

Abstract
Ectonucleotide pyrophosphatase phosphodiesterase (ENPP1) is a positional candidate gene at chromosome 6q23 where we previously detected strong linkage with fasting-specific plasma insulin and obesity in Mexican Americans from the San Antonio Family Diabetes Study (SAFDS). We genotyped 106 single-nucleotide polymorphisms (SNPs) within ENPP1 in all 439 subjects from the linkage study, and measured association with obesity and metabolic syndrome (MS)-related traits. Of 72 polymorphic SNPs, 24 were associated, using an additive model, with at least one of eight key metabolic traits. Three traits were associated with at least four SNPs. They were high-density lipoprotein cholesterol (HDL-C), leptin, and fasting plasma glucose (FPG). HDL-C was associated with seven SNPs, of which the two most significant P values were 0.0068 and 0.0096. All SNPs and SNP combinations were analyzed for functional contribution to the traits using the Bayesian quantitative-trait nucleotide (BQTN) approach. With this SNP-prioritization analysis, HDL-C was the most strongly associated trait in a four-SNP model (P = 0.00008). After accounting for multiple testing, we conclude that ENPP1 is not a major contributor to our previous linkage peak with MS-related traits in Mexican Americans. However, these results indicate that ENPP1 is a genetic determinant of these traits in this population, and are consistent with multiple positive association findings in independent studies in diverse human populations.
In a genome scan of Mexican Americans from the San Antonio Family Diabetes Study (SAFDS), we previously found a strong linkage signal with several obesity-related phenotypes in non-diabetic individuals, at the same chromosome 6q23 region that also contains the ectonucleotide pyrophosphatase phosphodiesterase (ENPP1) gene (1). Strong linkage was also found with bivariate traits composed of fasting-specific insulin and obesity measures (serum leptin concentration, sum of skinfold thickness, BMI, and log serum triglyceride concentration). Independent evidence for linkage at this locus with metabolic syndrome (MS)-related phenotypes has been reported in several studies in diverse populations (2–5).
Previous studies have shown genetic association of ENPP1 with obesity, insulin resistance, type 2 diabetes mellitus (T2DM), and related conditions including myocardial infarction (6–8). In a large European cohort, a three-variant haplotype containing the missense variant K121Q was strongly associated with severe obesity and T2DM (9). This haplotype was also associated with childhood obesity in both French and German children (8,10). In this and other studies, the Q121 allele was associated with increased insulin resistance and obesity. However, two recent studies reported association of the Q121 allele with decreased BMI, in whites and African Americans (11,12). These contradictory findings indicate a complex relationship between ENPP1 and metabolic disease, including the possibility that the K121Q variant is in linkage disequilibrium with other unknown functional variants. Similarly, contrasting and apparently incompatible results have been reported in several large association studies (13–16). Many ENPP1 association studies have included only a small number of DNA variants, often only the K121Q variant. Therefore, the relationship between genetic variation in the ENPP1 gene and common metabolic disease remains unclear.
We performed a comprehensive analysis of the association of genetic variation in ENPP1 with T2DM and related phenotypes in Mexican Americans from the SAFDS. Advantages of this study included, the analysis of all available single-nucleotide polymorphisms (SNPs) and a novel statistical assessment of the contributions of all SNPs, singly and in combination, to phenotypic association. We deliberately chose a comprehensive approach to SNP selection and genotyping of all available SNPs, including many SNPs with low minor allele frequency (<5%), to capture all variation potentially contributing to disease. Given recent evidence of the contribution of rare variants to complex multifactorial disease (17), we believe this is an appropriate safeguard against drawing false conclusions from insufficient data.
Clinical characteristics of the nondiabetic study participants are shown in Table 1. From the public databases, we identified 137 polymorphic variants within the gene. These variants included 133 SNPs (two in the promoter, seven in the 3′ untranslated region, nine in exons), with a mean SNP density of one SNP/625 bp. Of the nine exonic SNPs, one was synonymous (A/A) and the rest were nonconservative missense variants, including K121Q. We resequenced ∼31% of the ENPP1 gene in each of 20 SAFDS subjects from families that contributed most strongly to the linkage signal. Sequenced regions included all exons, intron/exon boundaries, the proximal promoter, 3′ and 5′ untranslated regions, plus all regions containing SNPs. Of 133 putative SNPs, 27 failed design criteria, 106 were genotyped in 439 subjects and, of these, 79 were polymorphic. A further seven SNPs were not in Hardy–Weinberg Equilibrium (P < 0.05). Thus 72 SNPs were used for association analysis. A substantial proportion of genetic variation in ENPP1 (69%) was found among the less common variants with allele frequencies <20%. Within this group, rare alleles with minor allele frequency <5% were the most common type and accounted for a substantial proportion (30%) of the low frequency variants.
Table 1. Characteristics of the nondiabetic SAFDS subjects distributed across 27 families.
| Distributional properties of the phenotypes | ||||
|---|---|---|---|---|
|
|
||||
| Variable | Sample size | Mean ± s.d. or % | Skewness | Kurtosis |
| Number | 323 | — | — | — |
| Females | 187 | 58% | — | — |
| Age (years) | 323 | 38.3 ± 15.4 | — | — |
| Fasting-specific insulin (pmol/l) | 293 | 138 ± 86 | 1.1 | 0.9 |
| Fasting glucose (mmol/1) | 323 | 4.9 ± 0.6 | 0.7 | 1.0 |
| ln HOMA-IR | 290 | 3.8 ± 0.7 | 0.0 | −0.9 |
| ln HOMA-β | 293 | 5.1 ± 0.5 | −0.0 | −0.6 |
| ln Triglycerides | 311 | 4.9 ± 0.6 | 0.4 | −0.3 |
| HDL-cholesterol (mmol/l) | 309 | 1.0 ± 0.3 | 0.6 | 0.3 |
| Leptin (ng/ml) | 292 | 22.2 ± 17.6 | 1.4 | 1.6 |
| BMI (kg/m2) | 317 | 29.4 ± 6.6 | 1.0 | 1.2 |
| Sum of skinfold thickness (mm) | 319 | 174.0 ± 56.3 | 0.2 | −0.4 |
| Waist circumference (mm) | 317 | 946 ± 171 | 0.7 | 0.5 |
β, beta-cell function (insulin secretion); HOMA, homeostasis model assessment (24); IR, insulin resistance; ln, natural log- transformed; SAFDS, San Antonio Family Diabetes Study.
The K121Q variant had a nominal minor allele frequency of 18.8 ± 2.4%. The structure of linkage disequilibrium within the ENPP1 gene, as measured by the absolute correlation (ρ) between genotypes, is shown in Figure 1. The average proportion of shared variation between SNPs, measured by the pairwise correlation (r2), was 0.047. On the basis of the evaluation of the eigenstructure of the correlation matrix among markers the effective number of SNPs was 67. Thus, these 72 SNPs behave statistically like 67 effectively independent markers. Conditional on the pedigree information, we estimated the haplotypes for each individual using the computer program MERLIN. We observed 298 unique haplotypes out of the sample theoretical maximum of 902. The observed haplotype diversity was 0.99. The most common haplotype had a frequency of 3.97 and 95.3% and the observed haplotypes had a frequency <1%.
Figure 1.
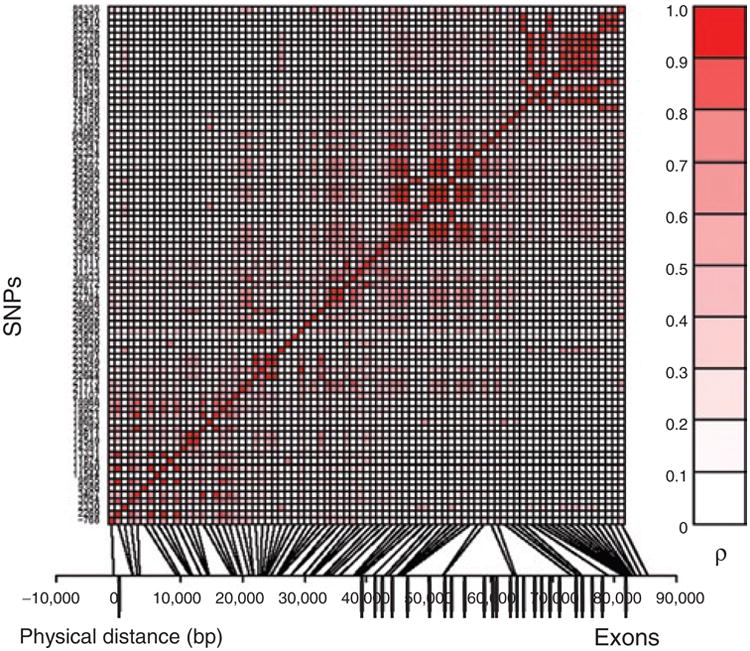
SNP linkage disequilibrium structure within ENPP1. SNPs are indicated by their physical order within the gene on the y-axis. The relative positions of the SNPs are indicated on the x-axis. Physical distance is denoted in bp. The degree of correlation (ρ) between SNP pairs is indicated by the intensity of shading in the small squares of the central figure, with perfect correlation between identical SNPs along the diagonal. Gene exonic structure is indicated by the vertical bars below the x-axis. A total of 79 SNPs are shown, including seven SNPs which were not in Hardy–Weinberg equilibrium.
We initially focused our association analyses on the traits where we found evidence of linkage. Using an additive model, all significant SNP association P values from single SNP analysis are included in Table 2, including results for T2DM as a discrete trait. The several obesity measures (ln leptin, sum of skinfold thickness, BMI, and log serum triglyceride concentration) were analyzed as quantitative traits to maximize statistical power. In addition, we also analyzed several discrete traits, including obesity (BMI ≥30 kg/m2) and the MS or its risk factors, using the National Cholesterol Education Program Adult Treatment Panel III criteria (18). All the findings from the additional analyses are included in Supplementary Tables 1 (discrete traits) and 2 (quantitative traits). Association analyses in the supplementary tables include dominant and recessive models of inheritance in addition to the additive model described below. In general, results from additive and dominant/recessive analyses were consistent.
Table 2. Significant (P < 0.05) association (measured genotype approach) of SNPs with key MS-related phenotypes.
| SNP | MAF (%) | Location (bp) | HDL-C | ln Leptin | FPG | FSI | SS | T2DM | BMI | ln TG | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs1800949 | 29.9 | −768 | 0.038 | 1 | |||||||
| rs876507 | 1.1 | 2,330 | 0.042 | 1 | |||||||
| rs4086314 | 28.8 | 3,407 | 0.027 | 1 | |||||||
| rs6918013 | 6.4 | 9,386 | 0.032 | 0.0047 | 2 | ||||||
| rs943004 | 4.4 | 11,544 | 0.046 | 1 | |||||||
| rs7756163 | 48.2 | 13,331 | 0.048 | 1 | |||||||
| rs6917903 | 30.7 | 18,284 | 0.010 | 1 | |||||||
| rs1830971 | 41.3 | 19,021 | 0.0096 | 1 | |||||||
| rs2021966 | 49.8 | 21,107 | 0.038 | 0.041 | 2 | ||||||
| rs9372999 | 10.6 | 23,820 | 0.0068 | 1 | |||||||
| rs7754609 | 7.9 | 25,598 | 0.020 | 1 | |||||||
| rs997509 | 1.8 | 38,645 | 0.045 | 1 | |||||||
| rs7769058 | 5.5 | 63,662 | 0.039 | 1 | |||||||
| rs7769712 | 9.2 | 64,063 | 0.037 | 1 | |||||||
| rs6928362 | 2.5 | 81,349 | 0.012 | 0.032 | 2 | ||||||
| rs1974201 | 43.3 | 81,789 | 0.045 | 1 | |||||||
| rs6929388 | 2.5 | 81,858 | 0.015 | 0.049 | 2 | ||||||
| rs7754586 | 44.9 | 83,410 | 0.013 | 1 | |||||||
| rs7754859 | 47.2 | 83,476 | 0.042 | 1 | |||||||
| rs1931009 | 5.8 | 85,336 | 0.044 | 1 | |||||||
| Total | 7 | 6 | 4 | 1 | 2 | 2 | 1 | 1 | 24 |
FPG, fasting plasma glucose; FSI, fasting-specific insulin; HDL-C, high-density lipoprotein cholesterol; ln, natural log-transformed; MAF, minor allele frequency; MS, metabolic syndrome; SNP, single-nucleotide polymorphisms; SS, sum of skinfold thickness; T2DM, type 2 diabetes mellitus; TG, triglyceride.
P values <0.01 are indicated in boldface.
We performed a marginal association analysis using a fixed effects measured genotype additive model. First, we tested for hidden population stratification using the quantitative transmission disequilibrium test approach in the computer program SOLAR (http://www.sfbr.org/). The tests of association were restricted to nondiabetics (n = 323, 74%), except for T2DM which included all subjects. Significantly associated SNPs are shown in Table 2, and their location within the gene is summarized in Figure 2. Nominal P values are shown uncorrected for multiple testing. For a statistical significance threshold of ≤0.05, the required experiment-wide significance threshold, using Bonferroni's correction per trait (with 67 statistically independent markers) accounting for multiple testing, was 0.00075. There were no additional attempts to correct for multiple testing because correcting for multiple testing with regard to multiple correlated traits is not straightforward, given that the corresponding models are not independent.
Figure 2.

Gene structure of ENPP1 and location of significantly associated SNPs. SNPs exhibiting significant nominal association (P < 0.05) are shown. The thick horizontal line represents the gene, with thick vertical lines representing the 25 exons, numbered below each exon.
Te most significant associations were with fasting plasma glucose (FPG) (P = 0.0047; rs6918013) and high-density lipoprotein cholesterol (HDL-C) (P = 0.0068 and 0.0096 for rs9372999 and rs1830971, respectively). Figure 3a shows the gene location and –log P values for all genotyped SNPs with HDL-C, which was significantly associated with seven SNPs. Interestingly, we previously found strong evidence for linkage of both HDL-C and BMI (univariate and bivariate analyses) to the 6q23 region using the Framingham study data (3). In this context, the present HDL-C findings are of interest, given the well-established connections between the MS, T2DM, obesity, and dyslipidemia. Figure 3b shows the gene location and –log P values for FPG, which were significantly associated with four SNPs. The genotype class specific mean values for the phenotypes that were most strongly associated (P < 0.01) are summarized in Table 3. These P values were not corrected for multiple testing because the SNPs were subsequently tested by a novel statistical procedure, Bayesian quantitative trait nucleotide (BQTN), which is robust to multiple testing.
Figure 3.

Association of ENPP1 SNPs with (a) HDL-C and (b) FPG. The physical location of each SNP is shown on the x-axis. The y-axis shows –log10 P values for association calculated using the measured genotype approach.
Table 3. Genotype class specific mean values for the traits that were associated (P < 0.01) with SNPs in ENPP1.
| Trait | SNP | MAF (%) | Major/major | Major/minor | Minor/minor | Direction of change | P value |
|---|---|---|---|---|---|---|---|
| HDL-C | rs1830971 | 41.3 | 32.3 ± 1.3 | 34.8 ± 1.2 | 37.2 ± 1.7 | ↑ | 0.0096 |
| HDL-C | rs9372999 | 10.6 | 34.7 ± 1.2 | 31.0 ± 1.5 | 27.4 ± 2.6 | ↓ | 0.0068 |
| FPG | rs6918013 | 6.4 | 92.8 ± 1.2 | 97.2 ± 1.8 | 101.6 ± 3.4 | ↑ | 0.0047 |
Measured genotype additive trait mean values ± s.e. are shown for the two most significantly associated traits, FPG (mg/dl) and HDL-C (mg/dl).
FPG, fasting plasma glucose; HDL-C, high-density lipoprotein cholesterol; MAF, mean allele frequency.
To predict statistically the causal variants responsible for the observed associations, we performed BQTN analysis (19) to examine all possible additive gene action models. BQTN analysis provides a rigorous approach to prioritization of all associated SNPs, to predict the causal variants responsible for the observed associations, by comparing all possible additive gene function models. It allows us to calculate the probability that any given SNP or combination of SNPs may be involved in a trait, and to compare that probability with the probabilities for all other SNP models. BQTN analysis provided modest evidence of a “true” signal. The most strongly associated trait was HDL-C, where a four-SNP model (with SNPs at 21,107, 25,663, 52,324, and 64,063 bp) gave a P value of 0.000080 and one SNP (21,107 bp) had a good overall posterior probability of 0.843, although the experiment-wide P value for this model was 0.081.
Interestingly, several recent studies have found genetic variants in ENPP1 interacting with obesity to influence variation in MS-related traits (7,8,16). Likewise, in our data we observed significant ENPP1-BMI interaction effects on fasting-specific insulin: (P = 0.00050 and 0.00070 for SNPs rs858342 and v24, respectively).
In summary, we found modest suggestive evidence of ENPP1 association with the MS-related phenotypes that contributed to our previous linkage finding in Mexican Americans. On the basis of our findings, after accounting for multiple testing, it appears that the ENPP1 gene is not a major contributor to the linkage signal. However, given the correlations between traits and between SNPs, the correction for multiple testing is conservative and sets a threshold for significance which may be overly stringent, with a corresponding increased risk of false negative findings (type II errors). A potential limitation of this study is that the power to detect significant associations between metabolic traits and rare genetic variants may be somewhat compromised by the moderate sample size. Taken together, it is reasonable to conclude that the observed associations of multiple ENPP1 variants with several MS-related traits, is consistent with positive association results from multiple independent studies in diverse populations. Furthermore, the strongest association signals came predominantly from intronic SNPs, potentially indicating a role of transcript regulation in disease causation. We are pursuing the 6q23 region further to identify DNA sequence variants that could relate to our original linkage findings.
Research Methods and Procedures
The SAFDS
All study participants were Mexican Americans from the SAFDS (20). In brief, probands for the SAFDS had T2DM identified in an earlier epidemiologic survey, the San Antonio Heart Study. All 1st, 2nd, and 3rd degree relatives of the probands, aged 18–98 years, were invited to participate in the study. Metabolic, anthropometric, demographic, and medical-history information was obtained on 439 (116 with T2DM) individuals distributed across 27 low-income Mexican-American pedigrees. All procedures were approved by the Institutional Review Board of the University of Texas Health Science Center at San Antonio, and all subjects gave informed consent before their participation.
The following eight MS-related phenotypes, which have been described in detail elsewhere (1), were considered for this study: FPG, fasting-specific insulin, fasting HDL-C, fasting triglyceride concentration, fasting leptin, BMI, sum of skinfold thickness as a measure of overall subcutaneous adiposity, and T2DM (as a discrete trait, see below). For these continuous traits we used phenotypic information from only nondiabetic subjects (N = 323 individuals depending on availability of data) to avoid metabolic effects secondary to T2DM or treatment effects. We also tested T2DM, as a discrete trait, in all subjects (N = 439). T2DM was defined using two widely accepted classifications: a FPG concentration ≥7.0 mmol/l (126 mg/dl) and/or a 2-h plasma glucose concentration after a 75-g oral glucose tolerance test ≥11.0 mmol/l (200 mg/dl). Patients who gave a history of physician diagnosed diabetes or who were on diabetic medication were also considered affected with diabetes. Phenotype-specific distinct outliers were excluded from the analyses (e.g., triglyceride values >9 mmol/l). Where indicated, the data were log transformed to normalize distributions. Selected clinical characteristics of nondiabetic SAFDS subjects are shown in Table 1.
DNA sequencing
The ENPP1 gene was amplified by the PCR in overlapping segments and sequenced using oligonucleotide primers designed to provide multiple overlapping sequences in both directions. Automated DNA sequencing was performed using BigDye fluorescent dye- terminator chemistry (Applied Biosystems, Foster City, CA). Contiguous sequences were assembled using Sequencher software (GeneCodes, Ann Arbor, MI). Polymorphisms were detected by direct comparison of electropherograms from 20 subjects, selected from families that contributed most strongly to linkage at 6q23 (see below). Target regions of genomic DNA were amplified by the PCR and sequenced with either the same primers used for amplification or nested internal primers. The ENPP1 sequence and SNPs from DbSNP can be located at http://www.ncbi.nlm.nih.gov/.
SNP genotyping
Genomic DNA was isolated from whole blood (Qiagen, Chatsworth, CA). SNPs were selected from resequencing data, the NCBI dbSNP database and from previous association studies. SNPs were genotyped according to the manufacturer's instructions (Applied Biosystems, Foster City, CA). In brief, in a 384-well plate, 2 μl of purified genomic DNA (2 ng/μl) was incubated with primers and probes for the SNP of interest (0.09 μl), 3.5 μl of TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA)—No AmpErase UNG, and 1.14 μl distilled water. Samples were PCR-amplified on the Applied Biosystems 9700HT Thermal Cycler under the following conditions: denatured for 10 min at 95 °C, denatured annealed and extended for 40 cycles of 15 s at 92 °C and 1 min at 60 °C. The 384-well microplates were scanned for fluorescence emission using a 7900HT sequence detector (Applied Biosystems) and alleles were scored using the allelic discrimination software Sequence Detection System v2.1 (Applied Biosystems). All SNPs were tested for Mendelian pedigree inconsistencies. For all SNPs genotyped, our mean rate of success for SNP genotyping was >98%. Primers and probe sequences used for the SNP genotyping are available on request.
Statistical genetic analysis
We performed association analysis in our complex pedigree-based data using the measured genotype approach within the variance components (VCs) analytical framework implemented in the program SOLAR (21). The VC-based approach accounts for nonindependence among family members. In this analytical technique, VCs are modeled as random effects (e.g., additive genetic effects and random environmental effects), whereas the effects of measured covariates such as age and sex are modeled as fixed effects on the trait mean. The VCs, the association parameters, and the other covariate effects (e.g., age and sex terms) were estimated, simultaneously, by maximum likelihood techniques. A likelihood function based on multivariate normal density was numerically maximized to obtain parameter estimates.
Before performing measured genotype approach, the quantitative transmission disequilibrium test (22), as implemented in SOLAR, was used to examine hidden population stratification, using the notation of within (w) and between (b) family components. In measured genotype approach, generally, the marker genotypes were incorporated in the mean effects model as a measured covariate assuming additivity of allelic effects (23). Using the notation of w and b family components, a significant test of b = w vs. b = 0 and w = 0 reflects significant difference between the genotypic means (Table 1). All analyses included age and sex terms as covariates, if found to be significant. The nested models were compared using the likelihood ratio test. Twice the difference between the log-likelihoods of these models yields a test statistic that is asymptotically distributed, approximating a χ2 distribution with one degree of freedom. A P value ≤0.05 is considered significant. Using the program SOLAR, linkage disequilibrium between SNP pairs was estimated using the absolute value of the correlation coefficient |ρ|. However, for the purpose of discussion, r2 values are reported to describe the pairwise linkage disequilibrium patterns.
The BQTN model
The BQTN analytical technique is employed to analyze SNPs to find the responsible nucleotide variants (the quantitative-trait nucleotides) influencing a given phenotype. This technique has been described in detail elsewhere (19). Given complete SNP data for a gene, this statistical technique can be used to identify the sequence variants that are either potentially functional or that exhibit the highest disequilibria with such potential functional sites. The BQTN model is a simple extension of the classical VC model that aims to disentangle the genetic architecture of a quantitative trait (19).
Supplementary Material
Acknowledgments
We thank Andrea Barrentine, Kathy Camp, and Sheila Taylor for excellent technical assistance, and John Kincaid, Jim King, Norma Diaz, and Tricia Wolfe for outstanding nursing assistance. Most important, we express our deep appreciation to the San Antonio Mexican-American community and all the volunteers who took part in this study. These studies were supported by the following grants: a Veterans Administration Merit Award (to C.P.J.); a Veterans Administration Genetic Epidemiology grant (to R.A.D.); the Veterans Affairs Medical Research Fund, and General Clinical Research Center Grant M01-RR-01346; a San Antonio Area Foundation Grant (to D.K.C.); and the following National Institutes of Health grants DK067690 (to C.P.J.), DK024092 (to R.A.D.), DK053889-06 (to R.D.), and DK047482 (to M.P.S.).
Footnotes
Supplementary Material: Supplementary material is linked to the online version of the paper at http://www.nature.com/oby
Disclosure: The authors declared no conflict of interest.
References
- 1.Duggirala R, Blangero J, Almasy L, et al. A major locus for fasting insulin concentrations and insulin resistance on chromosome 6q with strong pleiotropic effects on obesity-related phenotypes in nondiabetic Mexican Americans. Am J Hum Genet. 2001;68:1149–1164. doi: 10.1086/320100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghosh S, Watanabe RM, Valle TT, et al. The Finland-United States investigation of non-insulin-dependent diabetes mellitus genetics (FUSION) study. I An autosomal genome scan for genes that predispose to type 2 diabetes. Am J Hum Genet. 2000;67:1174–1185. [PMC free article] [PubMed] [Google Scholar]
- 3.Arya R, Lehman D, Hunt KJ, Schneider J, et al. Evidence for bivariate linkage of obesity and HDL-C levels in the Framingham Heart Study. BMC Genet. 2003;4(Suppl 1):S52. doi: 10.1186/1471-2156-4-S1-S52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atwood LD, Heard-Costa NL, Cupples LA, et al. Genomewide linkage analysis of body mass index across 28 years of the Framingham Heart Study. Am J Hum Genet. 2002;71:1044–1050. doi: 10.1086/343822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyre D, Lecoeur C, Delplanque J, et al. A genome-wide scan for childhood obesity-associated traits in French families shows significant linkage on chromosome 6q22.31-q23. 2 Diabetes. 2004;53:803–811. doi: 10.2337/diabetes.53.3.803. [DOI] [PubMed] [Google Scholar]
- 6.Pizzuti A, Frittitta L, Argiolas A, et al. A polymorphism (K121Q) of the human glycoprotein PC-1 gene coding region is strongly associated with insulin resistance. Diabetes. 1999;48:1881–1884. doi: 10.2337/diabetes.48.9.1881. [DOI] [PubMed] [Google Scholar]
- 7.Abate N, Chandalia M, Satija P, et al. ENPP1/PC-1 K121Q polymorphism and genetic susceptibility to type 2 diabetes. Diabetes. 2005;54:1207–1213. doi: 10.2337/diabetes.54.4.1207. [DOI] [PubMed] [Google Scholar]
- 8.Bacci S, Ludovico O, Prudente S, et al. The K121Q polymorphism of the ENPP1/PC-1 gene is associated with insulin resistance/atherogenic phenotypes, including earlier onset of type 2 diabetes and myocardial infarction. Diabetes. 2005;54:3021–3025. doi: 10.2337/diabetes.54.10.3021. [DOI] [PubMed] [Google Scholar]
- 9.Meyre D, Bouatia-Naji N, Tounian A, et al. Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat Genet. 2005;37:863–867. doi: 10.1038/ng1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Böttcher Y, Körner A, Reinehr T, et al. ENPP1 variants and haplotypes predispose to early onset obesity and impaired glucose and insulin metabolism in German obese children. J Clin Endocrinol Metab. 2006;91:4948–4952. doi: 10.1210/jc.2006-0540. [DOI] [PubMed] [Google Scholar]
- 11.Matsuoka N, Patki A, Tiwari HK, et al. Association of K121Q polymorphism in ENPP1 (PC-1) with BMI in Caucasian and African-American adults. Int J Obes (Lond) 2006;30:233–237. doi: 10.1038/sj.ijo.0803132. [DOI] [PubMed] [Google Scholar]
- 12.Prudente S, Chandalia M, Morini E, et al. The Q121/Q121 genotype of ENPP1/PC-1 is associated with lower BMI in non-diabetic whites. Obesity (Silver Spring) 2007;15:1–4. doi: 10.1038/oby.2007.636. [DOI] [PubMed] [Google Scholar]
- 13.Grarup N, Urhammer SA, Ek J, et al. Studies of the relationship between the ENPP1 K121Q polymorphism and type 2 diabetes, insulin resistance and obesity in 7,333 Danish white subjects. Diabetologia. 2006;49:2097–2104. doi: 10.1007/s00125-006-0353-x. [DOI] [PubMed] [Google Scholar]
- 14.Lyon HN, Florez JC, Bersaglieri T, et al. Common variants in the ENPP1 gene are not reproducibly associated with diabetes or obesity. Diabetes. 2006;55:3180–3184. doi: 10.2337/db06-0407. [DOI] [PubMed] [Google Scholar]
- 15.Weedon MN, Shields B, Hitman G, et al. No evidence of association of ENPP1 variants with type 2 diabetes or obesity in a study of 8,089 U.K. Caucasians. Diabetes. 2006;55:3175–3179. doi: 10.2337/db06-0410. [DOI] [PubMed] [Google Scholar]
- 16.Bochenski J, Placha G, Wanic K, et al. New polymorphism of ENPP1 (PC-1) is associated with increased risk of type 2 diabetes among obese individuals. Diabetes. 2006;55:2626–2630. doi: 10.2337/db06-0191. [DOI] [PubMed] [Google Scholar]
- 17.Blangero J. Localization and identification of human quantitative trait loci: king harvest has surely come. Curr Opin Genet Dev. 2004;14:233–240. doi: 10.1016/j.gde.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 19.Blangero J, Goring HH, Kent JW, Jr, et al. Quantitative trait nucleotide analysis using Bayesian model selection. Hum Biol. 2005;77:541–559. doi: 10.1353/hub.2006.0003. [DOI] [PubMed] [Google Scholar]
- 20.Stern MP, Duggirala R, Mitchell BD, et al. Evidence for linkage of regions on chromosomes 6 and 11 to plasma glucose concentrations in Mexican Americans. Genome Res. 1996;6:724–734. doi: 10.1101/gr.6.8.724. [DOI] [PubMed] [Google Scholar]
- 21.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abecasis GR, Cookson WO, Cardon LR. Pedigree tests of transmission disequilibrium. Eur J Hum Genet. 2000;8:545–551. doi: 10.1038/sj.ejhg.5200494. [DOI] [PubMed] [Google Scholar]
- 23.Almasy L, Blangero J. Exploring positional candidate genes: linkage conditional on measured genotype. Behav Genet. 2004;34:173–177. doi: 10.1023/B:BEGE.0000013731.03827.69. [DOI] [PubMed] [Google Scholar]
- 24.Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.