-TK with Bgl II endonuclease followed by ligation with T4 DNA ligase to join the 5′ region of MT-1 to the TK structural gene. The MT mRNA coding sequences are indicated by the large solid boxes, and the TK mRNA coding sequences by the large open boxes. Nontranscribed and intron regions of the two genes are shown by a single line. The pBR322 sequences are indicated by the narrow solid boxes. Restriction sites relevant to the construction are indicated. Also shown are the locations of the TATAA promoter sequences and ATG translation start codons for the two genes (Brinster et al., 1981b; Palmiter et al., 1982a).
Fig. 3.

Diagram of the technique used in making and studying transgenic mice. For detailed procedures see Brinster et al. (1985).
An obvious and exciting idea was to try and correct a genetic defect in mice by gene injection. The dwarf little (lit) mouse served as an ideal candidate because its small stature was known to reflect a deficiency of growth hormone (Eicher and Beamer, 1976; Beamer and Eicher, 1976). On the basis of earlier success with the MK construction, a metallothionein growth hormone (GH) fusion gene seemed a promising approach, and a fusion gene employing the rat growth hormone (rGH) gene was constructed, in a manner similar to that shown in Figure 2 and Figure 4 for other fusion genes. Because breeding sufficient homozygous (lit/lit) mice to provide eggs would take 6 to 12 months, a pilot experiment was performed. Our earlier observations indicated that linear DNA molecules integrated better than supercoiled DNA. These and other findings relative to the microinjection technique have recently been reviewed (Brinster et al., 1985). On the basis of these observations, we injected linear MTrGH molecules into the pronuclei of fertilized eggs collected from C57 × SJL F1 hybrid females mated to C57 × SJL F1 males. We anticipated that growth hormone would be produced in these mice and hoped that it would have an effect on the growth of the animals. In the first experiment, 21 mice were produced, seven of the mice contained the gene and six grew faster and became larger than control litter mates (Palmiter et al., 1982b). The dramatic difference in size is illustrated in Figure 5. The mice with the gene grew up to twice as large as controls. The visual impact of these experimental results was considerable, and they graphically dramatized the potential of the gene injection methodology.
Fig. 4.
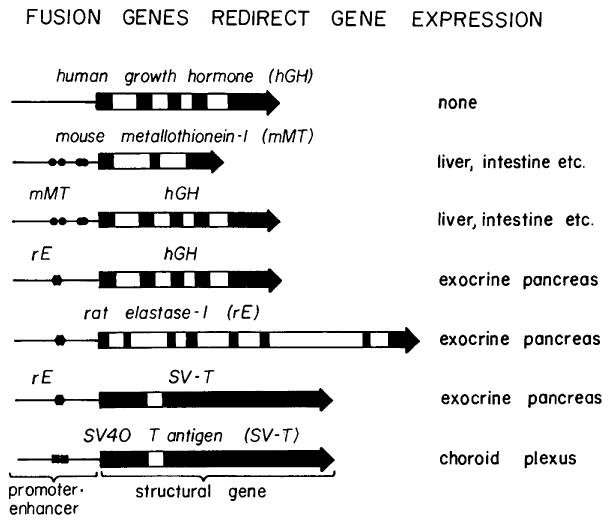
Fusion genes redirect gene expression in transgenic mice. The natural genes: human growth hormone, mouse metallothionein-I, rat elastase-1, and SV40 T antigen were expressed in the tissues indicated to the right. When their controlling elements (promoter/enhancer) were fused to different structural genes, then the expression of the structural gene was redirected as dictated by the promoter/enhancer specificity. The solid boxes represent exons, the open boxes introns, the line represents the 5′ flanking sequences including the promoter. The solid symbols in the promoter represent the enhancer elements; there are four functional metal regulatory elements in the mouse metallothionein promoter (circles), probably only one cell-specific enhancer element in the elastase promoter (hexagon), and two 72 bp repeats of the SV40 enhancer (squares). All fusions were made between the cap site (+ 1) and the initiation codon of the respective genes. The genes are not drawn to scale.
Fig. 5.

Expression of a microinjected gene in mice resulted in accelerated growth rate and increased size. The gene consisted of the mouse metallothionein promoter/regulator fused to the rat growth hormone structural gene. These mice were ten weeks old when the picture was taken. The large mouse weighed 44 grams. The small mouse was a litter mate control and weighed 30 grams. For details see Palmiter et al. (1982b).
In subsequent experiments, we fused the human growth hormone (hGH) structural gene to the MT promoter to facilitate detection of mRNA and GH (Fig. 4). Mice that carried and expressed the MThGH construct had high levels of growth hormone mRNA in several tissues (e.g. liver, kidney, and intestine) and human growth hormone was found in the serum (Palmiter et al., 1983). The hormone levels were 64 μg/ml in the blood of some animals, but levels as low as 100 ng/ml were sufficient to allow maximum growth. Although GH mRNA could be detected in the liver of 13-day-old fetuses and presumably growth hormone levels in serum were also elevated, increased growth rate commenced at about three weeks and plateaued at twelve weeks of age. In more recent experiments a similar construction employing the bovine growth hormone (bGH) gene has been shown to function in the same way as the rGH and hGH genes when introduced into mice (Hammer et al., 1985a). In addition, the human metallothionein II-A promoter/regulator region, which has been shown to contain a glucocorticoid receptor binding sequence (Karin et al., 1984), resulted in the expression of the hGH gene in transgenic mice (Hammer et al., 1985a). Induction of this fusion gene in animals occurred following either heavy metal or glucocorticoid stimulation. Thus, the general usefulness of the MT promoter/regulator region to obtain expression of structural genes seemed clear. Likewise the effectiveness of GH from several species in producing growth in mice was evident.
Because the lit/lit mice were difficult to breed, results from these studies developed slowly. However, it was demonstrated that introduction of the MTrGH gene into lit/lit mice resulted in accelerated growth, and the animals actually grew larger than normal mice (Hammer et al., 1984). An interesting aspect of these experiments was the improved fertility in lit/lit males that expressed the gene. Normally these mutant males have low fertility. However, the fertility of lit/lit females was impaired. In fact, female mice that expressed any of the MT growth hormone fusion genes had lowered fertility (Hammer et al., 1984). Males that express MT growth hormone genes, in contrast, exhibited essentially normal fertility.
Growth in animals results from a complex cascade of hormones acting at several levels (Fig. 6). Several studies indicated that introduction of the normal growth hormone structural gene did not increase growth rates (E. Wagner et al., 1983; Hammer et al., 1984). However, it was clear from our experiments with the metallothionein GH constructs that elevated levels of serum growth hormone did result in accelerated growth rates. In these animals the GH was produced primarily in tissues other than the pituitary (e.g. liver, kidney, intestines), and blood levels were not under normal endogenous regulatory controls. When the gene for human growth homone releasing factor (hGRF) was isolated (Mayo et al., 1985), it became possible to determine if enhanced growth could be obtained by stimulating production of endogenous GH from the pituitary. A fusion gene (MThGRF) was constructed and introduced into animals. The gene was expressed in the liver, kidney, and intestine as expected. Both GRF and GH levels were elevated in the serum of the animals that expressed the gene (Hammer et al., 1985b). The animals were clearly producing more endogenous growth hormone, and the pituitaries were up to eight times normal size. However, the maximum size of the animals appeared slightly less than with MT growth hormone genes. Perhaps because of the slightly lower stimulation of growth or because the endogenous GH may still be released in a pulsatile manner, the females expressing this gene had essentially normal fertility. Males also had normal fertility.
Fig. 6.
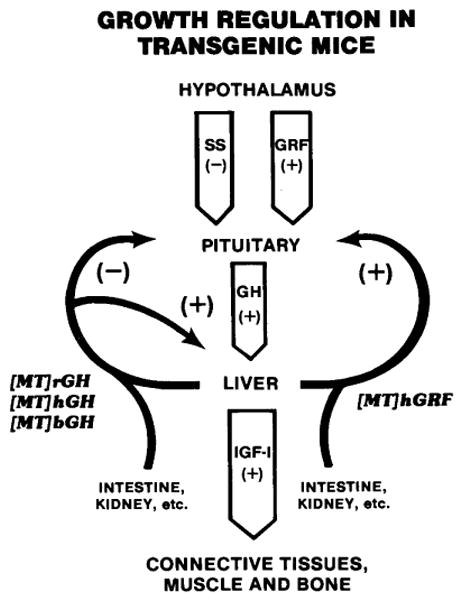
Growth hormone (GH) cascade. The hypothalamus produces the neuropeptides somatostatin (SS) and growth hormone releasing factor (GRF) that inhibit and stimulate, respectively, the release of GH. Growth hormone stimulates hepatocytes to produce insulinlike growth factor I (IGF-I) which acts on peripheral tissues to stimulate growth. Growth hormone also may stimulate peripheral tissues directly. Growth hormone and IGF-I are believed to influence the cascade by a negative feedback effect on the hypothalamus and pituitary. Metallothionein fusion genes are expressed in the liver and several other tissues. Therefore, their products are not controlled by the normal endogenous regulatory mechanism. [MT] indicates the MT promoter was responsible for production of the hormone. The origin of the hormone gene was rat (r), human (h), or bovine (b). (+) and (−) indicate positive and negative influences, respectively.
An unusual aspect of the expression of the MT growth hormone fusion genes was the appearance of growth hormone in specific neurons of the central nervous system (Swanson et al., 1985). The endogenous growth hormone gene is expressed only in somatotrophs of the anterior pituitary gland, and the metallothionein gene is expressed in astrocytes of the brain. However, immunohistochemical studies demonstrated expression of MTrGH or MThGH fusion genes in several highly localized regions of the brain, including the hypothalamus (especially the paraventricular and supraoptic nuclei), as well as areas in the neocortex, hippocampus, and olfactory cortex. These findings suggest that elements in the metallothionein and growth hormone sequences may interact to specify a location of expression unique and different from either of the original genes. This raises the possibility that selected regions of different genes may be combined to target gene expression in a manner not available within the existing genes of the animal. Furthermore, it is possible that similar phenomena act normally in vivo and that cell-specific expression is frequently influenced by more than a single DNA sequence. Such a multiplicity of regulatory elements would enhance the flexibility of gene expression. It is conceivable that the use of regulatory sequences by various genes in different formats may have evolved in a manner similar to the use of similar exon coding sequences to specify parts of different genes (Gilbert, 1985).
The metallothionein promoter has proven to be exceptionally useful in obtaining the expression of other genes. For instance, the human hypoxanthine phosphoribosyltransferase (hHPRT) cDNA has been inserted into an MT expression vector. Normal HPRT genes are constitutively expressed at low levels in most tissues and at higher levels in the CNS. Transgenic mice with the MThHPRT construct contained high levels of mRNA and enzyme in brain tissue (Stout et al., 1985). In contrast, the MTGH constructs were expressed at low levels in total brain. These results suggested that sequences within the HPRT influenced CNS expression by increased mRNA synthesis or stability.
Although transgenic mice have been used primarily to study gene regulation, they also make particularly good models to examine other physiological parameters. In a recent experiment, posttranslational processing was studied in transgenic mice that produced the precursor of somatostatin from a cDNA inserted into an MT expression vector (Low et al., 1985). Large quantities of the prohormone were made in the liver but processing was incorrect. Surprisingly, the concentration of the hormone was highest in the anterior pituitary where levels were 100 times greater than liver. In the pituitary correct processing to somatostatin-28 and somatostatin-14 occurred. Despite the expression and proper processing in some tissues and the high circulating levels of immunoreactive protein, no overt physiological effects were observed. For instance, the growth rates of the mice expressing the gene were normal.
The human hepatitis B virus (HBV) is widespread throughout the world and results in an increased incidence of liver cancer among carriers of the virus. In order to study the effects of viral proteins in the animal, the MT promoter has been fused to a DNA sequence that codes for the surface antigen of the virus (Chisari et al., 1985). The fusion gene was expressed in several tissues and high levels of HBV surface antigen were found in both cells and blood. The normal promoter of the HBV surface protein gene also resulted in expression of the gene in transgenic animals (Babinet et al., 1985; Chisari et al., 1985). The expression of individual genes from the virus in animals should provide a method to study the consequences of high levels of viral proteins on the etiology of the cancer.
III. Tissue Specificity and Developmental Control
While integration of the injected DNA into a chromosome of the animal is certainly an essential prerequisite, expression of the gene is the essence of the methodology. Only if expression is obtained will the wide range of questions dealing with developmental programming of gene expression be approachable with this system. Ideally the expression should mimic precisely the normal in vivo responses.
Certainly the early studies with metallothionein fusion genes were encouraging. In the first report, expression of the MK gene was found, and the highest levels were in the liver, an organ in which metallothionein is normally expressed at high levels (Brinster et al., 1981b). A further indication that expression of the fusion gene was similar to the endogenous metallothionein gene was its induction by cadmium, a normal transcriptional regulator of the MT gene (Palmiter et al., 1982a). In addition, the pattern of expression of the MThGH gene in a variety of tissues paralleled to a significant degree the pattern of the endogenous metallothionein gene (Palmiter et al., 1983).
The possibility of tissue-specific expression was also suggested by studies using a chicken transferrin gene that was regulated by its normal promoter region. Significant expression of this gene was found in transgenic mice, indicating that sequences specifying activity had been conserved between the mouse and chicken (McKnight et al., 1983). Furthermore, chicken transferrin mRNA levels were highest in the liver, the tissue that normally produces the greatest amounts of the protein. In more recent studies, it has been possible to demonstrate that estrogen treatment significantly increases the level of mRNA in the livers of mice expressing this gene (Hammer et al., 1986). These results and those with the human metallothionein-growth hormone fusion gene (see above) are the first examples of injected genes being regulated by steroids in transgenic animals.
The first clear demonstration of tissue-specific activity of a new gene in transgenic animals was provided by the kappa light chain immunoglobulin (Ig) gene (Brinster et al., 1983; Storb et al., 1984). These experiments will be discussed later.
The second demonstration of tissue-specific expression was obtained with the rat elastase gene (Swift et al., 1984a). The exocrine cells of the pancreas are highly specialized and produce a group of digestive enzymes including a set of serine proteases, e.g. trypsin, chymotrypsin, and elastase. Expression of the genes for these proteins is tightly controlled, since they are transcribed at high levels in acinar cells but only at extremely low levels or not at all in other tissues. The rat elastase 1 gene is one of these serine protease genes and has been well characterized (Swift et al., 1984b). To study the function of this gene in transgenic mice, a construct containing 7 kilobases (kb) of 5′ flanking sequence, 11 kb of exon and intron sequence, and 5 kb of 3′ flanking sequence was microinjected into fertilized eggs (Swift et al., 1984a). A total of five mice containing the gene were produced, and each animal expressed the rat elastase gene at high levels in the pancreas and at extremely low levels or not at all in other tissues. Levels of mRNA were approximately 10,000 times higher in the pancreas than in other tissues. The tissue-specific expression of the elastase gene in the transgenic mice mimicked the expression in the rat.
To identify more precisely the DNA elements involved in pancreas-specific expression, the 5′ flanking region of the elastase gene was fused to the human growth hormone structural gene (Fig. 4), and the gene was introduced into mice (Ornitz et al., 1985). When the tissues of animals containing the gene were analyzed, it was found that growth hormone mRNA was present at high levels in the pancreas but not in other tissues. Immunohistochemistry indicated that growth hormone was present in acinar cells of the exocrine pancreas but not in the islets of the endocrine pancreas (Fig. 7). The animals did not grow larger than normal, indicating that the growth hormone did not enter the blood. Immunoreactive GH was found in pancreatic ducts leading to the intestine, a location one would expect for the elastase enzyme. Deletions of the 5′ flanking region indicated that tissue specificity could be maintained with only 200 base pairs (bp) of the 5′ flanking sequence including the promoter. This area contains a 21 bp consensus sequence located between −250 and −90 bp that is common to the chymotrypsin gene, two trypsin genes, and two elastase genes (Swift et al., 1984b). This sequence may be important in providing the tissue-specific activity of these genes.
Fig. 7.
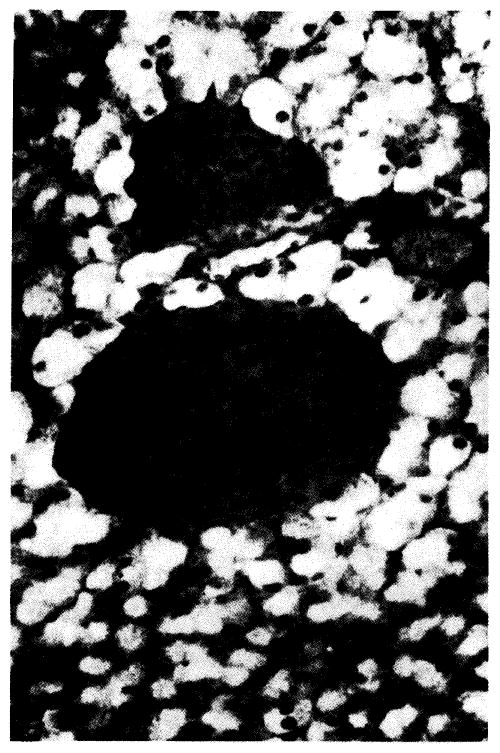
Immunofluorescent localization of human growth hormone in pancreas from a transgenic mouse containing a rat elastase-human growth hormone fusion gene (see Fig. 4). The exocrine acinar cells fluoresce brightly while the endocrine cells (two islets) are dark. For details see Ornitz et al. (1985).
It is not known whether the same DNA sequences that convey tissue specificity to a gene are also involved in the proper developmental timing of expression but it seems probable that they could be the same or, at least, overlap. The mouse α-fetoprotein (AFP) gene is a good candidate in which to examine both tissue-specific and developmental regulation. The gene is activated during fetal development and is expressed in extraembryonic yolk sac, fetal liver, and fetal gut. Following birth, transcription of the gene undergoes a 10,000-fold decrease during the first week of life. The AFP gene is located 3′ to the albumin gene, and although both genes have evolved from a common ancestral gene, their developmental regulation is different. Thus, the activating DNA sequences of AFP and albumin could yield information regarding the differences between signals for tissue specificity and developmental timing of gene expression. A minigene consisting of AFP exons 1, 2, 3, 14, and 15 was constructed and various lengths of 5′ flanking region were fused to the minigene. Deletion of the 5′ sequences to 7 kb allowed tissue-specific expression of the gene with correct developmental timing (Krumlauf et al., 1985). Thus, the gene was expressed in the correct tissues before birth and transcription declined following birth in parallel with the endogenous gene. However, the level of expression of the gene was never greater than 25% of the endogenous gene. Recent experiments indicate that levels of expression approximately equivalent to the endogenous gene can be obtained if the prokaryotic plasmid sequences are removed from the construct before injection into the egg (see later). It should now be possible to identify the sequences responsible for the developmental regulation of AFP and albumin and to ascertain how the regulatory sequences responsible for the activity of these genes have diverged during evolution.
The rat skeletal muscle light chain 2 gene is a member of a group of genes that is activated during terminal differentiation of muscle cells. Recently this gene was introduced into mice, and two of three animals that contained the gene expressed it in skeletal muscle cells. In one mouse a level similar to the endogenous gene was obtained, and in the other mouse the level was about 1% of the endogenous gene (Shani, 1985).
Genes that code for the globins are particularly interesting and important. The genes are developmentally regulated with different members of the family expressed in a sequential manner. Although a great deal is known about these genes, obtaining expression of them following introduction into transgenic mice has been difficult (E. Wagner et al., 1981; Costantini and Lacy, 1981; Lacy et al., 1983). Recently, Chada and colleagues (1985) obtained erythroid-specific expression of a fusion gene composed of the mouse 5′ and human 3′ β-globin gene. However, the highest level of expression was 2% of the endogenous β-globin gene. Subsequently, they demonstrated that the activation of the introduced gene and the endogenous gene occurred at the same time during fetal development (Magram et al., 1985).
We also have been studying the expression of the intact human β-globin gene and discovered that the gene did indeed function in a tissue specific manner in transgenic mice (Townes et al., 1985a). The levels of mRNA from the injected gene varied but in some cases were similar to those from the endogenous mouse β-globin gene. In an attempt to determine the region involved in tissue-specific expression of the gene, deletions of the 5′ region were made, and as little as 48 bp of 5′ sequence still allowed expression of the gene. Apparently a region within or 3′ to the gene was capable of directing significant expression of the gene in the absence of most of the 5′ region, in agreement with results obtained by others following transfection into Friend erythroleukemia cells (Wright et al., 1984; Charnay et al., 1984). A line of mice was established that express the human β-globin. In these mice the human β-globin was about 10–20% of the level of the mouse β-globin, but there were no abnormalities in standard hematological parameters. The production of transgenic mice capable of expressing foreign globin genes will provide a powerful tool to examine natural globin mutants as well as experimentally engineered globin genes to test various aspects of globin gene and protein function.
In another experiment, a construct containing both the metallothionein-human growth hormone fusion gene and the human β-globin (hβG) gene in a head-to-head configuration was introduced into mice. The β-globin gene was expressed, however, with a tissue specificity similar to that expected for the MThGH gene (Townes et al., 1985b). Thus, hβG message levels were high in intestine, but there was no MThGH or hβG mRNA in reticulocytes. In addition, there was evidence that the hβG gene inhibited expression of the MThGH gene, particularly in the liver. Thus, each gene influenced the expression of the other. These effects may be related to the interactions of enhancerlike elements that lie within the two genes. Separation of such elements may be one of the functions of the wide spacing of genes in chromosomes.
In the process of studying human β-globin gene expression, we found that plasmid vector sequences had a severe inhibitory effect on both the frequency and level of expression of the gene (Townes et al., 1985a). Similar inhibitory effects of vector sequences have been observed with MThGH genes and AFP genes (Hammer et al., 1985a). The location and mechanism of action of the poison sequences are unknown. Expression of some genes are less sensitive to the effects of plasmid DNA.
IV. The Immune System - A Complex Challenge
The immune system of the body is surpassed in complexity only by the nervous system. In the simplest terms there are two parts to the immune system: one involving humoral responses which consists of B-cells and the antibodies they produce; and one involving cell-mediated responses which consists of T-cells. Both the humoral and cell-mediated responses are influenced by proteins encoded by the major histocompatibility complex (MHC) genes. The introduction of genes involved in immune response into transgenic mice provides a unique approach to understanding not only the regulation of the individual genes but also the intricate relationships within the system.
Two identical light and two identical heavy chain immunoglobulin poly-peptides combine in a tetramer to form a functional antibody with unique antigen specificity. The genes coding for the light and heavy chain must undergo rearrangement before they can be expressed as a functional mRNA which will produce the appropriate protein. The first immunoglobulin gene that was introduced into animals was a rearranged immunoglobulin light chain kappa (k) gene from the myeloma MOPC-21. In this experiment all six of the mice that carried the new gene expressed it at high levels in B-cells but not in other tissues (Brinster et al., 1983; Storb et al., 1984). The level of kappa mRNA from the injected genes was 20–40% of total kappa mRNA in the spleens of the transgenic mice, and the mRNA was of correct length. Transcripts from the injected gene directed synthesis of a normal secreted light chain which comprised up to 50% of the light chain in these animals. In addition, evidence suggested that the new kappa chain gene was developmentally regulated since pre-B-cells existed that expressed heavy chain but did not express either endogenous or the new kappa chain gene (Storb et al., 1985). Furthermore, these experiments suggested that transcription activation signals for heavy chain genes preceded and were different from those for kappa genes. A functionally rearranged heavy chain immunoglobulin gene introduced into mice was likewise expressed in a tissue-specific manner (Grosschedl et al., 1984). The heavy chain from this injected gene, when combined with λ1 light chains, shows a specificity for binding the hapten nitrophenyl. Therefore, a functional antibody was formed. In a recent series of experiments the rearranged genes coding for both the kappa and heavy chain of an antibody with specificity against trinitrophenyl were injected simultaneously (Rusconi and Köhler, 1985). A mosaic mouse was obtained that contained both genes, and studies on the progeny demonstrated tissue-specific expression of both genes. Furthermore, a functional antibody was produced from the two injected genes. From the studies on microinjected immunoglobulin genes in transgenic mice, it seems clear that they are expressed in a tissue-specific manner and that functional antibody molecules are produced from the genes.
Mice that contain microinjected immunoglobulin genes have been used to help resolve questions about the regulation of the immune system. Accurate expression of the immunoglobulin genes requires a rearrangement of DNA sequences from several areas of the chromosome into a functional array. When a rearrangement becomes functional and a heavy or light chain is produced, no further rearrangement appears to occur of heavy or light chain genes. Thus, within an individual cell only one functional rearrangement of a heavy and light chain gene has been observed. This is known as allelic exclusion, and the mechanisms that control it are not understood. By constructing a series of hybridomas from transgenic mice with the injected k gene, Ritchie et al. (1984) were able to demonstrate that production of a complete immunoglobulin molecule appeared to turn off the endogenous kappa gene rearrangement. In some cases, for reasons that are unknown, the kappa chain from the new gene was not made. In those cases, the endogenous kappa chain genes were rearranged. These findings suggested that the production of a complete immunoglobulin molecule, not transcription or translation of the gene alone, terminated the rearrangement of light chain genes. The work of Rusconi and Köhler (1985) also suggested that there was a negative feedback inhibition of heavy and light chain products from the injected genes on the rearrangement of the endogenous immunoglobulin heavy chain. However, they did not find evidence for an effect of the injected genes on endogenous light chain gene rearrangement. This may be because an excess of transgenic heavy chain mRNA and protein occurred in the same cell, which mimicked the situation in normal B-cell development where the heavy chain is expressed first. The level of expression and production of complete immunoglobulin antibody may have to reach a critical level in order to block light chain gene rearrangement.
An interesting observation from the animals containing a new heavy chain gene was that transcription of the gene was almost as high in mature T-cells as in B-cells (Grosschedl et al., 1984). This suggests that all of the transacting factors necessary for high level expression of the heavy chain gene are present in T-cells and that rearrangement may be the mechanism that restricts heavy chain expression to B-cells.
One of the methods by which antibody diversity is achieved is somatic mutation of the variable region of the Ig genes. Two models, rearrangement and accumulation, have been proposed to account for these mutations. In the rearrangement model, it is suggested that the mutations are generated during the process of Ig gene rearrangement. In the alternative model, the mutations are thought to accumulate progressively as the specific B-cell clones age. Recently, transgenic mice (Brinster et al., 1983; Storb et al., 1984) have been used to examine this question. Since the introduced kappa gene has already been rearranged, no further rearrangements take place in the animal. A cDNA library from the spleens of these mice was screened, and clones of transcripts from the transgenic kappa gene were sequenced (O’Brien et al., unpublished observations). From 14 clones analyzed, 13 lacked mutations; the other clone was from an endogenous gene. These results indicate that no new mutations occurred in the microinjected gene.
The immune response in intact animals is controlled by several sets of genes. The immunoglobulin genes (above) give rise to the antibodies, while another set of genes, the immune response genes, are involved in interactions between macrophages, T-cells, and B-cells and regulate immune response to many different antigens. The immune response genes (Ir) belong to the Class II major histocompatibility complex (MHC), and they code for glycoproteins composed of two chains (α and β) that integrate into the cell membrane to form a functional dimer. In some mouse strains a deletion in the 5′ region of the Eα-chain gene results in loss of expression of a particular Class II product, I-E, since a complete α plus β dimer cannot form (Hyldig-Nielsen et al., 1983). Mouse strains of the b or s haplotype do not express I-E antigen because of this deletion. Since proteins from different haplotypes can combine, a functional Eα gene of the d haplotype was introduced into the germ line of b × s mice. The gene was expressed in a tissue-specific manner, and the I-Eαd antigen mRNA was found in the spleen (Pinkert et al., 1985; Yamamura et al., 1985). Furthermore, expression of the injected I-Eα gene was induced in macrophages by gamma interferon (Pinkert et al., 1985). Similar experiments have been performed with the I-Eαk gene (LeMeur et al., 1985). The I-Eαd mRNA resulted in a protein that appeared on the cell membrane with the endogenous Eβ protein. Of critical importance was that the Eαd gene product on the cell membrane functioned as an alloantigen, in the development of self-tolerance, and as a restriction element for the helper/inducer subset of T lymphocytes (Pinkert et al., 1985). Thus both normal expression and regulation of the gene occurred, and the product was functional. In this experiment we effectively corrected a genetic defect in a manner that allowed the mice to express functional I-E protein on the cell surface.
An intriguing finding in this study with the Eαd gene transfer was that male mice did not transmit the gene. One founder male was infertile and two founder males, although fertile, failed to transmit the gene to progeny. A founder female mouse transmitted the gene but her sons did not. Thus four founder animals, probably representing four different chromosomal integration sites, could not transmit the gene through the male germ line. The testis and epididymis of the infertile male contained only a few abnormal sperm, a condition also seen in male mice carrying several genes from the T locus that interfere with fertility (Sherman and Wudl, 1977). Further study of the transmission distortion in these mice may identify a relationship between MHC gene expression and male fertility.
Class I major histocompatability complex genes have also been introduced into mice. The Class I genes are involved in T-cell immune surveillance and are referred to as the transplantation antigens, since they are involved in graft rejection. A Class I gene from a pig was microinjected into eggs (Frels et al., 1985). One of the mice contained the pig gene, and it was expressed on the cell surface in the parent as well as in progeny. The level of expression of the pig gene did not appear to be correlated with expression of a similar endogenous Class I gene. However, only one line of animals was involved, and a larger number of lines representing different integrations might reveal a greater correlation. Interestingly, skin from this line of animals was rejected by normal C57Bl/10 animals, the strain from which the injected eggs were obtained. Clearly sufficient antigen was expressed on the cell surface to mediate this T-cell response.
V. The Cancer Riddle and 0nc Gene Models
In early 1981, when it was clear that the metallothionein-thymidine kinase fusion gene was expressed at significant levels in tissues of the mouse, we felt that several types of experiments offered promising opportunities for investigating classic problems in biology by this unique approach. One of these experimental directions was to introduce onc genes into mice to study their effect. Initially we chose the transforming gene called src from the Rous sarcoma virus, because it was the best characterized of available oncogenes. Since the MT promoter/regulator had worked well before, we fused it to the src structural gene and introduced the fused gene into mice. Although this fusion gene is capable of transforming cells in culture, transgenic mice carrying this gene remained healthy, although two expressed low levels of src mRNA in several tissues late in life. Results with the viral Harvey ras gene in a similar construction proved no better.
In another experiment, we placed the simian virus 40 (SV40) early region (Fig. 4), including the two 72 bp enhancers, in a head-to-head configuration with the MK transcription unit (Fig. 2) in an attempt to increase MK mRNA levels in transgenic mice. This construction did not affect TK activity in eggs as measured by a transient expression assay (see Brinster et al., 1982; Stuart et al., 1984; Chen et al., 1986). However, it decreased the percentage of transgenic animals expressing TK in liver from approximately 70% (Palmiter et al., 1982a) to less than 10% (Brinster et al., 1984). Surprisingly, three of 15 animals carrying the gene died before 3 months, and all were dead by 5 months of age. Because we did not expect pathological changes, it was difficult to explain the deaths. However, one of the animals had a distinctly bulged cranium and a tumor of the choroid plexus was found in the brain. To extend these findings, more transgenic mice were made in which the MThGH gene (Fig. 4) replaced the MK gene. These animals developed similar pathology to the first group, although some grew larger because of the MThGH gene. The primary pathology was again choroid plexus papilloma or carcinoma, but some animals also had thymic hyperplasia and kidney pathology. By examining both normal and tumor tissue it was possible to demonstrate that T-antigen mRNA and protein were very low or absent in normal tissue but were clearly present in tumor tissue or cell lines grown from tumors (Brinster et al., 1984). The SV40 transforming gene(s), although present in all tissues, were apparently activated primarily in choroid plexus tumor cells. It seemed probable that the early region genes were inactivated following injection into the egg and that an infrequent event activated the gene in a few cells during later development. Cell lines were readily established from tumors, and most other tissues in the body gave rise to transformed cell lines when placed in culture. Mice that contained these genes were used to establish lines of animals in which all of the progeny that inherited the foreign DNA developed tumors and died with a characteristic pattern of pathology. Small et al. (1985) have also reported the development of a choroid plexus tumor in a mouse that integrated a construct which contained the SV40 early region.
In order to determine which regions of the original constructs were responsible for the pathology, several modifications were made and introduced into mice (Palmiter et al., 1985). From these studies it was clear that the SV40 early region, without the companion MK or MThGH construct, was sufficient for the development of choroid plexus tumors (Fig. 4). The SV40 early region contains coding sequences for two proteins, large T-antigen and small t-antigen. To distinguish between the effects of these two, we tested two internal deletions, one encodes only an intact large T-antigen and the other enclodes only an intact small t-antigen. Animals containing the intact large T-antigen gene died with choroid plexus tumors, but those with only the intact small t-antigen gene had little or no unusual pathology. We also examined the effect of removing the SV40 72 bp enhancer from the original construct which contained both the SV40 early region and the MThGH fusion gene. This modification produced a major change in pathology. Choroid plexus tumors did not develop, indicating that the enhancer region was essential for targeting pathology to this area. However, the animals developed other pathology consisting of peripheral neuropathy, hepatocellular carcinoma, and insulinoma (Messing et al., 1985). The peripheral neuropathy was associated with hind limb paralysis, and microscopic examination of the peripheral nerves demonstrated normal axons but the Schwann cells were unable to form or maintain compact myelin. The simplest explanation of the enhancer deletion experiments, in which the MThGH gene was retained, was that the SV40 early region then came under the influence of the MT regulatory sequences which targeted the effects to peripheral nerves, hepatocytes, and β-cells.
Success with these experiments suggested the possibility of targeting the SV40 T-antigen transforming capability to other locations in the body. Previous experiments had demonstrated the highly selective activity of the elastase promoter region (see above); therefore, these sequences were fused to the SV40 early region (lacking enhancer and promoter), and this construct was introduced into mice (Fig. 4). The animals that carried this gene (El SV) all developed tumors of the pancreas between 3 and 7 months of age (Brinster and Palmiter, 1985; Ornitz et al., 1985). These tumors grew to an enormous size, in some animals they were 25–50% of body weight (Fig. 8). The tumors rarely metastasize. Other tissues of the body did not develop tumors and SV40 T-antigen could be demonstrated only in the acinar cells of the pancreas (Ornitz et al., 1985). Immunoreactive T-antigen was rarely seen in acinar cells of the pancreas from 4-day-old mice, and only isolated cells or small foci in the pancreas were reactive at 19 days of age. However, in fully developed tumors, all the cells contained high levels of T-antigen (Ornitz et al., 1985). Again it appeared that there was activation in a few cells followed by proliferation of these cells into a tumor.
Fig. 8.
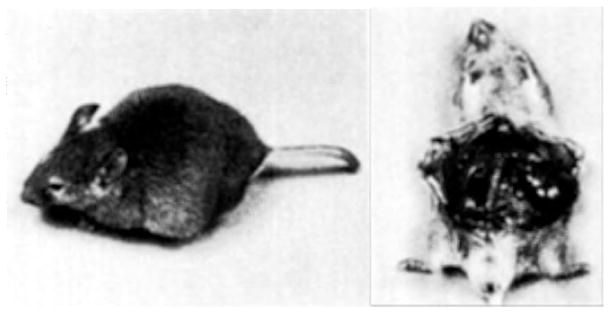
Transgenic mouse containing a rat elastase-simian virus 40 T-antigen fusion gene (see Fig. 4). The left picture shows the large abdominal swelling seen in advanced stages of tumor development. The right picture shows two large masses (arrows) which were found on histological examination to be adenocarcinomas of the exocrine pancreas.
In a similar fashion Hanahan (1985) fused the promoter region from the insulin gene to the early region of SV40 and introduced this construct into mice. He found immunoreactive T-antigen in the β-cells of the pancreatic islets that normally express the insulin gene. All of the islet cells expressed the T-antigen gene, which induced their proliferation. Oncogenic transformation occurred later in only a few of the islets, indicating that additional events were probably necessary for tumors to form. This sequence of events was different from that observed with the El SV construct where acinar cells rarely expressed the gene in early life (Ornitz et al., 1985).
Experiments in which the elastase and insulin promoters direct the oncogenic properties of the SV40 early region clearly indicate the possibility of targeting a variety of onc genes to selected tissues of the body. It should now be possible to examine the effect of the same oncogene acting in different cellular environments, or different oncogenes acting in the same cellular environments. Progress has been made in this direction by fusing the elastase promoter region to the normal human EC ras and the mutant EJ ras gene (Capon et al., 1983). Mice that contained the elastase promoter region fused to the EJ ras construct developed pancreatic tumors near the time of birth (unpublished observation), which was only about a week after the elastase gene is activated (Rutter et al., 1972) and was much earlier than the age when tumors develop from the El SV gene. Mice with the elastase promoter region fused to the normal EC ras construct have not shown abnormalities to date (9 months of age). Although El ras and El SV mice both developed tumors of the pancreas, the kinetics of tumor development were much different. The question is raised as to whether the difference is in gene activation, gene products, or secondary events necessary for tumor formation. Clearly the transgenic system will provide a unique opportunity to dissect the mechanisms involved in oncogenesis.
Introduction of various constructs including the c-myc gene has also resulted in the production of tumors in transgenic animals. In one series of experiments, the mouse mammary tumor virus (MMTV) promoter was fused to the c-myc structural gene (Stewart et al., 1984). A total of 13 mice with this fusion gene were made. In two lines of these mice, mammary adenocarcinomas developed after at least one pregnancy. Although expression of the MMTV-myc fusion gene occurred in a variety of tissues, including the salivary glands and normal mammary gland, tumors only developed randomly in individual mammary glands. The implication was that a second event, other than MMTV-myc activity, was required for tumorigenesis and that the environment of the hormonally activated mammary gland was essential for tumor formation from this construct. We have introduced a c-myc gene into mice that was isolated from a mouse plasma-cytoma and has the immunoglobulin heavy chain enhancer inserted 5′ to the first exon (Corcoran et al., 1985). Mice that contained this construct developed enlarged lymph nodes throughout the body and died at 2 to 6 months of age (Fig. 9). The pathology was inherited and was of B-cell origin (Adams et al., 1985).
Fig. 9.
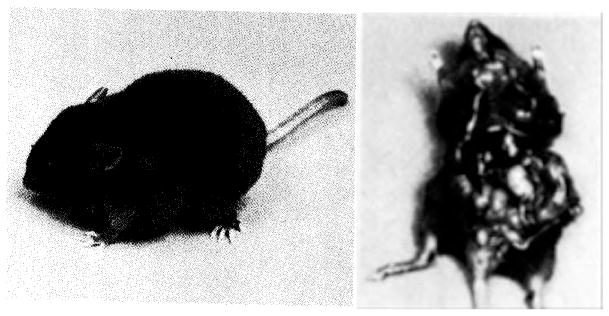
Transgenic mouse containing a c-myc gene isolated from a mouse plasmacytoma and reintroduced into the germ line by microinjection. In left picture note the greatly enlarged axillary lymph nodes. Right picture demonstrates that most or all lymph nodes (arrows) are enlarged. The tumor phenotype is inherited.
VI. Large Animal Species
Among mammals, the mouse is clearly the experimental animal of choice for most research involving gene regulation and basic questions in biology. However, transgenic animals in other species will be of considerable value: (1) to determine if common mechanisms are involved in gene regulation, (2) to study the physiologic effect of similar genes among species, and (3) to provide greater quantities of tissue for biochemical assays on cell functions, such as posttranslational processing of proteins. Furthermore, introduction of new genes into farm animals may result in increased weight gain, feed efficiency, milk production, or disease resistance.
To extend the transgenic mouse technique to other species, we undertook experiments in the rabbit, sheep, and pig. Studies with mouse eggs indicated that nuclear injection was superior to cytoplasmic injection for obtaining gene integration (Brinster et al., 1985). In the rabbit egg, pronuclear and nuclear structures are clearly visible, but they are difficult to see in the eggs of most farm animals. Initially, this prevented effective injection. However, we found that in sheep and goat eggs the nuclear structures could be clearly identified by interference-contrast microscopy. This technique did not work in pig and cow eggs, but centrifugation of the eggs stratified the cytoplasm allowing nuclear structures to be seen (Wall et al., 1985; Hammer et al., 1985c).
Because the MThGH gene had been effective in mice we chose this construct to inject into the eggs of larger animals. Approximately 5,000 eggs have been injected and transferred to foster females, and 500 of these resulted in fetuses or neonates. The integration frequency was very low in sheep. However, about 10% of rabbits and pigs born from injected eggs contained the gene (Hammer et al., 1985c). The MThGH gene was expressed in transgenic rabbits and pigs, and human growth hormone was found in the serum. The rabbit transmits the gene to progeny (Hammer et al., 1985a) as expected for genes integrated into the chromosome, and it is expected that the gene will also be transmitted in other species.
Although human growth hormone was present in the serum of about half the pigs, the levels were not as high as found in mice. In pigs we have measured levels of hGH as high as 4 μg/ml, which is lower than the maximum (64 μg/ml) found in transgenic mice but is more than 100 times endogenous pig serum GH. Much lower levels of hGH (100 ng/ml) accelerated growth and increased the size of mice (Palmiter et al., 1983). The number of transgenic rabbits is too small to assess the effect of the new gene on growth in this species, but it is clear in the pig that the elevated growth hormone levels do not result in a marked increase in growth rate or size (Hammer et al., 1985c). This may not be surprising since injections of hGH had no effect on growth (Baile et al., 1983), and exogenous pig GH only stimulated growth by 10% (Chung et al., 1985). The evidence suggests that the pig responds in a much less dramatic manner to increased levels of GH than does the mouse. A possible reason for the difference in physiological response to the activity of a similar gene in the two species is that the domestic pig has been selected for rapid growth. This may have resulted in a near maximum response in the normal animal to the growth regulating ability of GH. However, it seems quite possible that the elevated growth hormone in these transgenic pigs will result in an increased feed efficiency, since transgenic mice with an MThGH fusion gene have a feed efficiency at least 20% higher than controls (Cogburn et al., 1986). In addition, injection of growth hormone has been shown to increase the efficiency of converting feed to weight gain in pigs fed a restricted diet (Machlin, 1972).
VII. Insertional Mutagenesis
Introduction of DNA into the mouse embryo is thought to result in chromosomal integration at random sites. This assumption is based on DNA in situ hybridization studies in the mouse (Lacy et al., 1983), and by analogy, on the apparent randomness of P-element insertion in Drosophila (O’Hare and Rubin, 1983). As a consequence of random insertion one would expect that the function of endogenous genes would sometimes be disrupted by inserted foreign DNA. While it is conceivable that enhanced activity of an endogenous gene might occur as a result of the introduction of a strong enhancer element, abrogation of gene function seems more likely. Three separate examples of deleterious effects from introduced DNA have been reported in experiments with mice.
In the first example, Jaenisch et al. (1983) described a recessive lethal mutation that occurred in a strain of mice carrying the Moloney murine leukemia virus (MuLV) in the germ line. Mice that were homozygous for the MuLV gene died at 12 to 13 days of gestation. Since the viral DNA was foreign to the embryo, the inserted gene could be identified and the flanking sequences cloned. Surprisingly, in this case the MuLV integrated into the first intron of the well-known α1(I) collagen gene (Schnieke et al., 1983). The insertion blocked formation of the α1(I) collagen mRNA and led to embryonic death (Harbers et al., 1984). The ability to identify and recover the foreign DNA along with flanking DNA allows one to identify the endogenous gene involved in the mutation. Because the retrovirus integrated into a gene that was well studied, identification was facilitated; however, identification of the mutated gene may prove more difficult in other situations where it has not already been characterized.
The second reported example of insertional mutagenesis was based on experiments in which the hGH gene was microinjected into mouse eggs (E. Wagner et al., 1983). Six lines of animals developed, and in two lines, mating between heterozygote parents resulted in small litter sizes at birth and no homozygous offspring. Further examination indicated significant embryonic mortality early in gestation. In these two lines, the endogenous gene into which the foreign DNA integrated and the biological abnormality that resulted in the embryonic lethality have not been identified.
The third example of a deleterious effect of foreign DNA integration followed injection of the MK gene (Palmiter et al., 1984). A mosaic female developed that contained a low number of integrated genes. Breeding experiments established that both male and female progeny were fertile, but only females transmitted the gene to offspring. The evidence suggested that the foreign DNA had disrupted a gene that must be expressed during haploid stages of spermatogenesis. The inserted DNA was recovered, along with flanking sequences, and these are being used to identify the gene that was disrupted and to reconstruct the integration events.
VIII. Projections
While much has already been learned from transgenic animals (see Palmiter and Brinster, 1985), their potential use in biology is rapidly expanding. Although many advances can be imagined, they can be divided into two main classes of experiments, those that will improve or modify the methodology and those that will contribute to knowledge in a specific area of biology.
There are several areas where the methodology could obviously be improved.
An increase in the efficiency of the procedure would be valuable. Only about 10% of injected mouse eggs result in pups and approximately 25% of these contain the new gene (Brinster et al., 1985). Using current procedures, effective injection appears to be associated with decreased egg survival. One possibility is that integration efficiency might be improved by modifying the structure of the injected DNA. Another possibility is that other methods of DNA introduction would improve egg survival or DNA integration.
The ability to integrate single copies of the injected gene would be desirable. The tandem arrays that generally result from current methodology are difficult to analyze, and single integrants would facilitate studies on the relationship of gene structure (e.g., methylation) to function. It has been suggested that iontophoretic microinjection of DNA may lead to multiple integrations without tandem arrays, but more transgenic animals need to be created by this technique in order to assess the approach adequately (Lo, 1983). The use of retrovirus vectors would provide unit integration, but some viral sequences are known to influence expression and thus might obscure the normal regulatory processes associated with the test gene. A mammalian transposable element similar to the P-element in Drosophila (Spradling and Rubin, 1982; Rubin and Spradling, 1982) would be a valuable addition to the transgenic animal methodology.
An ultimate goal might be to target integration to a specific location in the genome. Since targeting depends on sequence recognition, homologous recombination is likely to be central to effective targeting. Although recombination among injected sequences clearly occurs, no one has succeeded in targeting a gene to a specific area of a chromosome. How this might be achieved is not clear. However, the injected genes apparently recombine by homologous recombination after injection to generate tandem arrays (Brinster et al., 1981). Furthermore, two partially overlapping gene fragments, if injected together, will recombine after injection, be expressed, and produce functional growth hormone in transgenic mice (Palmiter et al., 1985). Another aspect of targeting is the possibility of mutating or deleting specific genes. This would allow one to study the contribution of individual genes to development and body function (see below).
While the above improvements are exciting possibilities, the technique in its present form offers the opportunity to broaden our understanding in many areas, and these represent the second main class of experiments. Several areas appear promising to us.
Understanding gene regulation during development is perhaps the major question in biology today. Considerable progress has been made in identifying genes that code for specific products. To determine how these genes are regulated is the next step. It will be important to establish the location and sequence of the promoter and enhancer elements for these genes, and this may not be as simple as once hoped. There are several examples indicating that regulatory elements may be multiple and lie in locations other than 5’ to the gene. However, a significant advance will be the isolation of the genes that code for the trans-acting proteins which influence the enhancer elements. Then it will be possible to learn how the activity of these regulatory genes is controlled.
Growth in animals is a fundamental physiological process that is closely regulated and provides a system of considerable scientific as well as practical interest. The difference in animal size produced by the introduction of the MTGH genes into mice not only dramatized the value of transgenic animals in scientific research, but also established lines of mice to use in growth-related studies. Analysis of genes involved in growth regulation in the transgenic system should prove valuable in the exploration of how genes interact to affect this complex developmental process.
The immune system because of its complexity and importance provides an exciting area for experiments with transgenic animals. Two particularly valuable aspects of this system are the considerable wealth of biological information available about its function and the accessibility of the cell types through which the system operates. Transgenic animals that contain new immune response genes will provide a unique experimental system in which to examine the regulation of these genes and to determine how they affect health.
During the last five years there has been a rapid increase in our knowledge about oncogenesis. Particularly important has been the identification of genes that are known or thought to be involved in oncogenesis. These genes are identical to or related to endogenous genes of animals. Little is known about how they function in the intact animal to produce a tumor, and this is an area in which the transgenic animal will be extremely valuable. Animal models of cancer have already been created, and they will enable us to explore the effect of the oncogenes in different tissues and the effect of different oncogenes in the same tissues. The effect on tumor formation of structural modification in oncogenes can also readily be studied in transgenic animals.
The nervous system is the most complicated and intriguing system of the body. How to penetrate its complexity in higher animals is a difficult problem, but at least three avenues seem possible with transgenic animals. First, the introduction of genes that are thought or known to modify the function of the nervous system may provide information. Second, the introduction of dominant genes that cause abnormal function of the nervous system may create models for study (see below). Third, the switching of regulatory elements from one nervous system gene to another may create novel patterns of gene expression and CNS function.
Models for human genetic diseases may be created with transgenic animals. One technique would be to target a mutation to a selected gene that would make it defective. This approach is not currently possible but an alternative would be the introduction of genes with an antisense code. They would produce an RNA complementary to normal mRNA, and this antisense RNA could then bind and inactivate the endogenous mRNA (lzant and Weintraub, 1984). Another method of creating a model for genetic disease would be to introduce a gene that results in a dominant abnormal effect. For instance, when the gene for Huntington’s Chorea becomes available, its introduction into transgenic mice could create an animal model for the disease.
In projecting the use of the transgenic animal methodology, the possibility of gene transfer in humans must be considered. First, it should be clear that gene integration into somatic cells and subsequent use of those cells to correct medical problems will certainly be accomplished soon. Under these conditions gene transfer is much like many other medical treatments. The genes are not transmitted to offspring, and they do not become part of the human gene pool.
The introduction of new genes into human germ cells is a totally different matter. In this situation, the gene would become a permanent part of the gene pool of the species. While this is not of great concern in laboratory and livestock animals that we have discussed previously, it is of great concern in humans. Clearly an important ethical problem exists that needs to be discussed thoroughly before any human germ line gene transfers are attempted.
Regardless of the ethical questions, technical problems preclude the use of gene transfer into human eggs at the present time. On purely technical grounds, the system is now too inefficient. Under the best conditions only approximately 3% of mouse eggs injected result in animals carrying the gene (Brinster et al., 1985). In addition, deleterious effects of the injected DNA may occur in 10–20% of the animals that do integrate the foreign DNA (Schnieke et al., 1983; E. Wagner et al., 1983; Palmiter et al., 1984). Since injection of genes into human eggs is most frequently discussed in relation to correcting genetic defects, other considerations greatly detract from its use for this purpose. For instance, the gene integrates randomly into chromosomes. It is not possible to replace the defective gene with a normal gene. Therefore, in subsequent generations the injected gene will segregate separately from the defective endogenous gene, which will remain and continue to cause problems. In addition, most genetic diseases are a result of recessive genes and only one of four offspring are at risk, and it is not possible to tell which egg has the defective gene. These considerations argue against application of germ line gene transfer in humans on technical grounds alone.
IX. Conclusions
The past five years have witnessed an explosive growth in the number of laboratories working with transgenic animals from a few to dozens. The technique is extraordinarily powerful in its ability to provide a new approach of the utmost biological relevance to many questions. Improvements are sure to occur that will enhance the methodology by making it more precise and easier. It seems unlikely that a better method will be found than the transgenic animal to assess the activity of a gene in every cell of the body throughout development. The answers to questions surrounding that activity are among the most exciting and important to biologists. In addition, the technique has many practical applications for medicine and agriculture.
Acknowledgments
We are grateful to our colleagues, collaborators, and research assistants who made our contributions to this rapidly expanding field possible. Support for our research was obtained in part from the National Institutes of Health and the National Science Foundation.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Babinet C, Farza H, Morello D, Hadchouel M, Pourcel C. Science. 1985;230:1160–1163. doi: 10.1126/science.3865370. [DOI] [PubMed] [Google Scholar]
- Baile CA, Della-Fera M, McLaughlin CL. Growth. 1983;47:225–236. [PubMed] [Google Scholar]
- Beamer WG, Eicher EM. J Endocr. 1976;71:37–45. doi: 10.1677/joe.0.0710037. [DOI] [PubMed] [Google Scholar]
- Bradley A, Evans M, Kaufman MH, Robertson E. Nature. 1984;309:225–256. doi: 10.1038/309255a0. [DOI] [PubMed] [Google Scholar]
- Brinster RL. In: Growth, Nutrition and Metabolism of Cells in Culture. Rothblat G, Cristofalo V, editors. II. Academic Press; New York: 1972. pp. 251–286. [Google Scholar]
- Brinster RL. J Exp Med. 1974;104:1049–1056. doi: 10.1084/jem.140.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Messing A, van Dyke T, Levine AJ, Palmiter RD. Cell. 1984;37:367–379. doi: 10.1016/0092-8674(84)90367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME. Science. 1981a;211:396–398. doi: 10.1126/science.7194505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME, Avarbock MR. Nature. 1980;283:499–501. doi: 10.1038/283499a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME, Senear W, Warren R, Palmiter RD. Cell. 1981b;27:223–231. doi: 10.1016/0092-8674(81)90376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD. Proc Natl Acad Sci USA. 1985;82:4438–4442. doi: 10.1073/pnas.82.13.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Warren R, Sarthy A, Palmiter RD. Nature. 1982;296:39–42. doi: 10.1038/296039a0. [DOI] [PubMed] [Google Scholar]
- Brinster RL, Palmiter RD. Genetic Manipulation of the Mammalian Ovum and Early Embryo. In: Costantini E, Jaenisch R, editors. Banbury Report 20. Cold Spring Harbor Laboratory; New York: 1985. pp. 133–134. [Google Scholar]
- Brinster RL, Ritchie KA, Hammer RE, O’Brien RL, Arp B, Storb U. Nature. 1983;306:332–336. doi: 10.1038/306332a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capon DJ, Chen EY, Levinson AD, Seeburg PH, Goeddel DV. Nature. 1983;302:33–37. doi: 10.1038/302033a0. [DOI] [PubMed] [Google Scholar]
- Chada K, Magram J, Raphael K, Radice G, Lacy E, Costantini F. Nature. 1985;315:337–380. doi: 10.1038/314377a0. [DOI] [PubMed] [Google Scholar]
- Charnay P, Treisman R, Mellon P, Chao M, Axel R, Maniatis T. Cell. 1984;38:251–253. doi: 10.1016/0092-8674(84)90547-6. [DOI] [PubMed] [Google Scholar]
- Chen HY, Trumbauer ME, Ebert KM, Palmiter RD, Brinster RL. Molecular Developmental Biology. In: Bogorad L, editor. 43rd Symposium of the Society for Developmental Biology; New York: Alan R. Liss, Inc; 1986. pp. 149–159. [Google Scholar]
- Chisari FV, Pinkert CA, Milich DR, Filippi P, McLachlan A, Palmiter RD, Brinster RL. Science. 1985;230:1157–1160. doi: 10.1126/science.3865369. [DOI] [PubMed] [Google Scholar]
- Chung CS, Etherton TD, Wiggins JF. J Anim Sci. 1985;60:118–130. doi: 10.2527/jas1985.601118x. [DOI] [PubMed] [Google Scholar]
- Cogburn L, Hammer RE, Palmiter RD, Brinster RL. 1986 Submitted. [Google Scholar]
- Corcoran LM, Cory S, Adams JM. Cell. 1985;40:71–79. doi: 10.1016/0092-8674(85)90310-1. [DOI] [PubMed] [Google Scholar]
- Costantini F, Lacy E. Nature. 1981;294:92–94. doi: 10.1038/294092a0. [DOI] [PubMed] [Google Scholar]
- Durnam DM, Palmiter RD. J Biol Chem. 1981;256:5712–5716. [PubMed] [Google Scholar]
- Eicher EM, Beamer WG. J Hered. 1976;67:87–91. doi: 10.1093/oxfordjournals.jhered.a108682. [DOI] [PubMed] [Google Scholar]
- Frels WI, Bluestone JA, Hodes RJ, Capecchi MR, Singer DS. Science. 1985;228:557–580. doi: 10.1126/science.3885396. [DOI] [PubMed] [Google Scholar]
- Gardner RL. Nature. 1968;220:596–597. doi: 10.1038/220596a0. [DOI] [PubMed] [Google Scholar]
- Gilbert W. Science. 1985;228:823–824. doi: 10.1126/science.4001923. [DOI] [PubMed] [Google Scholar]
- Glanville N, Durnam DM, Palmiter RD. Nature. 1981;292:267–269. doi: 10.1038/292267a0. [DOI] [PubMed] [Google Scholar]
- Gordon JW, Ruddle FH. Science. 1981;214:1244–1246. doi: 10.1126/science.6272397. [DOI] [PubMed] [Google Scholar]
- Gordon JW, Scangos GA, Plotkin DJ, Barbosa JA, Ruddle FH. Proc Natl Acad Sci USA. 1980;77:7380–7384. doi: 10.1073/pnas.77.12.7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, van der Eb AJ. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Grosschedl R, Weaver D, Baltimore D, Costantini F. Cell. 1984;38:647–658. doi: 10.1016/0092-8674(84)90259-9. [DOI] [PubMed] [Google Scholar]
- Gurdon JB. Proc R Soc Lond B. 1977;198:211–247. doi: 10.1098/rspb.1977.0095. [DOI] [PubMed] [Google Scholar]
- Hammer RE, Brinster RL, Palmiter RD. Cold Spring Harbor Symp Quant Biol. 1985a;50:379–387. doi: 10.1101/sqb.1985.050.01.048. [DOI] [PubMed] [Google Scholar]
- Hammer RE, Brinster RL, Rosenfeld MG, Evans RM, Mayo KE. Nature. 1985b;315:413–416. doi: 10.1038/315413a0. [DOI] [PubMed] [Google Scholar]
- Hammer RE, Idzerda RL, Brinster RL, McKnight GS. Mol Cell Biol. 1986 doi: 10.1128/mcb.6.4.1010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer RE, Palmiter RD, Brinster RL. Nature. 1984;311:65–67. doi: 10.1038/311065a0. [DOI] [PubMed] [Google Scholar]
- Hammer RE, Pursel VG, Rexroad CE, Wall RJ, Bolt DJ, Ebert KM, Palmiter RD, Brinster RL. Nature. 1985c;315:680–683. doi: 10.1038/315680a0. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Nature. 1985;315:115–121. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Harbers K, Jahner D, Jaenisch R. Nature. 1981;293:540–542. doi: 10.1038/293540a0. [DOI] [PubMed] [Google Scholar]
- Harbers K, Kuehn M, Delius H, Jaenisch R. Proc Natl Acad Sci USA. 1984;81:1504–1508. doi: 10.1073/pnas.81.5.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyldig-Nielsen JJ, Schenning L, Hammerling U, Widmark E, Heldin E, Lind P, Serveius B, Lund T, Flavell R, Lee JS, Trowsdale J, Schreier PS, Zablizky F, Larhammar D, Peterson PA, Rask L. Nucleic Acids Res. 1983;11:5055–5071. doi: 10.1093/nar/11.15.5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illemensee K, Hoppe PC. Cell. 1981;23:9–18. doi: 10.1016/0092-8674(81)90265-8. [DOI] [PubMed] [Google Scholar]
- Izant JG, Weintraub H. Cell. 1984;36:1007–1015. doi: 10.1016/0092-8674(84)90050-3. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Harbers K, Schnieke A, Lohler J, Chumakov I, Jahner D, Grotkopp D, Hoffman E. Cell. 1983;32:209–216. doi: 10.1016/0092-8674(83)90511-1. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Mintz B. Proc Nat Acad Sci USA. 1974;71:1250–1254. doi: 10.1073/pnas.71.4.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Haslinger A, Holtgreve H, Richards RI, Krauter P, Westphal HM, Beato M. Nature. 1984;308:513–519. doi: 10.1038/308513a0. [DOI] [PubMed] [Google Scholar]
- Kleinsmith LJ, Pierce GB., Jr Cancer Res. 1964;24:1544–1551. [PubMed] [Google Scholar]
- Krumlauf R, Hammer RE, Tilghman SM, Brinster RL. Mol Cell Biol. 1985;5:1639–1648. doi: 10.1128/mcb.5.7.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy E, Roberts S, Evans EP, Burtenshaw MD, Costantini FD. Cell. 1983;34:343–358. doi: 10.1016/0092-8674(83)90369-0. [DOI] [PubMed] [Google Scholar]
- LeMeur M, Gerlinger P, Benoist C, Mathis D. Nature. 1985;316:38–42. doi: 10.1038/316038a0. [DOI] [PubMed] [Google Scholar]
- Lo CW. Mol Cell Biol. 1983;3:1803–1814. doi: 10.1128/mcb.3.10.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JM, Hammer RE, Goodman RH, Habener JF, Palmiter RD, Brinster RL. Cell. 1985;41:211–219. doi: 10.1016/0092-8674(85)90075-3. [DOI] [PubMed] [Google Scholar]
- McBride OW, Ozer HL. Proc Natl Acad Sci USA. 1973;70:1258–1263. doi: 10.1073/pnas.70.4.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Soltzer D. Science. 1983;220:1300–1302. doi: 10.1126/science.6857250. [DOI] [PubMed] [Google Scholar]
- McKnight G, Hammer RE, Kuenzal EA, Brinster RL. Cell. 1983;34:335–341. doi: 10.1016/0092-8674(83)90368-9. [DOI] [PubMed] [Google Scholar]
- McLaren A. Mammalian Chimaeras. Cambridge University Press; Cambridge: 1976. [Google Scholar]
- Machlin LJ. J Anim Sci. 1972;35:794–800. doi: 10.2527/jas1972.354794x. [DOI] [PubMed] [Google Scholar]
- Magram J, Chada K, Costantini F. Nature. 1985;315:338–340. doi: 10.1038/315338a0. [DOI] [PubMed] [Google Scholar]
- Mayo KE, Cerelli GM, Lebo RV, Bruce BD, Rosenfeld MG, Evans RM. Proc Natl Acad Sci USA. 1985;82:63–67. doi: 10.1073/pnas.82.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo K, Palmiter RD. J Biol Chem. 1981;256:2621–2624. [PubMed] [Google Scholar]
- Mayo KE, Warren R, Palmiter RD. Cell. 1982;29:99–108. doi: 10.1016/0092-8674(82)90094-0. [DOI] [PubMed] [Google Scholar]
- Messing A, Chen HY, Palmiter RD, Brinster RL. Nature. 1985;316:461–463. doi: 10.1038/316461a0. [DOI] [PubMed] [Google Scholar]
- Mintz B, Cronmiller C. Somatic Cell Genet. 1981;7:489–505. doi: 10.1007/BF01542992. [DOI] [PubMed] [Google Scholar]
- Mintz B, Illmensee K. Proc Natl Acad Sci USA. 1975;72(9):3585–3589. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RL, Selsing E, Storb U. 1985 Unpublished observations. [Google Scholar]
- O’Hare K, Rubin GM. Cell. 1983;34:25–35. doi: 10.1016/0092-8674(83)90133-2. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Palmiter RD, Hammer RE, Brinster RL, Swift GH, MacDonald RJ. Nature. 1985;313:600–603. doi: 10.1038/313600a0. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Palmiter RD, Messing A, Hammer RE, Pinkert CA, Brinster RL. Cold Spring Harbor Symp Quant Biol. 1985;50:399–409. doi: 10.1101/sqb.1985.050.01.050. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Brinster RL. Cell. 1985;41:343–345. doi: 10.1016/s0092-8674(85)80004-0. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Brinster RL, Hammer RE, Trumbauer ME, Rosenfeld MG, Birnberg NC, Evans RM. Nature. 1982b;300:611–615. doi: 10.1038/300611a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD, Chen HY, Brinster RL. Cell. 1982a;29:701–710. doi: 10.1016/0092-8674(82)90186-6. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Chen HY, Messing A, Brinster RL. Nature. 1985;316:457–460. doi: 10.1038/316457a0. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Hammer RE, Brinster RL. Genetic Manipulation of the Mammalian Ovum and Early Embryo. In: Costantini F, Jaenisch R, editors. Banbury Report 20. Cold Spring Harbor Laboratory; New York: 1985. pp. 123–134. [Google Scholar]
- Palmiter RD, Norstedt G, Gelinas RE, Hammer RE, Brinster RL. Science. 1983;222:809–814. doi: 10.1126/science.6356363. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Wilkie TM, Chen HY, Brinster RL. Cell. 1984;36:869–877. doi: 10.1016/0092-8674(84)90036-9. [DOI] [PubMed] [Google Scholar]
- Pinkert CA, Widera G, Cowing C, Heber-Katz E, Palmiter RD, Flavell R, Brinster RL. EMBO. 1985;4:2225–2230. doi: 10.1002/j.1460-2075.1985.tb03918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie KA, Brinster RL, Storb U. Nature. 1984;312:517–520. doi: 10.1038/312517a0. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Spradling AC. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Rusconi S, Kohler G. Nature. 1985;314:330–334. doi: 10.1038/314330a0. [DOI] [PubMed] [Google Scholar]
- Rutter WJ, Kemp JD, Bradshaw WS, Clark WR, Ronzio RA, Sanders TG. J Cell Physiol supplement. 1972;1:1–25. doi: 10.1002/jcp.1040720403. [DOI] [PubMed] [Google Scholar]
- Schnieke A, Barbers K, Jaenisch R. Nature. 1983;304:315–320. doi: 10.1038/304315a0. [DOI] [PubMed] [Google Scholar]
- Shani M. Nature. 1985;314:283–286. doi: 10.1038/314283a0. [DOI] [PubMed] [Google Scholar]
- Sherman MI, Solter D. Teratomas and Differentiation. Academic Press; New York: 1975. [Google Scholar]
- Sherman MI, Wudl LR. In: Concepts in Mammalian Embryogenesis. Sherman MI, editor. MIT Press; Cambridge, Mass: 1977. pp. 136–234. [Google Scholar]
- Small JA, Blair DG, Showalter SD, Scangos GA. Mol Cell Biol. 1985;5:642–648. doi: 10.1128/mcb.5.4.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM. Cell. 1983;34:47–57. doi: 10.1016/0092-8674(83)90135-6. [DOI] [PubMed] [Google Scholar]
- Stevens LC. Proc Natl Acad Sci USA. 1964;52:654–661. doi: 10.1073/pnas.52.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart TA, Pattengale PK, Leder P. Cell. 1984;38:627–637. doi: 10.1016/0092-8674(84)90257-5. [DOI] [PubMed] [Google Scholar]
- Stewart TA, Wagner EF, Mintz B. Science. 1982;217:1046–1048. doi: 10.1126/science.6287575. [DOI] [PubMed] [Google Scholar]
- Storb U, Denis KA, Brinster RL, Witte ON. Nature. 1985;316:356–358. doi: 10.1038/316356a0. [DOI] [PubMed] [Google Scholar]
- Storb U, O’Brien RL, McMullen MD, Gollahon KA, Brinster RL. Nature. 1984;310:238–241. doi: 10.1038/310238a0. [DOI] [PubMed] [Google Scholar]
- Stout JT, Chen HY, Brennand J, Caskey CT, Brinster RL. Nature. 1985;317:250–252. doi: 10.1038/317250a0. [DOI] [PubMed] [Google Scholar]
- Stuart GW, Searle PF, Chen HY, Brinster RL, Palmiter RD. Proc Natl Acad Sci USA. 1984;81:7318–7322. doi: 10.1073/pnas.81.23.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, Simmons DM, Arriza J, Hammer RE, Brinster RL, Rosenfeld MG, Evans RM. Nature. 1985;317:363–366. doi: 10.1038/317363a0. [DOI] [PubMed] [Google Scholar]
- Swift GH, Craik CS, Stary SJ, Quinto C, Lahaie RG, Rutter WJ, MacDonald RJ. J Biol Chem. 1984b;259:14271–14278. [PubMed] [Google Scholar]
- Swift GH, Hammer RE, MacDonald RJ, Brinster RL. Cell. 1984a;38:639–646. doi: 10.1016/0092-8674(84)90258-7. [DOI] [PubMed] [Google Scholar]
- Townes TM, Chen HY, Lingrel JB, Palmiter RD, Brinster RL. Mol Cell Biol. 1985b;5:1977–1983. doi: 10.1128/mcb.5.8.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townes TM, Lingrel JB, Chen HY, Brinster RL, Palmiter RD. EMBO. 1985a;4:1715–1723. doi: 10.1002/j.1460-2075.1985.tb03841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF, Covarrubias L, Stewart TA, Mintz B. Cell. 1983;35:647–655. doi: 10.1016/0092-8674(83)90097-1. [DOI] [PubMed] [Google Scholar]
- Wagner EF, Stewart TA, Mintz B. Proc Natl Acad Sci USA. 1981;78:5016–5020. doi: 10.1073/pnas.78.8.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner TE, Hoppe PC, Jollick JD, Scholl DR, Hodinka RL, Gault JB. Proc Natl Acad Sci USA. 1981;78:6376–6380. doi: 10.1073/pnas.78.10.6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall RJ, Pursel VG, Hammer RE, Brinster RL. Biol Reprod. 1985;32:645–651. doi: 10.1095/biolreprod32.3.645. [DOI] [PubMed] [Google Scholar]
- Wigler M, Silverstein S, Lee L, Pellicer A, Cheng Y, Axel R. Cell. 1977;11:223–232. doi: 10.1016/0092-8674(77)90333-6. [DOI] [PubMed] [Google Scholar]
- Wright S, Rosenthal A, Flavell R, Grosveld F. Cell. 1984;38:265–273. doi: 10.1016/0092-8674(84)90548-8. [DOI] [PubMed] [Google Scholar]
- Yamamura K, Kikutani H, Folsom V, Clayton L, Kimoto M, Akira S, Kashiwamura S, Tonegawa S, Kishimoto T. Nature. 1985;316:67–69. doi: 10.1038/316067a0. [DOI] [PubMed] [Google Scholar]