

Abstract
Krüppel-like factor 8 (KLF8) plays important roles in cancer and is strictly regulated by various post-translational modifications such as sumoylation, acetylation, ubiquitylation and PARylation. Here we report a novel phosphorylation of KLF8 by ERK2 responsible and critical for the stability of KLF8 protein. The full-length KLF8 protein displays a doublet in SDS-PAGE gel. The upper band of the doublet, however, disappeared when the N-terminal 50 amino acids were deleted. In its full-length the upper band disappeared upon phosphatase treatment or mutation of the serine 48 (S48) to alanine whereas the lower band was lost when the S48 was mutated to aspartic acid that mimics phosphorylated S48. These results suggest that S48 phosphorylation is responsible for the motility up-shift of KLF8 protein. Pharmacological and genetic manipulations of various potential kinases identified ERK2 as the likely one that phosphorylates KLF8 at S48. Functional studies indicated that this phosphorylation is crucial for protecting KLF8 protein from degradation in the nucleus and promoting cell migration. Taken together, this study identifies a novel mechanism of phosphorylation critical for KLF8 protein stabilization and function.
Keywords: KLF8, phosphorylation, ERK, protein stability
Introduction
Krüppel-like factor 8 (KLF8), a member of Krüppel-like transcription factor family, is upregulated and plays important roles in various cancer types [1-11]. KLF8 functions as a dual transcriptional factor and has been shown to repress or activate a variety of cancer-related genes such as E-cadherin [12], KLF4 [13], cyclin D1 [2,14], epidermal growth factor receptor (EGFR) [10], MMP9 and MMP14 [1,5] and epithelial-stromal interaction 1 [9,56]. In addition to regulating cancer-promoting processes including transformation [2], epithelial to mesenchymal transition [12] and metastasis [1,5,9,10], KLF8 also plays a role for DNA repair [15], adipogenesis [16] and Alzheimer’s disease [17]. Indeed, KLF8 is emerging as a critical factor for diverse diseases [7].
Post-translational modification (PTM) is one of the most important protein regulatory mechanisms. Previous studies showed that KLF8 undergoes sumoylation at lysine 67 [13], acetylation at lysine 93 and lysine 95, and potential phosphorylation at serine 165 and serine 80 [13,15,18]. The sumoylation, acetylation and their crosstalk play an important role in KLF8 function [13,18]. The serines 165 and 80 of KLF8 are critical for its nuclear localization and function such as DNA repair [15,19]. Interestingly, previous study with KLF8 truncation mutants revealed that the doublet of KLF8 protein became a single band when ≥ 50 amino acids were deleted from the N-terminus [19], suggesting that a PTM in this deleted region is responsible for the mobility shift and doublet formation. However, none of the PTM sites on KLF8 described above is located within this region. It has been mysterious how the mobility shift occurs and whether it has any impact on the function of KLF8.
In this study, we provide strong evidence that mobility shift of KLF8 protein is due to the phosphorylation at serine 48 by ERK2 and this phosphorylation is essential for maintaining the stability and function of KLF8 protein in the nucleus.
Materials and methods
Antibodies and reagents
Primary antibodies used for western blotting include mouse monoclonal to HA-probe (F-7) (sc-7392) (1:3000), mouse monoclonal to β-actin (C4) (sc-47778) (1:4000), Mouse monoclonal for c-Myc (9E10) (Sc-40) (1:2000), mouse monoclonal to pERK (E-4) (Sc-7383) (1:2000) and rabbit polyclonal to ERK (c-16) (Sc-93) (1:2000) (Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Secondary antibodies were horse radish peroxidase conjugated donkey anti-mouse (715-035-150) and donkey anti-rabbit IgG (711-035-152) (both 1:5000. Jackson ImmunoResearch laboratories, West Grove, PA, USA). Antibody used for co-immunoprecipitation was Anti-HA mouse monoclonal (IP0010) Immunoprecipitation Kit (Sigma-Aldrich, St. Louis, MO, USA). MEK inhibitor PD98059 (513000) and U0126 (662005) as well as the inhibitor of protein synthesis cycloheximide were from Calbiochem (San Diego, CA, USA). Glycogen synthase kinase 3 (GSK3) inhibitor SB216763 (S1075) was from Selleckbiochem (Boston, MA, USA). cyclin-dependent kinase 5 (CDK5) inhibitor Roscovitine (557360) was from Milipore (Billerica, MA, USA). c-Jun N-terminal kinase I (JNKI) inhibitor BI 78D3 (Cat. No. 3314) and JNKII inhibitor AEG 3482 (Cat. No. 2651) were from Tocris (Ellisville, MO, USA). All the inhibitors were reconstituted with DMSO. The alkaline phosphatase calf intestinal phosphatase (CIP. M0290) were purchased from New England Biolabs (Ipswich, MA, USA).
Plasmid construction
The mammalian expression vectors pKH3 (HA-tagged), pHAN (Myc-tagged), pKH3-KLF8, pKH3-KLF8-dN50, pHAN-KLF8 and pHAN-KLF8-dN50 were previously described [20]. KLF8 deletion and point mutants were constructed by site-directed mutagenesis PCR [21] using the pKH3-KLF8 vector as the template. We used the primer pKH3-F paired with mutant-R and primer mutant-F paired with pKH3-R for the mutagenesis PCR and the pKH3-F and pKH3-R primer pair for overlapping PCR. The mutant fragments were digested with HindIII and EcoRI to clone into pKH3 vector between the sate sites. To construct lentiviral vectors pLVPZ-KLF8-S48A and pLVZP-KLF8-S48D, we PCR-amplified the HA-KLF8-S48A and HA-KLF8-S48D fragments from pKH3-KLF8-S48A and pKH3-KLF8-S48D plasmids. These fragments were digested with Pst1/Not1 and inserted into the lentiviral vector pLVPZ [5]. All the constructs were verified by DNA sequencing. The human ERK2 cDNA was amplified using ERK2-SmaI-F and ERK2-ClaI-R primers. This amplified fragment was digested with SmaI/ClaI and cloned into pHAN vector between the same sites. The ERK2 dominant-negative (K54R in which the ATP binding activity is disabled [22,23]) and constitutively active (ERK2-CA, an ERK2 double mutant consisting of R67S that promotes ERK2 autophosphorylation and D321N that inhibits ERK2 phosphatase binding [23-25]) mutants were constructed by site-directed mutagenesis similarly. See Table 1 for sequences of all the primers and oligonucleotides used.
Table 1.
Sequences of primers and oligonucleotides used
| Primer name | Primer sequence (5’-3’) |
|---|---|
| pKH3-F | CCC AAG CTT CTG CAG GTC G |
| pKH3-R | GGA CAA ACC ACA ACT AGA ATG CAG |
| KLF8 d11-20-R | CCT GCA T TG AGT TGT T TA TGA GTT TAT CC |
| KLF8 d11-20-F | AAC AAC TCA ATG CAG GTA TTC AAG C |
| KLF8 d21-30-R | CGA ACA GAG CCA CCT TCT GAA TTA AGT T |
| KLF8 d21-30-F | AGG TGG CTC TGT TCG GAA CAG AGA TC |
| KLF8 d31-40-R | CTG TAT TCA GCA GTG ACC TGC TTG AAT A |
| KLF8 d31-40-F | TCA CTG CTG AAT ACA GAA GTA ATA TGA CT |
| KLF8 d41-50-R | ATC CAG GAG TAT CTC AGG GGG ATC TCT |
| KLF8 d41-50-F | TGA GAT ACT CCT GGA TGC CAA CCC CAT |
| KLF8 S31A-R | CCG AAC AGC AGC AGT GAC CTG |
| KLF8 S31A-F | ACT GCT GCT GTT CGG AAC AGA GAT C |
| KLF8 Y42A-R | ACT TCT GGC TTC TAT CTC AGG G |
| KLF8 Y42A-F | ATA GAA GCC AGA AGT AAT ATG ACT TCT CC |
| KLF8 S44A-R | CAT ATT AGC TCT GTA TTC TAT CTC AGG G |
| KLF8 S44A-F | TAC AGA GCT AAT ATG ACT TCT CCA ACA |
| KLF8 T47A-R | TGG AGA AGC CAT ATT ACT TCT GTA TTC |
| KLF8 T47A-F | AAT ATG GCT TCT CCA ACA CTC CTG |
| KLF8 S48A-R | TGT TGG AGC AGT CAT ATT ACT TCT |
| KLF8 S48A-F | ATG ACT GCT CCA ACA CTC CTG GAT |
| KLF8 T50A-R | CAG GAG TGC TGG AGA AGT CAT ATT |
| KLF8 T50A-F | TCT CCA GCA CTC CTG GAT GCC |
| KLF8 S48D-R | TGT TGG ATC AGT CAT ATT ACT TCT GTA |
| KLF8 S48D-F | ATG ACT GAT CCA ACA CTC CTG GAT |
| KLF8 d31-35-F | ACT GCT GAT CCC CCT GAG ATA GAA |
| KLF8 d31-35-R | AGG GGG ATC AGC AGT GAC CTG CTT |
| KLF8 d36-40-F | CGG A CGG AAC AGA GAA TAC AGA AGT AAT ATG AC AGA |
| KLF8 d36-40-R | TCT GTA TTC TCT GTT CCG AAC AGA AGC |
| KLF8 P37A-R | TAT CTC AGG GGC ATC TCT GTT CCG |
| KLF8 P37A-F | AGA GAT GCC CCT GAG ATA GAA TAC |
| KLF8 P38A-R | TAT CTC AGC GGG ATC TCT GTT CCG |
| KLF8 P38A-F | AGA GAT CCC GCT GAG ATA GAA TAC |
| KLF8 P37/38A-R | TAT CTC AGC GGC ATC TCT GTT CCG |
| KLF8 P37/38A-F | AGA GAT GCC GCT GAG ATA GAA TAC |
| KLF8 D36A-R | AGG GGG AGC TCT GTT CCG AAC AGA |
| KLF8 D36A-F | AAC AGA GCT CCC CCT GAG ATA GAA |
| KLF8 E39A-R | TTC TAT CGC AGG GGG ATC TCT GTT |
| KLF8 E39A-F | TCC CCC TGC GAT AGA ATA CAG AAG |
| KLF8 D36/E39A-R | TTC TAT CGC AGG GGG AGC TCT GTT |
| KLF8 D36/E39A-F | AAC AGA GCT CCC CCT GCG ATA GAA |
| KLF8 I40A-R | GTA TTC TGC CTC AGG GGG ATC TCT |
| KLF8 I40A-F | CCT GAG GCA GAA TAC AGA AGT AAT |
| KLF8 I40R-R | GTA TTC TCT CTC AGG GGG ATC TCT |
| KLF8 I40R-F | CCT GAG AGA GAA TAC AGA AGT AAT |
| ERK2-SmaI-F | TTT CCC GGG AGC AGC TGC GGC GGC GGC GGG CGC |
| ERK2-ClaI-R | CCC ATC GAT TTA AGA TCT GTA TCC TGG CTG GAA TC |
| ERK2 K54R-F | GCT ATC AGG AAA ATC AGC CCC TTT |
| ERK2 K54R-R | GAT TTT CCT GAT AGC TAC TCG AAC |
| ERK2 R67S-F | TGC CAG AGC ACC CTG AGG GAG ATA |
| ERK2 R67S-R | CAG GGT GCT CTG GCA GTA GGT CTG |
| ERK2 D321N-F | CCG AGT AAC GAG CCC ATC GCC GAA |
| ERK2 D321N-R | GGG CTC GTT ACT CGG GTC GTA ATA |
Cell culture, cell line generation and transfection
The HEK293 [21,26,27] and MCF-7 [2,12] cell lines were described previously. These cells were maintained in DMEM with 10% fetal bovine serum (FBS) and proper antibiotic supplements. MCF7 stable cells expressing HA-KLF8 (MCF7-K8) and its mutant S48A (MCF7-K8-S48A) and S48D (MCF7-K8-S48D) were generated by infecting the MCF7 cells with lentiviruses derived from the corresponding lentiviral vectors followed by puromycin selection. Selected cells were maintained in DMEM supplemented with 10% FBS and puromycin. GSK3α-/- and GSK3β-/- and matched wild-type mouse embryonic fibroblasts (MEFs) were kind gifts from Dr. Jim Woodgett of Lunenfeld-Tanenbaum Research Institute [28]. These cells were maintained in DMEM plus 10% FBS. Transfections were done using Lipofectamine 2000 (Invitrogen, Grand Island, NY, USA).
Phosphatase treatment
After 36-48 h of transfection, cells were washed with ice-cold phosphate-buffered saline, lysed with NP-40 buffer containing protease inhibitor cocktail (1 mM PMSF, 0.2 IU/ml approtini and 20 μg/ml leupeptin) for 30 minutes at 4°C and centrifuged (12,000 rpm, 10 min, 4°C) to obtain the cell lysate. A aliquot of 20 μg lysate was treated with 30 units of CIP in 40 μl reaction with NEBuffer 3 for 2 h at 37°C followed by SDS-PAGE and western blotting. Untreated lysate and lysate treated with CIP plus its inhibitor sodium orthovanadate (20 mM) were included as controls.
Western blotting and co-immunoprecipitation (Co-IP)
These assays were done as previously described [12,15]. Cells and antibodies used were described above. Western blots were quantified using Image Lab 3.0 (Bio-Rad, Hercules, CA) as previously described [15].
Protein lifespan assay
Cycloheximide (CHX) chase assay was performed essentially as previously described [15]. The HA-KLF8 and its mutant proteins overexpressed either transiently in HEK293 cells or stably in the MCF7 cell lines described above were treated with 50 μg/ml of CHX. In some experiment, the MEK inhibitor U0126 was included in the medium at 10 μM for 30 minutes prior to addition of CHX and at 3 μM during the CHX chase period of time. Lysates were collected at different time points for western blotting.
Cell migration assays
MCF7, MCF7-K8, MCF7-K8-S48A and MCF7-K8-S48D cells were seeded in a 12-well plate and grown to confluent state. Would closure (24 h) and Boyden Chamber (20 h) migrations were performed and analyzed quantitatively as previously described [12].
Statistical analysis
At least three observations per group were conducted. Data are presented as mean ± the standard deviation. Unpaired, paired or single sample Student’s t-test with the Bonferroni correction for the multiple comparisons was applied as appropriate. Statistical significance was determined using the alpha level of 0.05.
Results
Mobility shift of KLF8 is due to phosphorylation among its N-terminal 50 amino acids
The full-length wild-type KLF8 protein migrates as a doublet on SDS-PAGE and deletion of 50 or more amino acids from the N-terminus changes the doublet into a single band [19] regardless of the epitope types attached (Figure 1A). Since the known PTMs of KLF8 do not have evident effect on the mobility shift [13,15,18-20,29] and protein phosphorylation most frequently causes protein mobility up-shift [30,31], we sought to determine if phosphorylation plays a role for the KLF8’s mobility shift observed. We treated the cell lysates containing epitope-tagged KLF8 protein with calf intestine phosphatase (CIP) that removes phosphate group from phosphorylated serine, threonine or tyrosine. The result clearly showed that CIP treatment significantly reduced, if not abolished, the upper band of the doublet irrespective of the tag types (Figure 1B, compare middle to left lane). This is specific to the catalytic activity of CIP in that the loss of the upper band was prevented with the CIP inhibitor sodium orthovanadate (Na3VO4) [32] (Figure 1B, lane 3). Taken together, these results strongly suggest that phosphorylation of a residue among the N-terminal 50 amino acids is responsible for the mobility shift of KLF8 protein.
Figure 1.
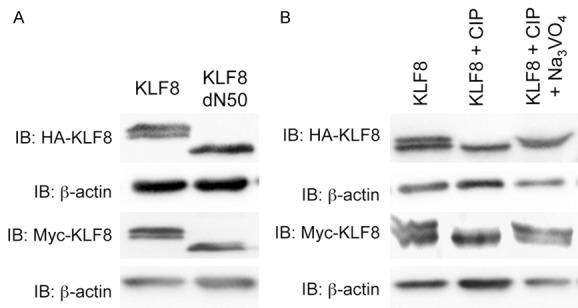
Mobility shift of KLF8 is due to phosphorylation in its N-terminal 50 amino acid residues. A. The N-terminal region of 50 residues is required for maintaining the doublet of KLF8 protein on SDS-PAGE gels. HA-KLF8, HA-KLF8-dN50, Myc-KLF8 or Myc-KLF8-dN50 was transfected into HEK293 cells. Whole cell lysates were collected after 48 hours and processed for western blotting with anti-HA (1:3000) or anti-Myc (1:2000) antibody with anti-β-actin as loading control. B. Treatment with alkaline phosphatase abolished the upper band of the KLF8 doublet. HA-KLF8 or Myc-KLF8 was oeverexpressed in HEK293 cells. Lysates prepared from the cells were treated with CIP without or with its inhibitor sodium orthovanadate (Na3VO4) as described in the Experimental Procedures and processed for western blotting as described above.
Phosphorylation of KLF8 at the serine 48 is responsible for the mobility up-shift
To determine the phosphorylation site, we first constructed small deletion mutants of KLF8 by deleting 10 amino acids at a time from the N-terminal 50 amino acid region (Figure 2A). Deletion of either the residues 31-40 or 41-50 resulted in a loss of the upper band, whereas other deletions did not affect the mobility (Figure 2B). This result suggested that the phosphorylated residue of KLF8 is somewhere between the amino acids 30 and 50. To identify the exact amino acid(s) that is phosphorylated, we mutated all the serine, threonine and tyrosine residues in the 31-50 region to alanine individually to prevent phosphorylation. We found that only the S48A mutant migrated as a single band with mobility identical to the lower band of the doublet (Figure 2C), whereas the S48D mutant, where the serine was mutated to aspartic acid to mimic the phosphorylated serine [33,34], migrated as a single band with mobility identical to the upper band of the doublet (Figure 2D). Thus, KLF8 is likely phosphorylated at S48, which is responsible for the mobility up-shift. Phylogenic analysis showed that the S48 residue is highly conserved across the species (Figure 2E). Taken together, these results provided strong evidence that the S48 can be phosphorylated which is potentially of functional importance for KLF8.
Figure 2.
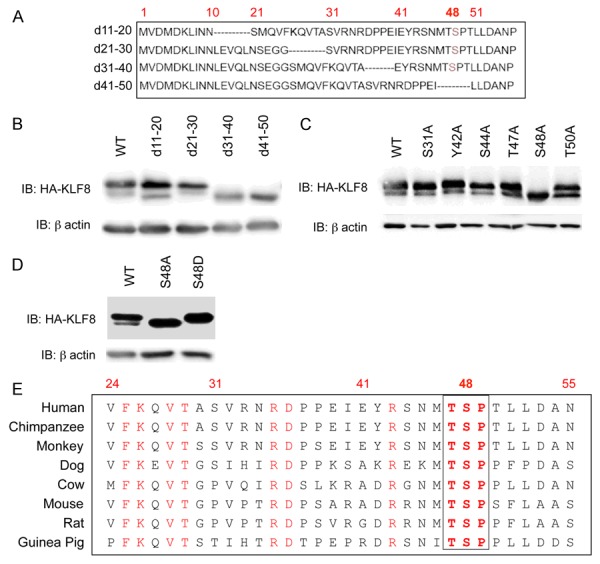
Phosphorylation of KLF8 at the serine 48 (S48) is responsible for the up-shift of KLF8 mobility. (A, B) Both KLF8 31-40 and 41-50 aa regions are essential for the phosphorylation (mobility upshift) of KLF8. Indicated small deletion mutants of KLF8 (A) were overexpessed for western blotting (B). (C, D) The mobility up-shift of KLF8 is due to the phosphorylation at the S48. All the potential phosphorylated residues including serine, threonine and tyrosine residues in the area of residues 31-50 were mutated to alanine and their band patterns were analyzed by overexpression and western blotting (C). Only the S48A mutant showed a single (lower) band. (D) Mutation of S48 to Aspartate results in the upper band only. The phosphorylation-mimicking mutant S48D was overexpressed for western blotting. (E) The S48 site of KLF8 is conserved across the species. Sequence homology was analyzed using the NCBI multiple sequence alignment.
The amino acid 31-40 region plays a regulatory role for the phosphorylation of KLF8 at S48
The loss of the upper band from the d31-40 mutant protein (Figure 2B) is not due to failure for any of the residues in this deleted region to be phosphorylated within this deleted region in that mutation of the serine 31, the only potential phosphorylation residue, to alanine did not alter the mobility of the protein (Figure 2C). This result suggested that, while this region does not contain a phosphorylation site, it might play a role in facilitating the phosphorylation at S48. We then made two smaller deletion mutants of KLF8, i.e., d31-35 and d36-40 and found that interestingly, the d31-35 mutant showed mostly the upper band whereas the d36-40 mutant migrated as only the lower band (Figure 3A). This result suggested that the amino acid 31-35 and 36-40 regions might respectively regulate the phosphorylation at S48 negatively and positively. Indeed, both phosphatase treatment and S48A mutation brought the d31-35 mutant protein to the lower band position, while the S48D-d31-35 mutant protein remained in the upper band position regardless of the phosphatase treatment (Figure 3B). We then mutated each residue to alanine in this region individually or in combination and found that only the R33A/R35A mutant showed a migration pattern closest to the d31-35 double-mutant (Figure 3C). This result suggested that this R-X-R motif at 33-35 position might be the one that mediates the negative effect on the phosphorylation at S48. To determine the core residue or motif among 36-40 residues, we similarly mutated each individual residue in this region to alanine. Isoleucine residue at position 40 was also mutated to arginine given that isoleucine and alanine share similar amino acid structure. All the single point-mutants migrated as a doublet similar to the wild-type KLF8. Because proline actively participates in protein folding to maintain protein tertiary structure, we constructed a P37A/P38A double-mutant of KLF8. It has been also reported that D-X-X-E motif is important for binding to metal ions [35] and proteins such as focal adhesion kinase [36]. Hence, we made a D36A/E39A mutant also to disrupt this motif. Interestingly, the D36A/E39A double mutant migrated as the lower band only whereas the P37A/P38A mutation did not alter the mobility (Figure 3D). These results suggested that the D-X-X-E motif at 36-39 positions mediates the positive effect on the phosphorylation at S48. Taken together, the amino acid 31-40 region appears to play an important counteracting role to balance a proper level of phosphorylation of KLF8 at S48 through mechanisms to be determined (Figure 3E).
Figure 3.
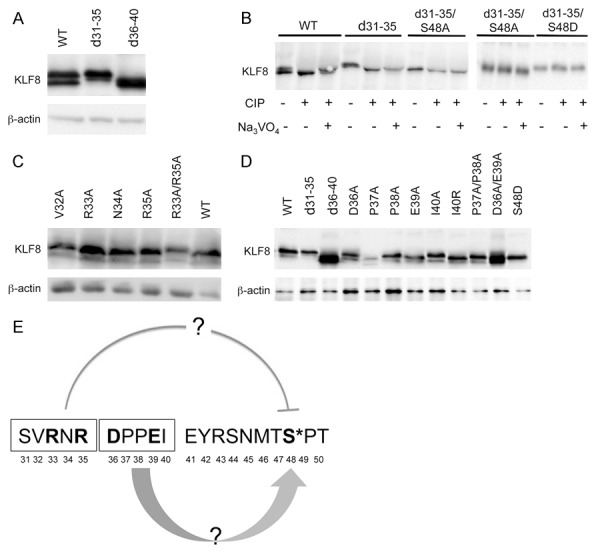
The region of residues 31-40 plays an essential regulatory role in the phosphorylation of KLF8 at the S48 site. The R-X-R motif in 31SVRNR35 of KLF8 plays an inhibitory role in the phosphorylation of KLF8 at the S48 (A-C) whereas the D-X-X-E motif in 36DPPEI40 of KLF8 has a positive effect on it (D). Indicated KLF8 mutants were overexpressed and lysates were prepared with or without CIP treatment in the presence or absence of the CIP inhibitor for western blotting. (E) The counteracting role of the R-X-R and D-X-X-E motifs was illustrated.
ERK2 phosphorylates KLF8 at S48
We then sought to determine the protein kinase responsible for the phosphorylation of KLF8 at S48. We first attempted to predict potential kinase(s) for the S48 site using the online application programs including NetPhosK [37] (http://www.cbs.dtu.dk/services/NetPhosK/), PhosphoNet (http://www.phosphonet.ca/) and Kinasephos (http://kinasephos.mbc.nctu.edu.tw/). The GSK3, CDK5, ERK and JNK stood out as the potential kinase candidates with GSK3β having the highest predicted score. Next, we treated the HEK293 cells overexpressing HA-KLF8 with specific inhibitors of these kinases. Only the MEK1/2 inhibitor treatment showed less KLF8 phosphorylation compared to DMSO treatment (Figure 4A). As the GSK3 inhibitor used was for both GSK3α and GSK3β, we transfected KLF8 into GSK3α knockout (GSK3α-/-) and GSK3β knockout (GSK3β-/-) mouse embryonic fibroblast (MEF) cells [28]. KLF8 protein mobility remained unchanged in these knockout cells as in the wild-type MEF cells (Figure 4B), thereby, excluding GSK3s. These results indicated that the MEK-ERK pathway might play a role in the phosphorylation of KLF8 at S48.
Figure 4.
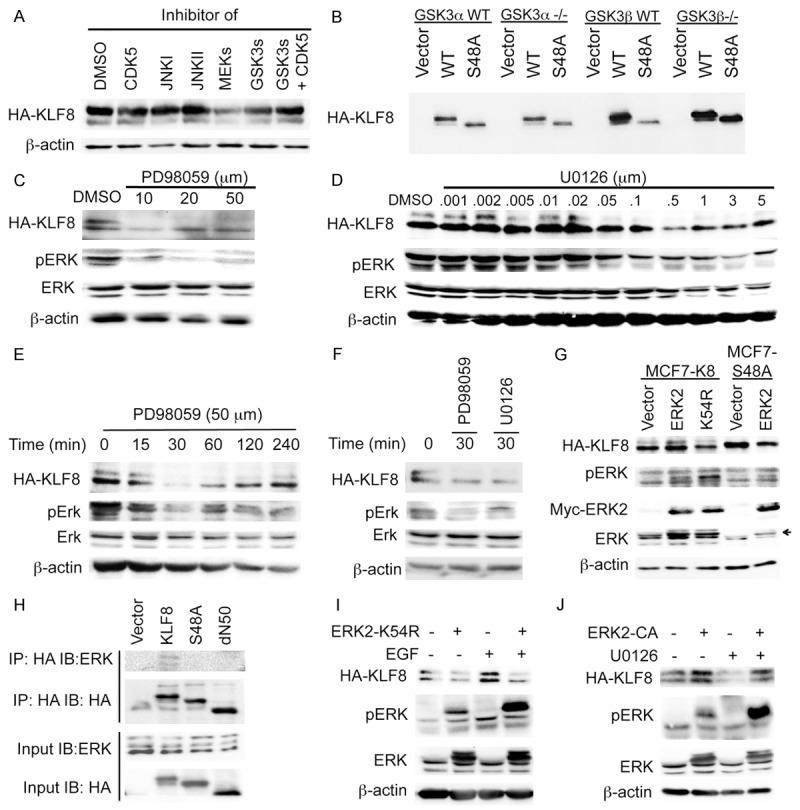
ERK2 is the kinase responsible for the phosphorylation of KLF8 at the S48 site. (A) Inhibition of MEKs, but not JNKs, GSK3s or CDK5, leads to a decrease in the phosphorylation of KLF8. HEK293 cells overexpressing KLF8 were treated with indicated inhibitors for 4 h. Cell lysates were prepared for western blotting. (B) Knockout of GSK3s does not affect mobility of KLF8 protein. Wild-type KLF8 (WT), its S48A mutant or vector control was overexpressed in the indicated cells for 48 h. Cell lysates were prepared for western blotting. (C-F) Dose- and time-dependent inhibition of the phosphorylation of KLF8 by MEK inhibitors. The MCF7-K8 cells at ~80% confluent growth were treated with PD98059 or U0126 as indicated, cell lysates were processed for western blotting for the band patterns of KLF8. Total ERK, pERK and β-actin were included as controls. For the dose-dependent study the treatment time was 4 hours. In (F) 50 µM PD98059 and 5 µM U0126 were used respectively. (G) The catalytic activity of ERK2 is the phosphorylation of KLF8 at the S48 site. Myc-tagged ERK2 or its dominant-negative mutant K54R was overexpressed in the MCF7-K8 or MCF-K8S48A cells. Lysates were prepared after 48 hours for western blotting. (H) ERK2 interacts with KLF8 in an S48-dependent manner. Indicated KLF8, its mutant or vector control was overexpressed in HEK293 cells and precipitated with anti-HA antibody from cell lysates prepared after 48 hours. Co-precipitated ERKs were analyzed by western blotting. (I) ERK2 activity is required for EGF-stimulated phosphorylation of KLF8. The MCF7-K8 cells were transfected with dominant-negative ERK2 mutant K54R plasmid or vector alone. After 36 h, the cells were serum-starved for 16 h and then treated with 50 ng/ml EGF or DMSO. Cell lysates were collected after 30 minutes for western blotting. (J) ERK2 activity is sufficient for the phosphorylation of KLF8. The MCF7-K8 cells were transfected with the ERK2 constitutive active mutant (ERK2-CA). After 36 h, the cells were treated with U0126 (5 μM) for 30 minutes. Cell lysates were then prepared for western blotting.
To determine cancer relevance of the aforementioned results, we generated MCF7 cell lines stably overexpressing the wild-type KLF8 (MCF7-K8) and the S48A mutant (MCF7-K8-S48A), respectively. Treating the MCF7-K8 cells with the MEK inhibitors, PD98059 or U0126, caused a dramatic reduction in the phosphorylation of KLF8 that is well correlated with the decrease in the phosphorylation of ERKs (Figure 4C-F). This result suggested that MEK(s) or/and ERK(s) could be responsible for the phosphorylation of KLF8 in the breast cancer cells. Since the above described computer programs predicted ERK(s) rather than MEKs as a potential KLF8 kinase, and the phosphorylation of ERK2 appeared to be inhibited more than ERK1 did by the MEK inhibitors in the cells (Figure 4C-F), we next tested the potential phosphorylation of KLF8 by ERK2. Overexpression of ERK2 increased the phosphorylation of KLF8 whereas its kinase dead, dominant negative mutant (ERK2-K54R) decreased the phosphorylation of KLF8 (Figure 4G). By contrast, the S48A mutant of KLF8 did not show any phosphorylated upper band upon ERK2 overexpression (Figure 4G). These results strongly suggested that ERK2 could phosphorylate KLF8 at S48. This notion was further supported by the co-IP assay that detected an interaction between ERK2 and KLF8 in the cells (Figure 4H). In addition, EGF stimulation of ERKs clearly increased the phosphorylation of KLF8, which was blocked by the ERK2 dominant negative mutant (Figure 4I). Furthermore, overexpression of the constitutively active mutant (ERK2-CA) prevented inhibition of the phosphorylation of KLF8 by the MEK inhibitor U0126 (Figure 4J). Taken together, these results identified ERK2 as the kinase likely responsible for the phosphorylation of KLF8 at the S48 site.
Phosphorylation of KLF8 at S48 maintains the stability of KLF8 protein
The phosphorylation of KLF8 appears to correlate with the total levels of KLF8 (Figure 4A and 4J), suggesting a potential protection of KLF8 protein from degradation by the phosphorylation at the S48 site. Indeed, both the phosphorylation-defective mutants, KLF8-S48A and KLF8-d41-50, expressed at a significant lower level than the wild-type KLF8 did (Figure 5A). We performed protein chase assays in transient transfected HEK 293 cells and found out that the wild-type KLF8 and the S48A mutant have the longest and shortest life-span, respectively (Figure 5B and 5C). Unexpectedly yet interestingly, the S48D and d31-35 mutants did not show a prolonged life over the wild-type KLF8 although the d36-40 mutant, like the S48A mutant, showed a shortened lifespan compared to the wild-type KLF8 (Figure 5B and 5C). Taken together, these results suggested that the phosphorylation at S48 is critical for maintaining the stability of KLF8 protein.
Figure 5.
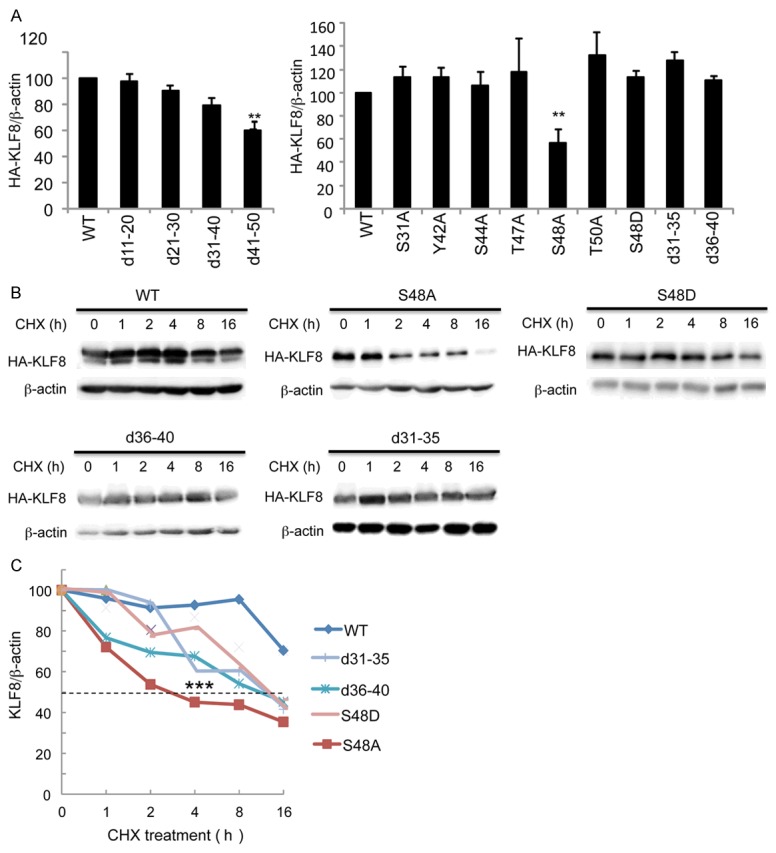
Phosphorylation of KLF8 at the S48 site is critical for KLF8 stability. A. The unphosphorylated KLF8 mutant proteins express at a decreased level. Indicated HA-tagged KLF8 and its mutants were overexpressed in HEK293 cells for 48 h prior to preparation of cell lysates for western blotting with HA and β-actin antibodies and quantification of protein expression. B, C. The unphosphorylated KLF8 mutant proteins have a shortened lifespan. Indicated HA-KLF8 and its mutants were overexpressed in HEK293 cells for 36 h. CHX drug was then added and cell lysates were collected at eh indicated time points for western blotting and protein quantification.
Phosphorylated KLF8 acts as a mask to protect the overall stability of KLF8 protein
In the MCF7 stable cell lines, we obtained stability patterns for the wild-type KLF8 and the S48A mutant similar to what was observed in the HEK293 cells (Figure 6A and 6B). Surprisingly, the unphosphorylated form (lower band) of the wild-type KLF8 stayed more stable than the phosphorylated form (upper band) (Figure 6B). These results raised an interesting possibility that the phosphorylation of KLF8 at S48 helps maintain the overall KLF8 protein levels by protecting the unphosphorylated form of KLF8 from degradation. This notion was further supported by the result showing that the lower band of KLF8 was degraded faster in the cells treated with the MEK inhibitor than that in the mock treated cells (Figure 6C and 6D). Therefore, the phosphorylated KLF8 may sacrifice itself to protect the unphosphorylated form of it.
Figure 6.
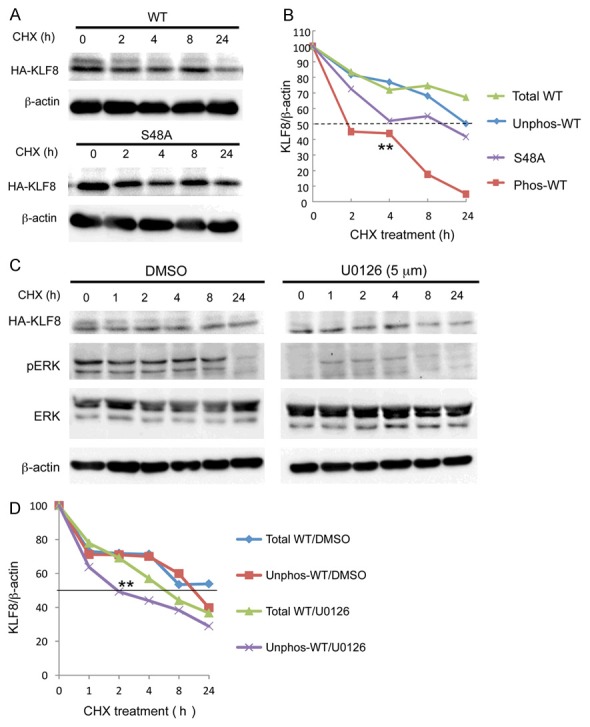
The phosphorylated form of KLF8 acts as a mask to protect the overall stability of KLF8 protein. A, B. The phosphorylated form of KLF8 protein degrades to protect the unphosphorylated form. The MCF7-K8 and MCF7-K8-S48 cells were subjected to CHX chase assay as described above. The upper and lower bands of the wild-type KLF8 were quantified separately and compared to total expression of the wild-type KLF8 as well as the S48A mutant protein. C, D. The lifespan of the unphosphorylated form of KLF8 protein decreases in the absence of the phosphorylated form. The CHX chase assay was carried out similarly except that the MCF7-K8 cells were treated with the MEK inhibitor U0126 inhibitor before and during the chase assay as described in the Experimental Procedures.
KLF8 requires the phosphorylation at the S48 site to promote cell migration
Lastly, we attempted to see if the phosphorylation of KLF8 at the S48 site plays a role for any known cellular function of KLF8. We performed two independent migration assays both of which demonstrated that the S48A, but not the S48D, mutant lost the capacity to promote the MCF7 cell migration (Figure 7). This result suggested that the phosphorylation of KLF8 at the S48 site by ERK2 is critical for KLF8 function.
Figure 7.
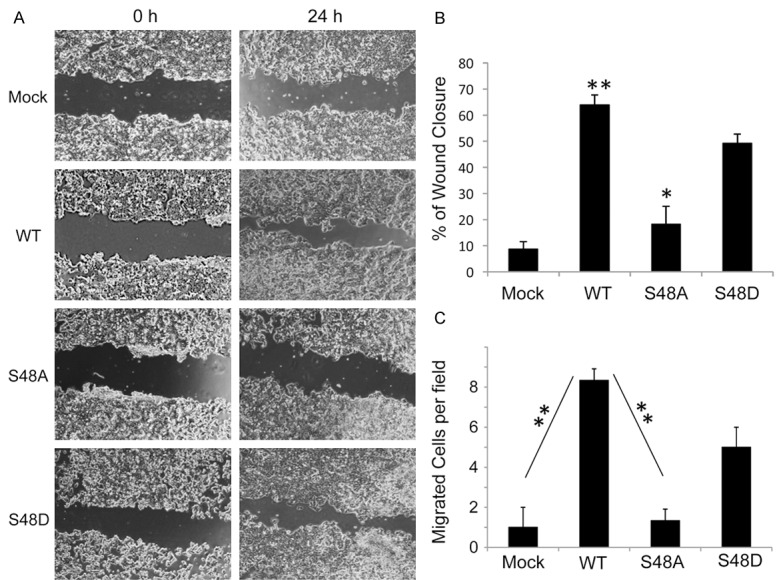
The phosphorylation at the S48 is essential for KLF8 to promote cell migration. The MCF7-K8 (WT), MCF7-K8-S48A, MCF7-K8-S48D alongside the mock control cells were grown and subject to wound closure (A, B) as well as Boyden chamber (C) migration assays as described in the Experimental Procedures.
Discussion
This study identified the serine 48 as a novel phosphorylation site on KLF8 responsible for its mobility shift and ERK2 as a novel kinase for the phosphorylation of KLF8 at this site. Based on the results of the study, we propose a model of mechanism of action (Figure 8) in which ERK2 phosphorylation of KLF8 at the serine 48 site gears the degradation of KLF8 protein from the unphosphorylated towards phosphorylated form of the protein to protect the stability of unphosphorylated form. The unphosphorylated form may be the primary functional form of KLF8. In addition, the phosphorylation at the serine 48 is critically balanced by N-terminal region of residues 31-40 where the regions of 31-35 and 36-40 have a negative and positive effect, respectively via unknown mechanisms.
Figure 8.
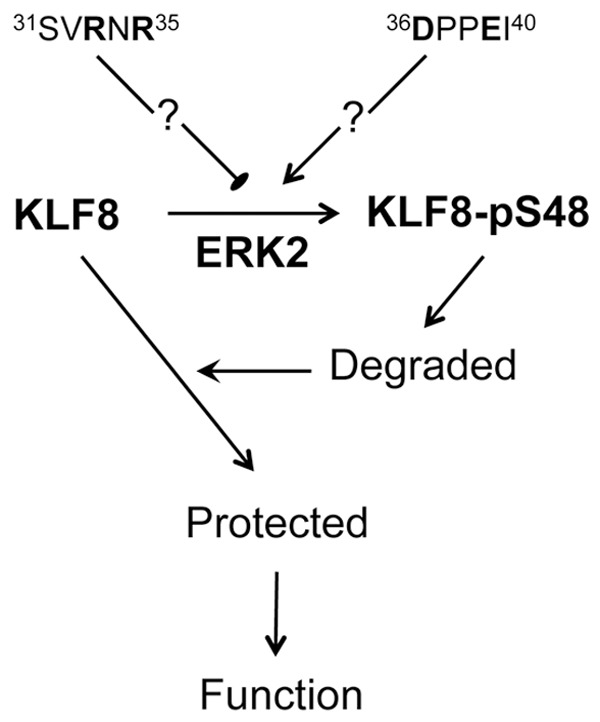
Proposed model of the mechanism of action. ERK2 phosphorylate KLF8 at the S48 site. This phosphorylation distracts KLF8 degradation of unknown mechanism away from the unphosphorylated form towards the phosphorylated form of the protein, resulting in an increase in overall stability of the KLF8 protein. On the other hand, the ERK2 phosphorylation of KLF8 is balanced by two potential counteracting mechanisms of regulation mediated by the inhibitory R-N-R motif and the supporting D-P-P-E motif.
In support of this study, it was reported that ERK2 phosphorylates c-Myc at serine 62 to increase the stability of c-Myc protein [38], and blocking this phosphorylation by inhibiting ERK2 leads to a marked decrease in the c-Myc protein level and tumor malignancy [39]. In addition to KLF8, other KLF family members such as KLF5 [40], KLF3 [41] and KLF2 [42] are also regulated by phosphorylation. Indeed, it has been reported that ERK2 phosphorylated KLF2 protein is more sensitive to proteasomal degradation [42], and ERK phosphorylation of KLF4 at serine 123 results in inhibition of KLF4’s function [43]. Another recent report showed that ERK inhibitor treatment decreased KLF8 expression and the ERK-KLF8 pathway might play a role in chemoresistance of colorectal cancer cells [4]. Our results provide mechanistic interpretation of this report. Our recent work has revealed that KLF8 upregulates EGFR in breast cancer cells [10]. Given that EGF stimulation enhances the ERK2-dependent phosphorylation of KLF8 (see Figure 4I), it is possible that a potential positive feedback loop of KLF8 to EGFR to ERK to KLF8 exists.
We showed that both the constitutively phosphorylation-mimicking and unphosphorylated mutants of KLF8 protein have a shortened lifespan compared to the total wild-type KLF8 (Figure 5). However, the phosphorylated form (upper band) of wild-type KLF8 has a much shorter lifespan than its unphosphorylated counterpart (lower band) (Figure 6). These seemingly paradoxical observations suggest a protective role of the phosphorylated form for the stability of the unphosphorylated form that might be the functional form of KLF8 in the nucleus.
How the amino acid 31-40 region regulates the phosphorylation of KLF8 at the serine 48 site remains to be determined. The R-X-R motif located in the 33-35 region has been shown to play an essential role in protein-protein interaction [44], endoplasmic reticulum (ER) retention of protein [45], and phosphatase binding [46]. This motif was first identified as an ER retention motif [47,48]. Proteins with this motif tend to show high ER retention and less ER to Golgi transport [49,50]. The positively charged arginine (R) residues can interact with other negatively charged residues to regulate protein tertiary structure [51]. This motif is also involved in co-factor binding [49]. Interestingly, two reports suggested that F-X-X-R-X-R motif is the binding site of the phosphatase PP1 [46,52]. It will be interesting to test if the KLF8’s 30ASVRNR35 motif plays a docking role for PP1 or PP1-like phosphatases. The D-X-X-E in the 36-39 region, unlike R-X-R motif, had a positive influence on the phosphorylation of KLF8. D-X-X-E motif was first found on β-integrin cytoplasmic tail critical for focal adhesion kinase binding [36]. Recent studies have also demonstrated the binding of D-X-X-E motif to the metal ion of magnesium, manganese or calcium [35,53,54]. Calcium has been shown to activate ERK through adynyl cyclase [55]. Therefore, it is possible that the KLF8’s 36DPPEI40 motif might play a role in metal binding required for the phosphorylation of KLF8 at the serine 48 site by ERK2.
Previous report showed that poly (ADP-ribose) polymerase 1 binds to KLF8 to promote its nuclear retention and prevent nuclear export and subsequent degradation of KLF8 protein in the cytoplasm [29]. Interestingly, the KLF8 S48A mutant is localized in the nucleus and the phosphorylation of KLF8 at serine 48 does not play any role in poly (ADP-ribose) polymerase 1 binding (data not shown). Therefore, it is likely that the stability of KLF8 protein is regulated in both the cytoplasm and nucleus via distinct molecular mechanisms. Additionally, cross talks between different PTM types on KLF8 have been reported. For instance, a switch between acetylation and sumoylation of KLF8 was found to be critical for its transcriptional activity [18]. Whether or not the phosphorylation of KLF8 at the serine 48 site plays a role for other PTM types is another interesting topic of future study.
Physiologically critical yet pathologically important proteins such as proto-oncogenic and tumor suppressor proteins are tightly regulated in normal cells to maintain their expression and function within the physiological window. Loss of the regulatory balance is a major cause of diseases. Both the S48 site of KLF8 and the function of ERK2 are well conserved across species. PTMs such as protein phosphorylation are among the most important protein regulatory mechanisms. These lines of evidence underscore the biological importance of the ERK2-dependent phosphorylation of KLF8 at the serine 48 site discovered in this study.
Acknowledgements
This work was supported by a grant from National Institute of Health (R01 CA123977) to JZ. We are grateful to Dr. J. Woodgett of Lunenfeld-Tanenbaum Research Institute for generously providing the GSK knockout MEFs.
Disclosure of conflict of interest
None.
References
- 1.Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, DiPersio CM, Feustel PJ, Zhao J. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene. 2011;30:1901–1911. doi: 10.1038/onc.2010.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Zhao J. KLF8 transcription factor participates in oncogenic transformation. Oncogene. 2007;26:456–461. doi: 10.1038/sj.onc.1209796. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Urvalek AM, Liu J, Zhao J. Activation of KLF8 transcription by focal adhesion kinase in human ovarian epithelial and cancer cells. J Biol Chem. 2008;283:13934–13942. doi: 10.1074/jbc.M709300200. [DOI] [PubMed] [Google Scholar]
- 4.Shi X, Luo X, Yan Q, Zhang W, Wu Y, Zhang M, Zhao J, Peng Y, Chen Y, Zhang Y, Chen C, Cheng T, Liu S, Bai Y, Wang J. Suppression of KLF8 induces cell differentiation and sensitizes colorectal cancer to 5-fluorouracil. Oncol Rep. 2015;34:1221–1230. doi: 10.3892/or.2015.4094. [DOI] [PubMed] [Google Scholar]
- 5.Lu H, Hu L, Yu L, Wang X, Urvalek AM, Li T, Shen C, Mukherjee D, Lahiri SK, Wason MS, Zhao J. KLF8 and FAK cooperatively enrich the active MMP14 on the cell surface required for the metastatic progression of breast cancer. Oncogene. 2014;33:2909–2917. doi: 10.1038/onc.2013.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan Q, Zhang W, Wu Y, Wu M, Zhang M, Shi X, Zhao J, Nan Q, Chen Y, Wang L, Cheng T, Li J, Bai Y, Liu S, Wang J. KLF8 promotes tumorigenesis, invasion and metastasis of colorectal cancer cells by transcriptional activation of FHL2. Oncotarget. 2015;6:25402–25417. doi: 10.18632/oncotarget.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lahiri SK, Zhao J. Kruppel-like factor 8 emerges as an important regulator of cancer. Am J Transl Res. 2012;4:357–363. [PMC free article] [PubMed] [Google Scholar]
- 8.Lu H, Wang X, Urvalek AM, Li T, Xie H, Yu L, Zhao J. Transformation of human ovarian surface epithelial cells by Kruppel-like factor 8. Oncogene. 2014;33:10–18. doi: 10.1038/onc.2012.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li T, Lu H, Shen C, Lahiri SK, Wason MS, Mukherjee D, Yu L, Zhao J. Identification of epithelial stromal interaction 1 as a novel effector downstream of Kruppel-like factor 8 in breast cancer invasion and metastasis. Oncogene. 2014;33:4746–4755. doi: 10.1038/onc.2013.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li T, Lu H, Mukherjee D, Lahiri SK, Shen C, Yu L, Zhao J. Identification of epidermal growth factor receptor and its inhibitory microRNA141 as novel targets of Kruppel-like factor 8 in breast cancer. Oncotarget. 2015;6:21428–21442. doi: 10.18632/oncotarget.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang T, Cai SY, Zhang J, Lu JH, Lin C, Zhai J, Wu MC, Shen F. Kruppel-like factor 8 is a new Wnt/beta-catenin signaling target gene and regulator in hepatocellular carcinoma. PLoS One. 2012;7:e39668. doi: 10.1371/journal.pone.0039668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung MC, Zhao J. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007;67:7184–7193. doi: 10.1158/0008-5472.CAN-06-4729. [DOI] [PubMed] [Google Scholar]
- 13.Wei H, Wang X, Gan B, Urvalek AM, Melkoumian ZK, Guan JL, Zhao J. Sumoylation delimits KLF8 transcriptional activity associated with the cell cycle regulation. J Biol Chem. 2006;281:16664–16671. doi: 10.1074/jbc.M513135200. [DOI] [PubMed] [Google Scholar]
- 14.Zhao J, Bian ZC, Yee K, Chen BP, Chien S, Guan JL. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol Cell. 2003;11:1503–1515. doi: 10.1016/s1097-2765(03)00179-5. [DOI] [PubMed] [Google Scholar]
- 15.Lu H, Hu L, Li T, Lahiri S, Shen C, Wason MS, Mukherjee D, Xie H, Yu L, Zhao J. A novel role of Kruppel-like factor 8 in DNA repair in breast cancer cells. J Biol Chem. 2012;287:43720–43729. doi: 10.1074/jbc.M112.418053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee H, Kim HJ, Lee YJ, Lee MY, Choi H, Kim JW. Kruppel-like factor KLF8 plays a critical role in adipocyte differentiation. PLoS One. 2012;7:e52474. doi: 10.1371/journal.pone.0052474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi R, Chen B, Zhao J, Zhan X, Zhang L, Liu X, Dong Q. Kruppel-like factor 8 ameliorates Alzheimer’s disease by activating beta-catenin. J Mol Neurosci. 2014;52:231–241. doi: 10.1007/s12031-013-0131-4. [DOI] [PubMed] [Google Scholar]
- 18.Urvalek AM, Lu H, Wang X, Li T, Yu L, Zhu J, Lin Q, Zhao J. Regulation of the oncoprotein KLF8 by a switch between acetylation and sumoylation. Am J Transl Res. 2011;3:121–132. [PMC free article] [PubMed] [Google Scholar]
- 19.Mehta TS, Lu H, Wang X, Urvalek AM, Nguyen KH, Monzur F, Hammond JD, Ma JQ, Zhao J. A unique sequence in the N-terminal regulatory region controls the nuclear localization of KLF8 by cooperating with the C-terminal zinc-fingers. Cell Res. 2009;19:1098–1109. doi: 10.1038/cr.2009.64. [DOI] [PubMed] [Google Scholar]
- 20.Urvalek AM, Wang X, Lu H, Zhao J. KLF8 recruits the p300 and PCAF co-activators to its amino terminal activation domain to activate transcription. Cell Cycle. 2010;9:601–611. doi: 10.4161/cc.9.3.10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao JH, Reiske H, Guan JL. Regulation of the cell cycle by focal adhesion kinase. J Cell Biol. 1998;143:1997–2008. doi: 10.1083/jcb.143.7.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seger R, Seger D, Reszka AA, Munar ES, Eldar-Finkelman H, Dobrowolska G, Jensen AM, Campbell JS, Fischer EH, Krebs EG. Overexpression of mitogen-activated protein kinase kinase (MAPKK) and its mutants in NIH 3T3 cells. Evidence that MAPKK involvement in cellular proliferation is regulated by phosphorylation of serine residues in its kinase subdomains VII and VIII. J Biol Chem. 1994;269:25699–25709. [PubMed] [Google Scholar]
- 23.Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, Counter CM. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509:492–496. doi: 10.1038/nature13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levin-Salomon V, Kogan K, Ahn NG, Livnah O, Engelberg D. Isolation of intrinsically active (MEK-independent) variants of the ERK family of mitogen-activated protein (MAP) kinases. J Biol Chem. 2008;283:34500–34510. doi: 10.1074/jbc.M806443200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu Y, Solski PA, Khosravi-Far R, Der CJ, Kelly K. The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J Biol Chem. 1996;271:6497–6501. doi: 10.1074/jbc.271.11.6497. [DOI] [PubMed] [Google Scholar]
- 26.Zhao J, Pestell R, Guan JL. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol Biol Cell. 2001;12:4066–4077. doi: 10.1091/mbc.12.12.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Zheng C, Guan J. Pyk2 and FAK differentially regulate progression of the cell cycle. J Cell Sci. 2000;113:3063–3072. doi: 10.1242/jcs.113.17.3063. [DOI] [PubMed] [Google Scholar]
- 28.Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314–6328. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu H, Wang X, Li T, Urvalek AM, Yu L, Li J, Zhu J, Lin Q, Peng X, Zhao J. Identification of poly (ADP-ribose) polymerase-1 (PARP-1) as a novel Kruppel-like factor 8-interacting and -regulating protein. J Biol Chem. 2011;286:20335–20344. doi: 10.1074/jbc.M110.215632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wegener AD, Jones LR. Phosphorylation-induced mobility shift in phospholamban in sodium dodecyl sulfate-polyacrylamide gels. Evidence for a protein structure consisting of multiple identical phosphorylatable subunits. J Biol Chem. 1984;259:1834–1841. [PubMed] [Google Scholar]
- 31.Wolf A, Rietscher K, Glass M, Huttelmaier S, Schutkowski M, Ihling C, Sinz A, Wingenfeld A, Mun A, Hatzfeld M. Insulin signaling via Akt2 switches plakophilin 1 function from stabilizing cell adhesion to promoting cell proliferation. J Cell Sci. 2013;126:1832–1844. doi: 10.1242/jcs.118992. [DOI] [PubMed] [Google Scholar]
- 32.Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 33.Leger J, Kempf M, Lee G, Brandt R. Conversion of serine to aspartate imitates phosphorylation-induced changes in the structure and function of microtubule-associated protein tau. J Biol Chem. 1997;272:8441–8446. doi: 10.1074/jbc.272.13.8441. [DOI] [PubMed] [Google Scholar]
- 34.Hao M, Lowy AM, Kapoor M, Deffie A, Liu G, Lozano G. Mutation of phosphoserine 389 affects p53 function in vivo. J Biol Chem. 1996;271:29380–29385. doi: 10.1074/jbc.271.46.29380. [DOI] [PubMed] [Google Scholar]
- 35.Aaron JA, Christianson DW. Trinuclear Metal Clusters in Catalysis by Terpenoid Synthases. Pure Appl Chem. 2010;82:1585–1597. doi: 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sorenson CM, Sheibani N. Focal adhesion kinase, paxillin, and bcl-2: analysis of expression, phosphorylation, and association during morphogenesis. Dev Dyn. 1999;215:371–382. doi: 10.1002/(SICI)1097-0177(199908)215:4<371::AID-AJA8>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 37.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 38.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marampon F, Ciccarelli C, Zani BM. Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol Cancer. 2006;5:31. doi: 10.1186/1476-4598-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Teng CT. Phosphorylation of Kruppel-like factor 5 (KLF5/IKLF) at the CBP interaction region enhances its transactivation function. Nucleic Acids Res. 2003;31:2196–2208. doi: 10.1093/nar/gkg310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dewi V, Kwok A, Lee S, Lee MM, Tan YM, Nicholas HR, Isono K, Wienert B, Mak KS, Knights AJ, Quinlan KG, Cordwell SJ, Funnell AP, Pearson RC, Crossley M. Phosphorylation of Kruppel-like factor 3 (KLF3/BKLF) and C-terminal binding protein 2 (CtBP2) by homeodomain-interacting protein kinase 2 (HIPK2) modulates KLF3 DNA binding and activity. J Biol Chem. 2015;290:8591–8605. doi: 10.1074/jbc.M115.638338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeo JC, Jiang J, Tan ZY, Yim GR, Ng JH, Goke J, Kraus P, Liang H, Gonzales KA, Chong HC, Tan CP, Lim YS, Tan NS, Lufkin T, Ng HH. Klf2 is an essential factor that sustains ground state pluripotency. Cell Stem Cell. 2014;14:864–872. doi: 10.1016/j.stem.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Kim MO, Kim SH, Cho YY, Nadas J, Jeong CH, Yao K, Kim DJ, Yu DH, Keum YS, Lee KY, Huang Z, Bode AM, Dong Z. ERK1 and ERK2 regulate embryonic stem cell self-renewal through phosphorylation of Klf4. Nat Struct Mol Biol. 2012;19:283–290. doi: 10.1038/nsmb.2217. [DOI] [PubMed] [Google Scholar]
- 44.Okamoto Y, Shikano S. Phosphorylation-dependent C-terminal binding of 14-3-3 proteins promotes cell surface expression of HIV co-receptor GPR15. J Biol Chem. 2011;286:7171–7181. doi: 10.1074/jbc.M110.199695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nasu-Nishimura Y, Hurtado D, Braud S, Tang TT, Isaac JT, Roche KW. Identification of an endoplasmic reticulum-retention motif in an intracellular loop of the kainate receptor subunit KA2. J Neurosci. 2006;26:7014–7021. doi: 10.1523/JNEUROSCI.0573-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Remenyi A, Good MC, Lim WA. Docking interactions in protein kinase and phosphatase networks. Curr Opin Struct Biol. 2006;16:676–685. doi: 10.1016/j.sbi.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Schutze MP, Peterson PA, Jackson MR. An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. EMBO J. 1994;13:1696–1705. doi: 10.1002/j.1460-2075.1994.tb06434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zerangue N, Malan MJ, Fried SR, Dazin PF, Jan YN, Jan LY, Schwappach B. Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc Natl Acad Sci U S A. 2001;98:2431–2436. doi: 10.1073/pnas.051630198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gassmann M, Haller C, Stoll Y, Abdel Aziz S, Biermann B, Mosbacher J, Kaupmann K, Bettler B. The RXR-type endoplasmic reticulum-retention/retrieval signal of GABAB1 requires distant spacing from the membrane to function. Mol Pharmacol. 2005;68:137–144. doi: 10.1124/mol.104.010256. [DOI] [PubMed] [Google Scholar]
- 50.Shikano S, Li M. Membrane receptor trafficking: evidence of proximal and distal zones conferred by two independent endoplasmic reticulum localization signals. Proc Natl Acad Sci U S A. 2003;100:5783–5788. doi: 10.1073/pnas.1031748100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borders CL Jr, Broadwater JA, Bekeny PA, Salmon JE, Lee AS, Eldridge AM, Pett VB. A structural role for arginine in proteins: multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci. 1994;3:541–548. doi: 10.1002/pro.5560030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia A, Cayla X, Caudron B, Deveaud E, Roncal F, Rebollo A. New insights in protein phosphorylation: a signature for protein phosphatase 1 interacting proteins. C R Biol. 2004;327:93–97. doi: 10.1016/j.crvi.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 53.Lewit-Bentley A, Rety S. EF-hand calcium-binding proteins. Curr Opin Struct Biol. 2000;10:637–643. doi: 10.1016/s0959-440x(00)00142-1. [DOI] [PubMed] [Google Scholar]
- 54.Mohanta TK, Mohanta N, Mohanta YK, Bae H. Genome-Wide Identification of Calcium Dependent Protein Kinase Gene Family in Plant Lineage Shows Presence of Novel D-x-D and D-E-L Motifs in EF-Hand Domain. Front Plant Sci. 2015;6:1146. doi: 10.3389/fpls.2015.01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sindreu CB, Scheiner ZS, Storm DR. Ca2+ -stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron. 2007;53:79–89. doi: 10.1016/j.neuron.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gray J, Zhao J. The implications of Epithelial Stromal Interaction 1 in Diseases Associated with Inflammatory Signaling. Cell Communication Insights. 2016;8:1–6. [Google Scholar]