

SUMMARY
Long non-coding RNAs (lncRNAs) have been implicated in numerous physiological processes and diseases, most notably cancers. However, little is known about the mechanism of many functional lncRNAs. We identified an abundantly-expressed lncRNA associated with decreased melanoma patient survival. Increased expression of this lncRNA, SLNCR1, mediates melanoma invasion through a highly-conserved sequence similar to the lncRNA SRA1. Using a sensitive technique we term RATA (RNA-associated transcription factor array), we show that the brain-specific homeobox protein 3a (Brn3a) and the androgen receptor (AR) bind within and adjacent to SLNCR1’s conserved region, respectively. SLNCR1, AR, and Brn3a are specifically required for transcriptional activation of matrix metalloproteinase 9 (MMP9) and increased melanoma invasion. Our observations directly link AR to melanoma invasion, possibly explaining why males experience more melanoma metastases and have an overall lower survival as compared to females.
Keywords: long non-coding RNA, hormone receptor, melanoma, MMP9, invasion, metastasis
INTRODUCTION
The incidence of melanoma world-wide has been on the rise for the past 30 years. In the United States, it is estimated that 73,870 new cases will be diagnosed and 9,940 individuals will die of melanoma in 2015 (Siegel et al., 2015). Most early melanomas are easily treated by surgical excision; once metastasized, melanoma is a highly-lethal cancer with a 5-year survival rate of 16% (Howlader N, 2015). Thus, prevention of metastasis is key to melanoma survival and identification of molecular drivers would enable development of novel melanoma therapies.
There is mounting evidence that non-coding RNAs are critical determinants of tumor progression. A major portion (>70,000 genes) of the human non-coding transcriptome is comprised of long non-coding RNAs (lncRNAs) (Zhao et al., 2015). Dysregulated lncRNA expression has recently been linked to many cancers (Li et al., 2013a). LncRNAs may act as oncogenes or tumor suppressors, with an emerging role specifically in cancer metastasis (Serviss et al., 2014). Recent studies implicate lncRNAs in melanomagenesis, though their exact role in melanoma etiology is poorly understood because their molecular and biological functions are obscure (Flockhart et al., 2012; Khaitan et al., 2011; Tang et al., 2013; Tian et al., 2014; Wu et al., 2013).
We profiled lncRNAs expressed in patient-derived melanomas and identified a lncRNA (XLOC_012568) critical for melanoma invasion. This lncRNA contains a conserved ~300 nucleotide region with significant similarity to steroid receptor RNA activator 1 (SRA1), and is hence named SLNCR (SRA-like non-coding RNA). Survival analysis of The Cancer Genome Atlas (TCGA) melanomas indicates decreased patient survival in patients expressing high levels of SLNCR. Knockdown of the most prevalent isoform SLNCR1 results in the differential expression of 111 transcripts and decreases melanoma invasion. We show that the brain-specific homeobox protein 3a (Brn3a) and the androgen receptor (AR) bind to SLNCR1’s conserved sequence and an adjacent sequence, respectively, and that SLNCR1 coordinates the transcriptional activities of these transcription factors to upregulate the gene encoding the gelatinase MMP9 and increase melanoma invasion. SLNCR1 binds to and functions with AR, implicating a hormone-responsive transcription factor in melanoma invasion. Thus, our results may reconcile the long-established gender bias in melanoma in which males have a higher frequency of metastases compared to females.
RESULTS
SLNCR expression is associated with melanoma survival outcome
To identify melanoma-associated lncRNAs, we performed RNA-Sequencing (RNA-seq) on three melanoma short-term cultures (MSTCs) and fibroblast short-term cultures (FSTCs) derived from the tumor microenvironment (unpublished data from Charles Yoon, Brigham and Woman’s Hospital, Boston, MA). MSTCs have undergone relatively few passages outside of the patient and closely reflect the genetics of patient melanomas and provide a tractable system to study disease-relevant transcriptional changes. Of the 137 lncRNAs expressed in human melanomas (FPKM > 1, Table S1), the third most abundant lncRNA (XLOC_012568; linc00673, Refseq NR_036488.1; average FPKM = 55.33) is expressed in MSTCs but not FSTCs. Moreover, this lncRNA is located within a chromosomal region commonly amplified in melanoma, lung and ovarian cancers (www.broadinstitute.com/tumorscape, Table S2). We confirmed increased expression of XLOC_012568 in eight MSTCs compared to three normal melanocyte controls by RT-qPCR (Tables S1 and S3, Figure 1A). In addition to melanomas, the MiTranscriptome database (mitranscriptome.org) reveals that XLOC_012568 is increased in lung adenocarcinoma and squamous cell carcinomas compared to corresponding normal tissues, while it is decreased in stomach cancers compared to normal tissues (Iyer et al., 2015) (Figure S1A). XLOC_012568 is also expressed in cervical, ovarian, and pancreatic cancers, low-grade glioma and glioblastoma multiforme. Collectively, these data suggest a broader role for this lncRNA in human tumorigenesis.
Figure 1. SLNCR is expressed in melanomas and is associated with worse overall survival.

(A) Relative expression of SLNCR across multiple melanocytes and melanomas, as measured by RT-qPCR, compared to A375 after normalization to GAPDH. Error bars represent standard deviations calculated from 3 reactions. (B) Schematic presentation of SLNCR1’s exon structure. The highly-conserved and SRA1-like sequences are highlighted. (C) Box plot of SLNCR expression from 150 TCGA melanomas categorized based on tumor thickness at diagnosis. Data are represented as mean ± SEM. Significance was calculated using the Student’s t-test: * p-value < 0.05. (D) Kaplan-Meier survival analysis of high or low SLNCR expressing TCGA melanomas, defined by the median SLNCR expression (RPKM = 14.1). See also Figure S1.
There are three XLOC_012568 isoforms expressed in melanomas (Figure S1B). The most prevalent isoform, SLNCR1, is 2257 nucleotides and composed of 4 exons spanning chr17:70399463-70588943 (Figure 1B). Isoforms 2 and 3 contain an additional short or long exon, respectively, located between exon 3 and 4. Despite the fact that most lncRNAs display only modest sequence conservation due to their rapid evolution, XLOC_012568 includes a highly-conserved region across mammals (Figures 1B and S1C) (Necsulea et al., 2014). This conserved region is located within a region of high identity (54%) to the steroid receptor RNA Activator-1 (SRA1; Figures 1B and S1D). While the SRA1 locus expresses both protein-coding and functional non-coding transcripts, none of the 3 SLNCR isoforms exhibit protein-coding potential (coding potential scores SLNCR1: 0.12, SLNCR2: 0.10, SLNCR3: 0.37) (Chooniedass-Kothari et al., 2004; Kong et al., 2007; Lanz et al., 1999). The conservation of SLNCR1, its similarity to another functional non-coding RNA, and its abundant expression across multiple cancers suggests a functionally important role for SLNCR1.
To annotate SLNCR expression to clinically-relevant parameters, we assessed SLNCR expression across 150 randomly-selected human melanomas from TCGA. It is important to note that this analysis does not distinguish between SLNCR isoforms. In agreement with results from patient-derived melanomas, SLNCR is expressed in 146 out of 150 randomly selected human melanomas (RPKM > 1, Table S1). Tumor depth, as described by Breslow’s thickness (T, measured in millimeters), is one of the most important prognostic factors in melanoma treatment. Specifically, while thin tumors (≤1 mm thick) are typically treatable by surgical excision, thicker tumors (>1 mm thick) have a greater possibility of reaching blood vessels and are thus more likely to metastasize, requiring more aggressive treatment. SLNCR expression is significantly higher in tumors at least 1 mm thick, correlating with severity of the melanoma (AJCC staging classification TX/Tis/T0/T1 versus T2/T3/T4; Figure 1C).
To investigate whether SLNCR expression is related to disease outcome in TCGA melanomas, we performed a Kaplan-Meier survival analysis comparing melanoma patients expressing high (n = 72, red line) or low (n = 70, blue line) levels of SLNCR1 defined by the median SLNCR expression (Figure 1D). High expression of SLNCR is associated with shorter overall survival in melanoma patients (p-value = 0.0426). The median survival for the low SLNCR group was 14.3 years, while the high SLNCR group had a median survival of only 5.3 years. Additionally, the pooled hazard ratio shows an 84% increase in the risk of death for the high SLNCR group (logrank HR = 1.84, 95% confidence interval 1.03 to 3.60). Together, these data suggest a role for SLNCR at a clinically-critical stage of melanomagenesis.
SLNCR1 increases melanoma invasion by transcriptionally upregulating MMP9
To gain insights into the role of SLNCR in melanoma formation, we used global transcriptional profiling before and after knockdown of the most abundant isoform, SLNCR1, in the MSTC WM1976. Two custom-designed siRNAs directed against exon 3–4 junction resulted in ~80–90% knockdown of SLNCR1 (Figures 2A and S2A). Due to toxicity and apparent off-target effects of si-SLNCR1 (2) 48 hours post-transfection, only differentially expressed transcripts from duplicate knockdown using si-SLNCR1 (1) were used for RNA-seq analyses (Figure S2B). Knockdown of endogenous SLNCR1 resulted in the differential expression of 111 transcripts (adjusted p-value < 0.05, fold change >2, Table S4), indicating that SLNCR1 regulates expression of numerous genes in trans.
Figure 2. SLNCR’s highly-conserved sequence increases melanoma invasion.

(A) Schematic presentation of SLNCR1-specific siRNAs targeting the exon 3–4 junction. Right: relative expression of SLNCR1 upon siRNA knockdown in the MSTC WM1976. RT-qPCR data is represented as the fold change compared to scramble siRNA control, normalized to GAPDH. Error bars represent standard deviations calculated from 3 reactions. (B–C) Matrigel invasion assays of WM1976 (B) or WM1575 (C) cells transfected with the indicated siRNA. Invasion is calculated as the percent of invading cells compared to mobile cells as counted in 8 fields of view. Top panels show representative images of the indicated invading and mobile cells. Quantification from 3 independent replicates, represented as mean ± SD, is shown at the bottom. (D) As in (B and C) but with A375 melanoma cells transfected with the indicated plasmids. The schematic presentation (top) denotes the SLNCR1 sequences expressed from the indicated plasmids. The bottom left panel shows representative images, while quantification from 3 independent replicates is shown at the right. Significance was calculated using the Student’s t-test: * p-value < 0.05, ** p-value < 0.005, *** p-value < 0.0005, ns = not significant. See also Figure S2.
Next, we assessed whether SLNCR1 promotes cancer phenotypes in melanoma cells in vitro. First, SLNCR1 expression did not affect viability or proliferation of melanoma cells, as quantified using the metabolic reagent WST-1 (Figures S2B–S2C). Second, SLNCR1 expression did not significantly alter cell motility, as measured by transwell migration assays (Figure S2D). Finally, we measured invasion using a matrigel invasion assay. SLNCR1 knockdown significantly decreased invasion in WM1976 (~80%) and WM1575 (~60%) MSTCs, suggesting that endogenous SLNCR1 plays a critical role in melanoma invasion (Figures 2B and 2C).
To independently validate a role for SLNCR1 in melanoma invasion, we over-expressed SLNCR1 in the A375 melanoma cell line. A375 cells express lower levels of SLNCR1 compared to patient melanomas (Figure 1A), providing a tractable system for mechanistic studies of SLNCR1 function. As expected, over-expression of SLNCR1 increased invasion of A375 cells (~200%; Figures 2D and S2E). Over-expression of a SLNCR1 mutant lacking the highly-conserved sequence (SLNCR1Δcons, nucleotides 462–572 deleted) did not increase invasion, while over-expression of the conserved sequence, including ~100 nucleotides of flanking sequences to ensure proper RNA folding (SLNCR1cons, nucleotides 372–672), increased invasion to the same degree as full-length SLNCR1 (~200%; Figure 2D). Collectively, these data indicate that the conserved region is necessary and sufficient for SLNCR1-mediated melanoma invasion.
To identify genes that mediate increased melanoma invasion, we over-expressed SLNCR1, SLNCR1Δcons, or SLNCR1cons in A375 melanoma cells and performed unbiased transcriptional profiling by RNA-seq. Expression of SLNCR1 resulted in the differential expression of 110 genes (adjusted p-value < 0.05, fold change > 2, Table S5 and Figure S3A). Because the conserved sequence is necessary and sufficient for SLNCR1-mediated melanoma invasion, we searched for transcripts differentially expressed upon over-expression of SLNCR1 and SLNCR1cons, but not SLNCR1Δcons. Using a less stringent criteria (p-value < 0.05, fold change > 1.5) to ensure identification of all potential candidates, we identified two transcripts significantly upregulated by SLNCR1’s conserved sequence: RARRES2P8, a pseudogene of the retinoic acid receptor responder, and MMP9, a gene that encodes matrix metallopeptidase 9, also known as gelatinase B (Figure 3A). MMP9 contributes to early melanoma invasion through remodeling of the extracellular matrix (Hofmann et al., 2005; MacDougall et al., 1999; MacDougall et al., 1995; van den Oord et al., 1997). Consistent with a role in early tumor dissemination, analysis of our TCGA melanoma cohort revealed that MMP9 expression is significantly higher in regional metastases compared to primary tumors (p-value = 0.0003, Figure S3B). Furthermore, SLNCR1 expression in primary melanomas precedes increased MMP9 expression, supporting SLNCR1-regulated expression of MMP9 (Figure S3B).
Figure 3. SLNCR1 increases melanoma invasion by transcriptionally upregulating MMP9.

(A) Heat map of differentially expressed genes significantly regulated by SLNCR1 and SLNCR1cons, but not SLNCR1Δcons, in the melanoma cell line A375. The shading represents the log2 fold change compared vector only control. (B) Relative MMP9 expression in A375 cells transfected with the indicated plasmids. RT-qPCR data is represented as the fold change compared to a vector control, normalized to GAPDH. Error bars represent standard deviations calculated from 3 reactions. (C–E) MMP9 activity from supernatants of cells transfected with the indicated plasmids or siRNAs were quantified using gelatin zymography. Percent MMP9 activity is represented as fold change compared to the vector or scramble control, normalized to MMP2 activity. Error bars represent standard deviations from three independent replicates. (C) Percent MMP9 activity in supernatants of A375 cells transfected with the indicated plasmids. (D) Percent MMP9 activity of WM1976 supernatant upon knockdown of SLNCR1. (E) Percent MMP9 activity of WM1575 supernatant upon knockdown of SLNCR1. (F) Matrigel invasion assay of A375 melanoma cells transfected with the indicated plasmids and siRNAs, as in Figure 2 (B–D). (G) A375 cells, grown in steroid-deprived conditions, were transfected with a MMP9p-firefly (FL) reporter plasmid, a CMV-RL (renilla luciferase) control, and the indicated SLNCR1 expression plasmids. Luciferase activity was measured 24 hours post-transfection. Relative FL activity was calculated as a fold-change compared to vector only control cells, after normalization to RL activity. Shown is one representative assay from at least three independent replicates. Error bars represent standard deviation from four reactions. Significance was calculated using the Student’s t-test: * p-value < 0.05, ** p-value < 0.005, *** p-value < 0.0005, ns = not significant. See also Figure S3.
Because SLNCR1cons regulates expression of MMP9 and mediates SLNCR1-induced invasion, we hypothesized that MMP9 is responsible for SLNCR1-induced invasion. First, we confirmed that SLNCR1cons is necessary and sufficient for increasing MMP9 mRNA (~2.5-fold), as well as MMP9 enzymatic activity (~50%) (Figures 3B and 3C). Over-expression of lncRNAs, like proteins, may force non-physiological interactions and subsequently cause artefactual downstream affects. To confirm that endogenous SLNCR1 regulates MMP9, we quantified MMP9 expression and activity in MSTCs following SLNCR1 knockdown. Consistent with a role in regulating MMP9, SLNCR1 knockdown decreased MMP9 expression (Log2 fold change = −0.74, Table S4) in WM1976 and decreased MMP9 activity (40–50%) in WM1575 and WM1976 cells (Figures 3D and 3E).
If SLNCR1 increases melanoma invasion by upregulating MMP9, depleting MMP9 should block SLNCR1-mediated invasion. To test this hypothesis, we transfected A375 cells with empty or SLNCR1-expressing vectors, along with control or MMP9-specific siRNAs (Figure S3C). As expected, MMP9 knockdown blocked the SLNCR1-mediated increase in MMP9 activity and invasion (Figures S3D, S3E, and 3F). These data demonstrate that SLNCR1 increases melanoma invasion by upregulating MMP9.
LncRNAs can transcriptionally or post-transcriptionally regulate gene expression. To test if SLNCR1 transcriptionally upregulates MMP9, we generated a firefly luciferase (FL) reporter under control of the 2 kilobase MMP9 promoter (MMP9p-FL) and monitored expression in A375 cells (Figure 3G). When normalized to expression of renilla luciferase from a co-transfected control reporter plasmid, SLNCR1 expression resulted in a significant (~3.5-fold) increase in FL activity. To further validate the requirement of SLNCR1cons, we generated deletion mutants of SLNCR1 and monitored FL activation. In agreement with previous results showing a requirement of SLNCR1cons, expression of SLNCR1Δcons did not increase FL activity. Interestingly, deletion of 70 bases immediately 3′ to the conserved region (SLNCR1Δ568-637) also failed to increase FL activity, indicating an additional requirement for this sequence in MMP9 regulation. It is important to note that this region is included in the sequence over-expressed in SLNCR1cons (Figure 2D). Furthermore, because serum-containing media contains exogenous hormones and steroids that affect activity of steroid hormone receptors, the assay was performed in the absence of steroids. SLNCR1 increased FL activity in steroid-deprived cells indicating that SLNCR1-mediated regulation of MMP9 is not dependent on exogenous hormones contained in the media. Collectively, these data confirm that nucleotides 462–637 of SLNCR1 upregulates the MMP9 promoter in a ligand-independent manner.
AR and Brn3a bind to adjacent regions of SLNCR1
Previously characterized lncRNAs ‘fine-tune’ gene expression through a range of mechanisms (Geisler and Coller, 2013). We hypothesized that SLNCR1 binds TFs because SLNCR1 (1) is expressed in the nucleus (Figure S4A), (2) transcriptionally upregulates the MMP9 promoter (Figure 3G), and (3) has sequence similarity to SRA1 (Figure S1D), which has been shown to bind to multiple TFs (Colley and Leedman, 2011). Importantly, TCGA patient melanomas express SLNCR1 at comparable levels to SRA1 in other tissues (~0–60 RPKM, or approximately up to 100 copies per cell) (http://gdac.broadinstitute.org) (Harvard, 2015; Kellis et al., 2014; Mortazavi et al., 2008).
Identifying TFs using standard techniques is challenging because of their low expression. We therefore designed a novel method for identifying RNA-bound TFs that we term RATA (RNA-associated transcription factor array). This technique couples an RNA pulldown with a high-throughput TF activation array, enabling highly-sensitive and unbiased identification of TFs bound to an RNA of interest (Figure 4A). The bacteriophage coat protein MS2 interacts with high-affinity to a specific stem-loop structure in the phage genome and has been widely adapted for biochemical purification of mammalian RNAs (Gong and Maquat, 2015). SLNCR1 constructs containing 12 copies of the MS2 binding sites were co-expressed with a plasmid expressing nuclear FLAG-tagged MS2 protein and immunoprecipitation with anti-FLAG antibodies routinely showed ~30–100 fold enrichment of SLNCR1 or SLNCR1Δcons (Figure S4B). For subsequent use in the TF activation array, bound RNAs and proteins were eluted from beads using FLAG peptide under non-denaturing conditions (Figure 4B). The eluate was then subjected to a TF Activation Profiling Plate Array (Signosis), allowing for quantitative analysis of multiple TFs in a single assay. Pulldowns were repeated in triplicate, and TFs showing specific >7-fold enrichment compared to the untagged control in at least 2 experiments were considered potential candidates.
Figure 4. AR and Brn3a bind to adjacent SLNCR1 sequences.

(A) Schematic presentation of the RATA method for identifying TFs associated with SLNCR1. SLNCR1-MS2 RNP complexes were immunoprecipitated with α-FLAG antibody, eluted from beads using FLAG peptide, and the eluate was immediately subjected to the TF Activation Profiling Plate Array I (Signosis). TF-bound probes were isolated through column separation and analyzed through hybridization with a plate whose wells are pre-coated with complementary DNA. (B) Ectopically expressed FLAG-tagged MS2 was immunoprecipitated from A375 cells transfected with the indicated MS2-loop containing SLNCR1 construct, compared to control cells expressing untagged SLNCR1 constructs. Left panel: total protein input or bound proteins following IP with α-FLAG antibody was subjected to Western blot analysis. The blot was probed with α-FLAG and α-GAPDH antibodies. Middle and right: relative enrichment of the indicated transcripts as measured by RT-qPCR compared to RNA enriched from cells expressing SLNCR1 without MS2 stem loops. Bound FLAG-MS2 RNPs were eluted using FLAG peptide. (C) Fold enrichment of TF-specific probes with MS2-based purification of SLNCR1 or SLNCR1Δcons from A375 cells. Probe enrichment is represents fold enrichment compared to an untagged RNA control IP, after normalization to the signal of GATA-specific probes. Shown is one representative assay of TF-specific probes showing significant (>7-fold) enrichment in at least two out of three replicates. (D) Immunoprecipitations from HEK293T transfected with GFP-tagged AR and the indicated SLNCR1 expressing plasmids using either α-AR antibody or an IgG nonspecific control. Top panel: western blot analysis of input (I), IgG bound (IgG) or α-AR bound (AR) proteins. Bottom panels: relative enrichment of the indicated transcripts from AR-IPs, compared to an IgG nonspecific control. HEK293T cells were transfected with GFP-tagged AR and either SLNCR1 (bottom left panel) or SLNCR1 Δcons (bottom middle panel) or SLNCR1 Δ568-637(bottom right panel) expression plasmids. (E) Immunoprecipitations from UV-crosslinked HEK293T transfected with Brn3a and the indicated SLNCR1 expression plasmid using either anti-Brn3a antibody or an IgG nonspecific control. Top panel: western blot of input (I), IgG bound (IgG) or α-Brn3a bound (Brn3a) proteins. Bottom panels: relative enrichment of the indicated transcripts from Brn3a-IPs. HEK293T cells were transfected with Brn3a and either SLNCR1 (bottom left panel) or SLNCR1 Δcons (bottom right panel). To control for differences in the efficiency of proteinase K digestion, enrichment was calculated compared to input transcript levels after normalization to levels of the 18s RNA. All RT-qPCR are represented as mean ± SD from three replicates. See also Figure S4.
To identify TFs binding to the conserved region of SLNCR1, we compared probes enriched from SLNCR1 versus SLNCR1Δcons immunoprecipiations. Probes for Brn3a (Pou4F1) routinely showed strong enrichment with SLNCR1, but showed negligible enrichment upon deletion of the conserved sequence, suggesting that Brn3a binds to the conserved region. Although Brn3a has not previously been shown to bind RNA, the TF contains a predicted RNA-binding motif in amino acid position 143–175 (MOTIF Search, http://www.genome.jp/tools/motif/). Importantly, Brn3a has been implicated in melanoma cell cycle progression (Hohenauer et al., 2013).
We also observed modest enrichment of probes specific for the androgen receptor (AR), EGR, E2F-1, ATF2, and AP2. Of these, we focused on AR for several reasons: (1) AP2, ATF2, E2F-1, and EGR directly or indirectly interact with AR (Altintas et al., 2012; Jorgensen and Nilson, 2001; Verger et al., 2001; Zhang et al., 2010), suggesting that enrichment of these probes is a consequence of protein-protein interactions rather than direct interaction with SLNCR1. (2) Transcriptional network analysis of melanomas over-expressing or knocking down SLNCR1 reveals significant enrichment of AR-regulated genes (Table S4: SLNCR1 knockdown, p-value = 1.45e-59, z-score = 134.17; Table S5: SLNCR1 over-expression, p-value 5.070E-63, z-score = 160.15; MetaCore™, Thomson Reuters). (3) AR directly binds to other lncRNAs (Yang et al., 2013; Zhang et al., 2015). (4) AR positively regulates MMP9 in other cancers, including gastric, bladder, and prostate cancers (Ergun et al., 2007; Hara et al., 2008; Wang et al., 2013; Wu et al., 2010; Zhang et al., 2014). Notably, (5) melanomas express AR (Allil et al., 2008; Morvillo et al., 2002).
To validate SLNCR1cons binding to AR and Brn3a, we performed RNA immunoprecipitation (RIP) assays in HEK293T (human embryonic kidney) cells, which express very low levels of endogenous SLNCR1. SLNCR1 is significantly enriched (~120-fold) in RNAs immunoprecipitating with ectopically expressed AR (Figure 4D). Surprisingly, SLNCR1Δcons is still enriched in AR immunoprecipitates (~50-fold). RNA secondary structure is often critical to RNA function; thus, deletion of the conserved sequence may disrupt adjacent secondary structures and weaken the interaction of AR and SLNCR1 in the RATA assay. We note that SLNCR1568-637 is required for MMP9 upregulation (Figure 3G), suggesting that AR binds to this region adjacent to the conserved region. Consistent with this hypothesis, SLNCR1Δ568-637 is not enriched above background levels of endogenous SLNCR1 (Figure 4D and data not shown). Taken together, these data confirm that AR binds to SLNCR1568-637.
To capture specific Brn3a-RNA interactions, we used ultraviolet (UV) light to crosslink HEK293T cells prior to immunoprecipitation of Brn3a. SLNCR1 is significantly enriched in Brn3a immunoprecipitates (~1500-fold) while SLNCR1Δcons shows no enrichment (Figure 4E), confirming that Brn3a binds directly to SLNCR1462-572.
Upregulation of MMP9 requires SLNCR1, AR and Brn3a
Because Brn3a binds to SLNCR1’s conserved region and AR binds to an adjacent sequence, respectively, we hypothesized that all 3 components are required for upregulation of MMP9 and melanoma invasion. If SLNCR1 coordinates transcriptional activity of both TFs, MMP9 should not significantly contribute to melanoma invasion in the absence of any single component. It is important to note that in A375 cells, SLNCR1 is required for MMP9-mediated invasion because knockdown of MMP9 does not decrease invasion of low SLNCR1-expressing A375 cells (Figure 3F, vector only samples). To confirm a specific requirement for AR and Brn3a, we repeated gelatin zymography, MMP9p-FL reporter and matrigel invasion assays after over-expressing SLNCR1 and simultaneously knocking down of either TF in the A375 melanoma cell line. Consistent with our hypothesis that AR is required for regulating MMP9 activity, depleting AR prevented MMP9 activation and promoter upregulation, as well as melanoma invasion, after SLNCR1 over-expression (Figures 5A–C and S5A–D). This confirms that AR is required for SLNCR1-mediated invasion, even when both Brn3a and SLNCR1 are present (Figure 5C).
Figure 5. SLNCR1 mediated invasion requires AR and Brn3a.

(A and D) MMP9 activity of A375 cells transfected with the indicated plasmids and siRNAs, as in Figure 3 (C–E). (B and E) Relative luciferase activity of A375 cells transfected with an MMP9-RL reporter, as well as the indicated plasmids and siRNAs. Quantification was performed as in Figure 3G. (C and F) Matrigel invasion assay of A375 melanoma cells transfected with the indicated plasmids and siRNAs. The top panel shows representative images of the indicated invading or mobile cells, and the quantification from 3 independent replicates is shown at the bottom. Significance was calculated using the Student’s t-test: * p-value < 0.05, n.s. = not significant. See also Figure S5.
To test if Brn3a is also required for SLNCR1-mediated invasion, we repeated the above assays using Brn3a-specific siRNAs. Similar to results seen with AR, depleting Brn3a prevented SLNCR1-mediated upregulation of MMP9 activity and promoter upregulation (Figures 5D–E and S5E–G). We also performed invasion assays of A375 melanoma cells expressing vector alone or SLNCR1 in the presence of scramble or Brn3a-specific siRNAs (Figure 5F). Interestingly, knockdown of Brn3a increased melanoma invasion. However, this increased invasion occurred independently of an increase in MMP9 (Figures 5D and 5E). Most importantly, expressing SLNCR1 does not increase melanoma invasion when Brn3a is depleted (Figure 5F, compare bars 3 to 4 and 5 to 6). Thus, our data indicate that Brn3a regulates melanoma invasion through two pathways: one independent of SLNCR1 and MMP9, and another that requires AR and SLNCR1 to upregulate MMP9. Because knockdown of AR or Brn3a completely abrogates upregulation of MMP9 and SLNCR1-mediated melanoma invasion, our data demonstrates a functional requirement for SLNCR1, AR and Brn3a, and suggests formation of a ternary complex composed of SLNCR1, AR and Brn3a.
SLNCR1 increases AR binding to the MMP9 promoter
LncRNAs may direct TFs to target regions in the chromosome through direct binding to DNA and formation of an RNA-DNA complex, or by acting as scaffolds to assemble a complex of multiple TFs and regulatory proteins (Geisler and Coller, 2013; Wang and Chang, 2011). We observe no significant similarity between SLNCR1 and the MMP9 promoter, arguing against a direct interaction between SLNCR1 and the DNA. Supporting a model of direct TF binding to MMP9 promoter elements, the MMP9 promoter contains multiple functional AREs (androgen response elements), as well as a near perfect consensus Brn3a binding site (gcAT[A/T]A[T/A]T[A/T]AT) (Figure 6A) (Gruber et al., 1997; Zhang et al., 2014). To test if these TF binding sites (TFBSs) are required for SLNCR1-mediated transcriptional upregulation of MMP9, we generated MMP9p-FL reporter constructs harboring mutations within the predicted ARE (MMP9p-FL ARE mut) or the Brn3a binding site (MMP9p-FL BBS mut). While over-expression of SLNCR1 in A375 cells significantly increased luciferase expression from the wild-type MMP9p-FL reporter, mutation of either the predicted Brn3a binding site or the ARE abolished the ability of SLNCR1 to increase luciferase activity (Figure 6B). These data further suggest formation of a ternary complex composed of SLNCR1, AR and Brn3a at the MMP9 promoter.
Figure 6. AR and Brn3a binding sites are required for SLNCR1-induced upregulation of the MMP9 promoter.

(A) Schematic presentation of the 2 KB MMP9 promoter cloned upstream of the firefly luciferase reporter. The black box denotes a predicted Brn3a binding site. The wild-type and mutated sequences are shown below. The black circle denotes a functional ARE, with wild-type and mutated sequences below. The grey circles denote additional predicted AREs. (B) Mutation of either the Brn3a binding site (MMP9p-FL BBS mut) or the ARE (MMP9p-FL ARE mut) prevents SLNCR1-mediated upregulation of the MMP9 promoter. Assay was completed as in Figure 3G. Error bars represent standard deviation from four reactions. (C) AR-ChIP from A375 cells transfected with either vector or SLNCR1-expressing plasmid. qPCR was performed using primers specific to the regions indicated, including primers corresponding to a gene desert (negative control). * p-value < 0.05, ** p-value < 0.005, n.s. = not significant. See also Figure S6.
Next, we used chromatin immunoprecipitation and PCR (ChIP-PCR) to test if AR directly binds to the MMP9 promoter. Compared to a vector only control, AR is significantly enriched (~2-fold) at the MMP9 promoter in the presence of SLNCR1 (Figure 6C). Collectively, these data indicate that binding of AR, and likely Brn3a, to the MMP9 promoter is required for transcriptional activation of MMP9.
Finally, we examined our TCGA melanoma cohort for in vivo evidence of AR-mediated regulation of MMP9. Expression of AR and MMP9 are significantly correlated (r = 0.41, p-value = 0.0003) in high-SLNCR melanomas (RPKM ≥ 14.1), but not in low-SLNCR1 melanomas (RPKM < 14.1), suggesting that AR is bound to the MMP9 promoter only when SLNCR1 expression reaches a certain threshold (Figure S6). These data are consistent with AR- and SLNCR1-mediated regulation of MMP9 in patient melanomas.
DISCUSSION
LncRNAs are emerging as important players in cancer biology; however, the mechanistic details of most lncRNA functions remain unknown. Here, we identify SLNCR as a robustly-expressed lncRNA associated with worse overall melanoma survival. SLNCR1 increases melanoma invasion by transcriptionally upregulating MMP9. Our work provides direct biochemical evidence that SLNCR1 physically interacts with both AR and Brn3a, and that all three components are required for upregulating MMP9. The Brn3a and AR binding sites are located approximately 100 nucleotides apart, an orientation consistent with cooperative TF binding. Our data therefore supports a model in which SLNCR1 mediates formation of a nuclear SLNCR1/AR/Brn3a ternary complex with high affinity for the proximal TFBSs in the MMP9 promoter (Figure 7). Taken together, this study identifies SLNCR1 as a novel oncogenic lncRNA with a critical role in melanomagenesis. This finding is in agreement with a recent report in which linc00673 (SLNCR1) was identified as a possible oncogene in non-small-cell lung cancer (Shi et al., 2016).
Figure 7. Model for SLNCR1-induced MMP9 transcriptional upregulation.
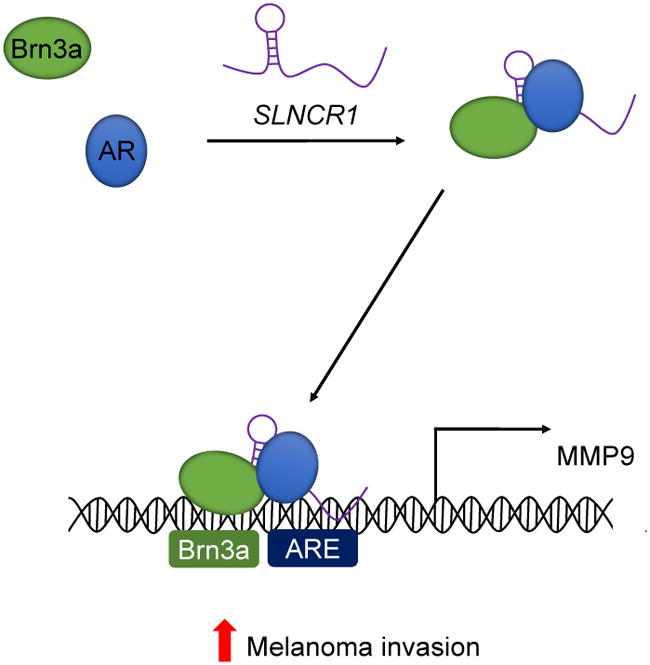
When SLNCR1 increases, AR and Brn3a bind to conserved, adjacent regions of SLNCR1. The SLNCR1/AR/Brn3a ternary complex has high affinity for adjacent Brn3a and AR binding sites located upstream of the MMP9 transcriptional start site. Cooperative binding of AR and Brn3a to its promoter increases MMP9 expression and activity and thus increases invasion of melanoma cells. See also Figures S7.
Although canonical AR activation occurs upon binding of an androgenic hormone ligand, translocation to the nucleus, and AR-dimerization, our data is most consistent with ligand-independent activation of AR and formation of an AR/Brn3a heterodimer. First, SLNCR1 upregulates the MMP9 promoter in the absence of exogenous steroids (Figure 3G) or increased nuclear localization of AR (Figure S7A), consistent with ligand-independent activation of AR. Second, if SLNCR1 induced canonical AR dimerization, over-expression of the lncRNA would increase global activity of AR as opposed only a subset of AR-target genes (Table S5). Third, other AR-associated lncRNAs have been shown to induce AR in a ligand-independent manner (Yang et al., 2013; Zhang et al., 2015). Fourth, AR forms functional heterodimers with other TFs, and was previously shown to directly bind Brn3a in mouse ND7 cells (Berwick et al., 2010; Chen et al., 1997; Lee et al., 1999). These data are consistent with formation of a heterodimer TF complex which significantly increases TF affinity for the tandem TFBSs than either TF alone. An AR monomer has only low affinity for AREs. Similarly, Brn3a has lower affinity for non-consensus TFBSs, such as the one located upstream of MMP9 (Gruber et al., 1997). Formation of an AR/Brn3a heterodimer likely allows for cooperative binding of the TFs to their respective TFBSs, effectively concentrating the nuclear AR in melanoma cells to specific promoters, including the MMP9 promoter.
It is very difficult to predict lncRNA function based on sequence analysis because lncRNAs are not as conserved as protein-coding genes (Necsulea et al., 2014). However, SLNCR1 contains a region highly-conserved among mammals that is also similar to a region of SRA1, allowing us to extrapolate the function of SLNCR1cons from a wide range of possibilities. In addition to SRA1, the lncRNAs HOTAIR, and possibly PRNCR1 and PCGEM1, bind to AR (Agoulnik and Weigel, 2009; Prensner et al., 2014; Yang et al., 2013; Zhang et al., 2015). We note a significant sequence similarity between regions of PCGEM1, HOTAIR, SLNCR1 and SRA1 (Figure S7B), suggesting that it is feasible to predict lncRNA function from these sequences. Identification of functional lncRNA sequences may enable determination of the mechanisms of uncharacterized lncRNAs.
While this work has focused on a region of SLNCR1 with sequence and functional similarities to SRA1, there are several important differences between the lncRNAs. (1) SRA1 has not been implicated in melanoma, and is not differentially expressed between normal melanocytes and melanomas (data not shown). (2) While SRA1 binds to multiple steroid receptors, including AR, there is no evidence that it interacts with Brn3a or regulates MMP9 (Lanz et al., 1999). (3) Most importantly, siRNA-mediated knockdown of SLNCR1 does not affect levels of SRA1 (Figure S2A), indicating that the decrease in invasion (Figures 2B and 2D) and differential gene expression patterns (Table S4) are attributable to depletion of SLNCR1 and not SRA1. SLNCR1 therefore functions independently of SRA1 to regulate melanoma invasion. Identification and analysis of additional functional regions of SLNCR1 outside of the SRA1-like sequence will be an important topic for future studies. For example, SLNCR1cons is required for only a small fraction of SLNCR1’s gene-regulatory function (Figure S3A). A candidate SLNCR1-interacting TF binding outside of its conserved sequence is PAX5, a TF involved in B-cell differentiation (Figure 4C). In addition to TF-dependent functions, SLNCR1 may have TF-independent functions, such as through interactions with mRNAs, microRNAs or splicing factors, some of which may occur in the cytoplasm (Figure S4A) (Geisler and Coller, 2013).
A gender bias in melanoma biology favoring females was first considered over 40 years ago (Clark et al., 1969). In the years since this study, other studies have confirmed that females have a significant survival advantage compared to males (38%), fewer and delayed metastases, longer delay before relapse, and higher cure rates than males, strongly suggesting a biological basis for the observed gender bias (Bidoli et al., 2012; de Vries et al., 2007; Fisher and Geller, 2013; Gamba et al., 2013; Geller et al., 2002; Joosse et al., 2012; Joosse et al., 2011; Schwartz et al., 2002; Swetter et al., 2009). Differences in expression or activity of sex-hormone receptors, including the estrogen receptor or AR, have long been considered a plausible explanation for the melanoma gender bias (de Giorgi et al., 2011; Morvillo et al., 2002). Recent studies favor a deleterious role for AR and androgens in melanoma, as (1) the female advantage persists even in post-menopausal women, suggesting against an estrogen-related advantage (de Vries et al., 2008; Joosse et al., 2011; Micheli et al., 2009) and (2) there is an increased risk of melanoma following prostate cancer, and vice-versa, suggesting an AR-based connection between the two cancers (Li et al., 2013b; Spanogle et al., 2010). Our results directly implicate AR in melanoma invasion by interacting with SLNCR1 and upregulating MMP9, possibly reconciling the observation that males suffer an increased number of melanoma metastases.
EXPERIMENTAL PROCEDURES
Cell culture
Primary human melanocytes were derived from neonatal foreskins and cultured as previously described (Yokoyama et al., 2008). All other cells were cultured as adherent cells in DMEM (Dulbecco’s modified eagle medium, Invitrogen) without glutamine supplemented with 10% fetal bovine serum (FBS). A375 cells were purchased from ATCC, HEK293T cells were a gift from Ronny Drapkin, ‘CY’ melanomas were a gift from Charles Yoon, and ‘WM’ melanomas were from collections of the Wistar Institute (Philadelphia, PA). ‘CY’ melanomas were derived by Charles Yoon.
For luciferase assays, cells were cultured in phenol-red free DMEM without glutamine (Invitrogen), supplemented with 5% charcoal stripped FBS. Luciferase activity was measured using Promega Dual-Glo® Luciferase Assay system. For fractionation experiments, cells were grown to ~80% confluency in 10 cm tissue culture treated dishes and fractionated using Thermo Scientific™ NE-PER™ Nuclear and Cytoplasmic Extraction Kit, according to manufacturer’s instructions. Nuclear and cytoplasmic fractions were split for protein and RNA analysis. For proliferation assays, cells were transfected with the indicated siRNAs 24 hours post-seeding and proliferation was measured every 24 hours using WST-1 reagent (Roche) according to the manufacturer’s instructions. For chromatin immunoprecipitation and PCR (ChIP-PCR), A375 cells were cultured in phenol-red free DMEM without glutamine (Invitrogen), supplemented with 5% charcoal stripped FBS, and transfected with the indicated plasmid 24 hours post-seeding. Cells were crosslinked in 1% formaldehyde for 15 minutes 48 hours port-transfection, and the reaction was quenched by addition of 0.125 M glycine. ChIP and PCR were performed by Active Motif (Carlsbad, CA). The FKBP5 control primers amplified a region roughly +87015 to the gene, and MMP9 specific primers amplified a region −2254 to the gene.
Invasion Assays
Cells were plated in either BD BioCoat™ matrigel inserts or uncoated control inserts (Corning) in serum-free media and placed into DMEM with 30% FBS. The number of invaded or migrant cells were imaged on 20x magnification in 8 fields of view for 3 independent replicates.
Plasmid construction
SLNCR1 and a codon-optimized Brn3a were synthesized by Biomatik Corporation and cloned into pCDNA3.1 (-). The simian virus nuclear localization signal (SV40-NLS) was cloned upstream of the MS2 ORF in a FLAG-tagged, hMS2-expressing vector, a gift from Dr. Lynne Maquat, University of Rochester Medical Center. Nuclear localization of tagged MS2 was confirmed via fractionation and western blotting. pEGFP-C1-AR was a gift from Michael Mancini (Addgene plasmid # 28235).
Reagents and antibodies
Lipofectamine® RNAiMax (Life Technologies) was used for all siRNA transfections, and Lipofectamine® 2000 (Life Technologies) was used for all plasmid transfections and siRNA/plasmid cotransfections. Protein G Dynabeads® (Life Technologies) were used for FLAG-MS2 IPs, and Protein A Dynabeads® (Life Technologies) were used for AR and Brn3a IPs. The following antibodies were used: Sigma Monoclonal ANTI-FLAG® M2 antibody; Santa Cruz AR (N-20), Brn3a (14A6), Hsp90 (4F10), and rabbit IgG control; Cell Signaling GAPDH (14C10); Abcam SNRNP70 (ab83306); and BD Pharmingen™ mouse IgG control. Santa Cruz AR (H-280) was used for ChIP PCR. Sequences for all siRNAs and oligos used in this study can be found in Table S7.
RNA pulldowns
A375 cells were grown to ~80% confluency in 10 cm dishes, transfected with 10 μg of the plasmid encoding nuclear MS2 and 8 μg of the indicated 3′ MS2 stem-loop tagged SLNCR1, and harvested 36–48 hours post-transfection. MS2-tagged SLNCR1 was confirmed functional by RT-qPCR of selected SLNCR1 transcriptional targets (data not shown). MS2 RNA pull-downs were completed from non-crosslinked cells a slightly modified protocol from Gong and Maquat (Gong and Maquat, 2015). For samples immediately subjected to western blot analysis, beads were resuspended in 25 μl 2X Laemmli sample buffer and incubated at 95°C for 5 minutes. For pulldown extracts subjected to Transcription Factor array analysis, 25 μl of wash buffer containing flag peptide at final concentration of 0.1 mg/ml was added and beads were rotated for 30 minutes at 4°C. Twelve μl of eluate was incubated with biotinylated DNA probe mixture from the Signosis® TF Activation Profiling Plate Array I and subjected to downstream analysis, according to manufacturer’s instructions. The signal corresponding to each TF was normalized to that of GATA, and represented as a fold enrichment compared to a cells transfected with a plasmid encoding SLNCR1 without the MS2 stem loop tag.
RIP assays were performed from HEK293T cells co-transfected with pEGFP-C1-AR or pCDNA-Brn3a and the indicated SLNCR1 expressing plasmids.
RNA extraction and cDNA library preparation
RNA was isolated using Trizol® (Life Technologies) and Qiagen RNeasy® Mini Kit and treated with DNase. cDNA was generated using SuperScript III (Invitrogen) reverse transcriptase. The indicated transcripts were quantified using Platinum® SYBR® Green qPCR SuperMix-UDG mix on a CFX384 Touch™ Real-Time PCR Detection System.
The T-test statistics, Pearson correlations, hazard ratio and Kaplan-Meier survival analysis were performed using GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla California USA). Image quantifications were performed using ImageJ software.
Please see Extended Experimental Procedures for complete information.
Supplementary Material
Acknowledgments
We gratefully acknowledge T. Benjamin, M. Hemler, M. Brown and the members of our laboratory, especially J. Carroll, for technical advice and critical discussions; D. Fisher for melanocytes; M. Herlyn for melanomas; and R. Rubio, Y. Wang, A. Holman and the entire team of the Dana-Farber Center for Computational Cancer Biology for RNA-Sequencing. This work was supported by funding from NIH R01 CA140986 and R01 CA185151 and a Claudia Adams Barr Award (to C.D.N.) and K.S. was supported by funding from T32 AI007386.
Footnotes
AUTHOR CONTRIBUTIONS
K.S., C.E.J., F.B., and C.H.Y. conducted experiments, C.E.J. and A.B. performed bioinformatic analyses, and J.R. was consulted for statistical analyses. Experiments were designed by K.S., R.J.D., and C.D.N. The manuscript was written by K.S. and C.D.N.
ACCESSION NUMBERS
The data discussed in this publication have been deposited to the NCBI GEO (Edgar et al., 2002), and the accession number for the data reported in this paper is GEO: GSE77903. Sequencing of patient-derived melanoma and fibroblast STCs can be accessed through the NCBI dbGaP Database of Genotypes and Phenotypes under accession number phs001115.v1.p1.
Supplemental Information includes Extended Experimental Procedures, 7 figures and 6 tables and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agoulnik IU, Weigel NL. Coactivator selective regulation of androgen receptor activity. Steroids. 2009;74:669–674. doi: 10.1016/j.steroids.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allil PA, Visconti MA, Castrucci AM, Isoldi MC. Photoperiod and testosterone modulate growth and melanogenesis of s91 murine melanoma. Med Chem. 2008;4:100–105. doi: 10.2174/157340608783789185. [DOI] [PubMed] [Google Scholar]
- Altintas DM, Shukla MS, Goutte-Gattat D, Angelov D, Rouault JP, Dimitrov S, Samarut J. Direct cooperation between androgen receptor and E2F1 reveals a common regulation mechanism for androgen-responsive genes in prostate cells. Mol Endocrinol. 2012;26:1531–1541. doi: 10.1210/me.2012-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berwick DC, Diss JK, Budhram-Mahadeo VS, Latchman DS. A simple technique for the prediction of interacting proteins reveals a direct Brn-3a-androgen receptor interaction. The Journal of biological chemistry. 2010;285:15286–15295. doi: 10.1074/jbc.M109.071456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidoli E, Fratino L, Bruzzone S, Pappagallo M, De Paoli P, Tirelli U, Serraino D. Time trends of cancer mortality among elderly in Italy, 1970–2008: an observational study. BMC cancer. 2012;12:443. doi: 10.1186/1471-2407-12-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang J, Yu G, Liu W, Pearce D. Androgen and glucocorticoid receptor heterodimer formation. A possible mechanism for mutual inhibition of transcriptional activity. The Journal of biological chemistry. 1997;272:14087–14092. doi: 10.1074/jbc.272.22.14087. [DOI] [PubMed] [Google Scholar]
- Chooniedass-Kothari S, Emberley E, Hamedani MK, Troup S, Wang X, Czosnek A, Hube F, Mutawe M, Watson PH, Leygue E. The steroid receptor RNA activator is the first functional RNA encoding a protein. FEBS letters. 2004;566:43–47. doi: 10.1016/j.febslet.2004.03.104. [DOI] [PubMed] [Google Scholar]
- Clark WH, Jr, From L, Bernardino EA, Mihm MC. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer research. 1969;29:705–727. [PubMed] [Google Scholar]
- Colley SM, Leedman PJ. Steroid Receptor RNA Activator - A nuclear receptor coregulator with multiple partners: Insights and challenges. Biochimie. 2011;93:1966–1972. doi: 10.1016/j.biochi.2011.07.004. [DOI] [PubMed] [Google Scholar]
- de Giorgi V, Gori A, Grazzini M, Rossari S, Scarfi F, Corciova S, Verdelli A, Lotti T, Massi D. Estrogens, estrogen receptors and melanoma. Expert Rev Anticancer Ther. 2011;11:739–747. doi: 10.1586/era.11.42. [DOI] [PubMed] [Google Scholar]
- de Vries E, Houterman S, Janssen-Heijnen ML, Nijsten T, van de Schans SA, Eggermont AM, Coebergh JW. Up-to-date survival estimates and historical trends of cutaneous malignant melanoma in the south-east of The Netherlands. Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2007;18:1110–1116. doi: 10.1093/annonc/mdm087. [DOI] [PubMed] [Google Scholar]
- de Vries E, Nijsten TE, Visser O, Bastiaannet E, van Hattem S, Janssen-Heijnen ML, Coebergh JW. Superior survival of females among 10,538 Dutch melanoma patients is independent of Breslow thickness, histologic type and tumor site. Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2008;19:583–589. doi: 10.1093/annonc/mdm498. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergun A, Lawrence CA, Kohanski MA, Brennan TA, Collins JJ. A network biology approach to prostate cancer. Molecular systems biology. 2007;3:82. doi: 10.1038/msb4100125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher DE, Geller AC. Disproportionate burden of melanoma mortality in young U.S. men: the possible role of biology and behavior. JAMA dermatology. 2013;149:903–904. doi: 10.1001/jamadermatol.2013.4437. [DOI] [PubMed] [Google Scholar]
- Flockhart RJ, Webster DE, Qu K, Mascarenhas N, Kovalski J, Kretz M, Khavari PA. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome research. 2012;22:1006–1014. doi: 10.1101/gr.140061.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba CS, Clarke CA, Keegan TH, Tao L, Swetter SM. Melanoma survival disadvantage in young, non-Hispanic white males compared with females. JAMA dermatology. 2013;149:912–920. doi: 10.1001/jamadermatol.2013.4408. [DOI] [PubMed] [Google Scholar]
- Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nature reviews Molecular cell biology. 2013;14:699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller AC, Miller DR, Annas GD, Demierre MF, Gilchrest BA, Koh HK. Melanoma incidence and mortality among US whites, 1969–1999. Jama. 2002;288:1719–1720. doi: 10.1001/jama.288.14.1719. [DOI] [PubMed] [Google Scholar]
- Gong C, Maquat LE. Affinity Purification of Long Noncoding RNA-Protein Complexes from Formaldehyde Cross-Linked Mammalian Cells. Methods in molecular biology. 2015;1206:81–86. doi: 10.1007/978-1-4939-1369-5_7. [DOI] [PubMed] [Google Scholar]
- Gruber CA, Rhee JM, Gleiberman A, Turner EE. POU domain factors of the Brn-3 class recognize functional DNA elements which are distinctive, symmetrical, and highly conserved in evolution. Mol Cell Biol. 1997;17:2391–2400. doi: 10.1128/mcb.17.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Miyazaki H, Lee A, Tran CP, Reiter RE. Androgen receptor and invasion in prostate cancer. Cancer research. 2008;68:1128–1135. doi: 10.1158/0008-5472.CAN-07-1929. [DOI] [PubMed] [Google Scholar]
- Harvard, B.I.o.M.a. Broad Institute TCGA Genome Data Analysis Center: Firehose. 2015. [Google Scholar]
- Hofmann UB, Houben R, Brocker EB, Becker JC. Role of matrix metalloproteinases in melanoma cell invasion. Biochimie. 2005;87:307–314. doi: 10.1016/j.biochi.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Hohenauer T, Berking C, Schmidt A, Haferkamp S, Senft D, Kammerbauer C, Fraschka S, Graf SA, Irmler M, Beckers J, et al. The neural crest transcription factor Brn3a is expressed in melanoma and required for cell cycle progression and survival. EMBO molecular medicine. 2013;5:919–934. doi: 10.1002/emmm.201201862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlader N, NA, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z,Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA. SEER Cancer Statistics Review, 1975–2012. National Cancer Institute; Bethesda, MD: 2015. http://seer.cancer.gov/csr/1975_2012/), pp. based on November 2014 SEER data submission, posted to the SEER web site. [Google Scholar]
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et al. The landscape of long noncoding RNAs in the human transcriptome. Nature genetics. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosse A, Collette S, Suciu S, Nijsten T, Lejeune F, Kleeberg UR, Coebergh JW, Eggermont AM, de Vries E. Superior outcome of women with stage I/II cutaneous melanoma: pooled analysis of four European Organisation for Research and Treatment of Cancer phase III trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:2240–2247. doi: 10.1200/JCO.2011.38.0584. [DOI] [PubMed] [Google Scholar]
- Joosse A, de Vries E, Eckel R, Nijsten T, Eggermont AM, Holzel D, Coebergh JW, Engel J, Munich Melanoma G. Gender differences in melanoma survival: female patients have a decreased risk of metastasis. The Journal of investigative dermatology. 2011;131:719–726. doi: 10.1038/jid.2010.354. [DOI] [PubMed] [Google Scholar]
- Jorgensen JS, Nilson JH. AR suppresses transcription of the alpha glycoprotein hormone subunit gene through protein-protein interactions with cJun and activation transcription factor 2. Mol Endocrinol. 2001;15:1496–1504. doi: 10.1210/mend.15.9.0690. [DOI] [PubMed] [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, et al. Defining functional DNA elements in the human genome. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitan D, Dinger ME, Mazar J, Crawford J, Smith MA, Mattick JS, Perera RJ. The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer research. 2011;71:3852–3862. doi: 10.1158/0008-5472.CAN-10-4460. [DOI] [PubMed] [Google Scholar]
- Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic acids research. 2007;35:W345–349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, O’Malley BW. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. doi: 10.1016/s0092-8674(00)80711-4. [DOI] [PubMed] [Google Scholar]
- Lee YF, Shyr CR, Thin TH, Lin WJ, Chang C. Convergence of two repressors through heterodimer formation of androgen receptor and testicular orphan receptor-4: a unique signaling pathway in the steroid receptor superfamily. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14724–14729. doi: 10.1073/pnas.96.26.14724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Xuan Z, Liu C. Long non-coding RNAs and complex human diseases. International journal of molecular sciences. 2013a;14:18790–18808. doi: 10.3390/ijms140918790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WQ, Qureshi AA, Ma J, Goldstein AM, Giovannucci EL, Stampfer MJ, Han J. Personal history of prostate cancer and increased risk of incident melanoma in the United States. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013b;31:4394–4399. doi: 10.1200/JCO.2013.51.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall JR, Bani MR, Lin Y, Muschel RJ, Kerbel RS. ‘Proteolytic switching’: opposite patterns of regulation of gelatinase B and its inhibitor TIMP-1 during human melanoma progression and consequences of gelatinase B overexpression. British journal of cancer. 1999;80:504–512. doi: 10.1038/sj.bjc.6690385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall JR, Bani MR, Lin Y, Rak J, Kerbel RS. The 92-kDa gelatinase B is expressed by advanced stage melanoma cells: suppression by somatic cell hybridization with early stage melanoma cells. Cancer research. 1995;55:4174–4181. [PubMed] [Google Scholar]
- Micheli A, Ciampichini R, Oberaigner W, Ciccolallo L, de Vries E, Izarzugaza I, Zambon P, Gatta G, De Angelis R, Group EW. The advantage of women in cancer survival: an analysis of EUROCARE-4 data. European journal of cancer. 2009;45:1017–1027. doi: 10.1016/j.ejca.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Morvillo V, Luthy IA, Bravo AI, Capurro MI, Portela P, Calandra RS, Mordoh J. Androgen receptors in human melanoma cell lines IIB-MEL-LES and IIB-MEL-IAN and in human melanoma metastases. Melanoma research. 2002;12:529–538. doi: 10.1097/00008390-200212000-00002. [DOI] [PubMed] [Google Scholar]
- Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grutzner F, Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505:635–640. doi: 10.1038/nature12943. [DOI] [PubMed] [Google Scholar]
- Prensner JR, Sahu A, Iyer MK, Malik R, Chandler B, Asangani IA, Poliakov A, Vergara IA, Alshalalfa M, Jenkins RB, et al. The IncRNAs PCGEM1 and PRNCR1 are not implicated in castration resistant prostate cancer. Oncotarget. 2014;5:1434–1438. doi: 10.18632/oncotarget.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JL, Wang TS, Hamilton TA, Lowe L, Sondak VK, Johnson TM. Thin primary cutaneous melanomas: associated detection patterns, lesion characteristics, and patient characteristics. Cancer. 2002;95:1562–1568. doi: 10.1002/cncr.10880. [DOI] [PubMed] [Google Scholar]
- Serviss JT, Johnsson P, Grander D. An emerging role for long non-coding RNAs in cancer metastasis. Frontiers in genetics. 2014;5:234. doi: 10.3389/fgene.2014.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Ma C, Zhu Q, Yuan D, Sun M, Gu X, Wu G, Lv T, Song Y. Upregulation of long intergenic noncoding RNA 00673 promotes tumor proliferation via LSD1 interaction and repression of NCALD in non-small-cell lung cancer. Oncotarget. 2016;7(18):25558–25575. doi: 10.18632/oncotarget.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- Spanogle JP, Clarke CA, Aroner S, Swetter SM. Risk of second primary malignancies following cutaneous melanoma diagnosis: a population-based study. Journal of the American Academy of Dermatology. 2010;62:757–767. doi: 10.1016/j.jaad.2009.07.039. [DOI] [PubMed] [Google Scholar]
- Swetter SM, Layton CJ, Johnson TM, Brooks KR, Miller DR, Geller AC. Gender differences in melanoma awareness and detection practices between middle-aged and older men with melanoma and their female spouses. Archives of dermatology. 2009;145:488–490. doi: 10.1001/archdermatol.2009.42. [DOI] [PubMed] [Google Scholar]
- Tang L, Zhang W, Su B, Yu B. Long noncoding RNA HOTAIR is associated with motility, invasion, and metastatic potential of metastatic melanoma. BioMed research international. 2013;2013:251098. doi: 10.1155/2013/251098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Zhang X, Hao Y, Fang Z, He Y. Potential roles of abnormally expressed long noncoding RNA UCA1 and Malat-1 in metastasis of melanoma. Melanoma research. 2014 doi: 10.1097/CMR.0000000000000080. [DOI] [PubMed] [Google Scholar]
- van den Oord JJ, Paemen L, Opdenakker G, de Wolf-Peeters C. Expression of gelatinase B and the extracellular matrix metalloproteinase inducer EMMPRIN in benign and malignant pigment cell lesions of the skin. The American journal of pathology. 1997;151:665–670. [PMC free article] [PubMed] [Google Scholar]
- Verger A, Buisine E, Carrere S, Wintjens R, Flourens A, Coll J, Stehelin D, Duterque-Coquillaud M. Identification of amino acid residues in the ETS transcription factor Erg that mediate Erg-Jun/Fos-DNA ternary complex formation. The Journal of biological chemistry. 2001;276:17181–17189. doi: 10.1074/jbc.M010208200. [DOI] [PubMed] [Google Scholar]
- Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lee SO, Xia S, Jiang Q, Luo J, Li L, Yeh S, Chang C. Endothelial cells enhance prostate cancer metastasis via IL-6-->androgen receptor-->TGF-beta-->MMP-9 signals. Mol Cancer Ther. 2013;12:1026–1037. doi: 10.1158/1535-7163.MCT-12-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CF, Tan GH, Ma CC, Li L. The non-coding RNA llme23 drives the malignant property of human melanoma cells. Journal of genetics and genomics = Yi chuan xue bao. 2013;40:179–188. doi: 10.1016/j.jgg.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Wu JT, Han BM, Yu SQ, Wang HP, Xia SJ. Androgen receptor is a potential therapeutic target for bladder cancer. Urology. 2010;75:820–827. doi: 10.1016/j.urology.2009.10.041. [DOI] [PubMed] [Google Scholar]
- Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, Merkurjev D, Ohgi KA, Meng D, Zhang J, et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013;500:598–602. doi: 10.1038/nature12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Feige E, Poling LL, Levy C, Widlund HR, Khaled M, Kung AL, Fisher DE. Pharmacologic suppression of MITF expression via HDAC inhibitors in the melanocyte lineage. Pigment Cell Melanoma Res. 2008;21:457–463. doi: 10.1111/j.1755-148X.2008.00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Zhao JC, Kim J, Fong KW, Yang YA, Chakravarti D, Mo YY, Yu J. LncRNA HOTAIR Enhances the Androgen-Receptor-Mediated Transcriptional Program and Drives Castration-Resistant Prostate Cancer. Cell Rep. 2015;13:209–221. doi: 10.1016/j.celrep.2015.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BG, Du T, Zang MD, Chang Q, Fan ZY, Li JF, Yu BQ, Su LP, Li C, Yan C, et al. Androgen receptor promotes gastric cancer cell migration and invasion via AKT-phosphorylation dependent upregulation of matrix metalloproteinase 9. Oncotarget. 2014;5:10584–10595. doi: 10.18632/oncotarget.2513. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J, Gonit M, Salazar MD, Shatnawi A, Shemshedini L, Trumbly R, Ratnam M. C/EBPalpha redirects androgen receptor signaling through a unique bimodal interaction. Oncogene. 2010;29:723–738. doi: 10.1038/onc.2009.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, Li Z, Bu D, Sun N, Zhang MQ, et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic acids research. 2015 doi: 10.1093/nar/gkv1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.