

Abstract
We have reported that the recombinant avirulent Newcastle disease virus (NDV) LaSota strain expressing the rabies virus glycoprotein (rL-RVG) could induce autophagy and apoptosis in gastric carcinoma cells. In the present study, we explored the upstream regulators, endoplasmic reticulum (ER) stress that induce autophagy and apoptosis and the relationships among them. For this purpose, SGC-7901 and HGC cells were infected with rL-RVG. NDV LaSota strain and phosphate-buffered saline (PBS) were treated as the control groups. Western blotting and immunofluorescence microscopy were used to detect the expression of the ER stress-related proteins glucose-regulated protein 78 (GRP78) and the transcription factor GADD153 (CHOP), among others. The expression of beclin-1 and the conversion of light chain (LC) 3-I were used to determine the occurrence of autophagy, and flow cytometry (FCM) and western blotting were used to examine apoptosis-related protein expression. Transmission electron microscopy was also performed to monitor the ultrastructure of the cells. Moreover, small interfering (si) RNA was used to knock down CHOP expression. rL-RVG treatment increased the expression of ER stress-related proteins, such as GRP78, CHOP, activating transcriptional factor 6 (ATF6), X-box-binding protein 1 (XBP-1), and phosphorylated eukaryotic initiation factor 2 (p-eIF2α), in a time- and concentration-dependent manner, and knockdown of CHOP reduced LC3-II conversion and beclin-1 expression. When ER stress was inhibited with 4-PBA, the expression of both autophagy-related proteins and apoptosis-related proteins markedly decreased. Interestingly, inhibition of autophagy with 3-methyladenine (3MA) decreased not only apoptosis-related protein expression but also ER stress-related protein expression. Moreover, we found that downregulation of the c-Jun N-terminal kinase (JNK) pathway by SP600125 reduced LC3-II conversion, beclin-1 expression and caspase-3 activation. Collectively, the results suggest that rL-RVG increased ER stress in three branch pathways (ATF6, inositol-requiring enzyme 1 (IRE1), and PKR-like ER protein kinase (PERK)) that are upstream regulators of autophagy and apoptosis. Moreover, the IRE1-JNK pathway played an important role in switching ER stress to autophagy. These findings will provide molecular bases for developing rL-RVG into a drug candidate for the treatment of gastric carcinoma.
Keywords: Recombinant Newcastle disease virus, endoplasmic reticulum stress, autophagy, apoptosis
Introduction
Despite unceasing progress in surgery, chemotherapy, radiotherapy, gene therapy and immune therapy, cancer is still the second leading cause of death and is expected to surpass heart diseases as the leading cause of death in the next few years [1,2]. In particular, gastric cancer is dominant among the causes of cancer-related deaths and is the third most common form of cancer worldwide [1]. Currently, many studies have demonstrated that oncolytic virotherapy is a means of biological treatment with great potential for the treatment of cancer [3,4].
Newcastle disease virus (NDV), which was explored as an oncolytic agent in the early 1950s [5], contains six open reading frames encoding six structural proteins, including a nucleocapsid protein (NP), phosphoprotein (P), matrix protein (M), fusion protein (F), hemagglutinin-neuraminidase (HN) and RNA-dependent RNA polymerase (L) [6]. To generate recombinant NDV expressing rabies virus glycoprotein (RVG), an infectious recombinant genomic cDNA clone of NDV LaSota has been constructed by inserting the RVG gene between the P and the M genes. It has been shown that this recombinant NDV (rL-RVG) is safe in mice, poultry, dogs and cats, and it has served as a vaccine against rabies in animals [7]. We have also reported that NDV and rL-RVG can induce autophagy and apoptosis in gastric carcinoma cells [8].
In eukaryotic cells, the endoplasmic reticulum (ER) is an organelle where membrane proteins are modified, folded and assembled prior to secretion [8]. Conditions interfering with the function of the ER, such as hypoxia [9]; Ca2+ depletion; viral infections; and agents that affect the Ca2+ balance, protein glycosylation, and ER-Golgi vesicular transport, lead to accumulation of misfolded and unfolded proteins, termed the unfolded protein response (UPR) [10,11]. Upon UPR activation, three branches of signaling pathways, named after transmembrane ER stress sensors, are differentially or selectively regulated: PKR-like ER protein kinase (PERK), activating transcriptional factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1). Activated PERK in turn phosphorylates the α-subunit of eukaryotic initiation factor 2 (eIF2α), IRE1 catalyzes the splicing of the mRNA encoding the transcription factor X-box-binding protein 1 (XBP-1), and ATF6 is sequentially proteolyzed by the site-1 and site-2 proteases (S1P and S2P) to release its amino terminus (ATF6-N). All of these active transcription factors transduce death or survival signals [12-14]. In general, if the stress is persistent or intense, ER stress-induced apoptotic cell death ensues [15]. Moreover, numerous reports have shown that ER stress induces cell death by initiating autophagy [12,16]. Therefore, it is worth investigating the roles of ER stress and autophagy in virus-induced apoptosis.
In this study, we focused on the changes in ER and molecular mechanisms after infection of gastric carcinoma SGC-7901 and HGC cells with rL-RVG. Our findings provide molecular bases for developing rL-RVG into a drug candidate for the treatment of gastric carcinoma.
Materials and methods
Materials
The material sources were as follows: rL-RVG and NDV were kindly provided by Harbin Veterinary Research Institute (Harbin, Heilongjiang, China). The human cancer cell lines SGC-7901 and HGC were purchased from the Cancer Cell Repository (Shanghai Cell Bank, 2010-02-20) and the American Type Culture Collection (Manassas, VA, USA). 3-Methyladenine (3MA), a specific inhibitor of c-Jun N-terminal kinase (JNK) (SP600125), sodium phenylbutyrate (4-PBA), and Hoechst 33342 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rabbit polyclonal anti-beclin-1, anti-PERK, anti-eIF2α and anti-JNK were from Boster (Wuhan, Hubei, China). Rabbit monoclonal anti-light chain (LC) 3 antibody and anti-p-JNK antibody were from Cell Signaling Technology (Beverly, MA, USA), and anti-p-eIF2α, anti-ATF6, anti-GRP78, anti-CHOP and anti-XBP-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-cleaved-caspase-3, p-bcl-2 and p-PERK were from ImmunoWay (Newark, DE, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit, HRP-conjugated goat anti-mouse and FITC-conjugated goat anti-mouse antibodies were purchased from CWBio (Shanghai, China). Alexa Fluor 488-conjugated goat anti-mouse antibody and Cy3-conjugated goat anti-mouse antibody were purchased from KPL (Washington, DC, USA). A short interfering (si) RNA specific for human CHOP was obtained from GenePharma (Shanghai, China). The PCR primers were purchased from Shanghai Sangon Biological Engineering Technology & Services Co. Ltd. (Shanghai, China). TRIzol and Lipofectamine 2000 were from Invitrogen (Carlsbad, CA, USA).
Cell culture and interference test
SGC-7901 and HGC cells were maintained in RPMI 1640 medium with 10% (v/v) fetal bovine serum (HyClone, Logan, UT, USA) and antibiotics (100 U/ml penicillin and 100 U/ml streptomycin) at 37°C with 5% CO2 and 100% humidity. When the cells reached 50-70% confluence, they were infected with NDV or rL-RVG.
Western blot assay
After the cells were infected for a certain time, the medium was removed, and the cells were then washed three times with ice-cold phosphate-buffered saline (PBS; pH 7.4). The cells were subsequently lysed in RIPA buffer (2% Nonidet P-40, 0.2% sodium dodecyl sulfate (SDS), 50 mM Tris buffer (pH 7.4), and 150 mM sodium chloride) containing a protease inhibitor cocktail and a phosphatase inhibitor cocktail for 30 min on the ice. The protein concentrations were quantified using a BCA kit (Thermo Fisher Scientific, USA). Equivalent amounts of protein were then separated by 8-12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, CA, USA). The membrane was incubated in TBST with 5% BSA for 2 h and then probed with primary antibody at an appropriate dilution overnight at 4°C. The next day, the membrane was incubated with HRP-conjugated secondary antibodies at room temperature for 1 h. Protein bands were visualized using a Typhoon 9400 Variable Mode Imager (Amersham Biosciences, UK) and detected using Pierce ECL Plus Substrate (Thermo Fisher Scientific).
Immunofluorescence
After the indicated treatment, cells in a 24-well plate were fixed in 4% paraformaldehyde for 20 min at room temperature, washed three times in PBS for 5 min, permeabilized using 0.3% TritonX-100 in PBS for 10 min, washed three additional times in PBS, and sequentially probed with primary antibodies against GRP78 and secondary 488-conjugated goat antibodies against mouse IgG. The stained cells were then examined using immunofluorescence microscopy.
Transmission electron microscopy
After the indicated treatment, cells on a 100-mm dish were fixed in 2.5% glutaraldehyde in a 0.1 M phosphate buffer (pH 7.4) at room temperature for 1 h. Thin sections were then cut and were examined under an H-600 transmission electron microscope at 200 kV.
XBP-1 reverse transcriptase PCR splicing assay
Cells were treated as described previously. Total RNA was extracted from the cells above using TRIzol reagent (Sigma-Aldrich) and was reverse transcribed using primer sequences for amplification of homo XBP-1: 5’-GGGA ATGAAGTGAGGCCAGT-3’ and 5’-TGAAGAGTCAATAC CGCCAGA-3’. The thermal cycle consisted of 94°C for 10 min and 28 cycles of 94°C for 1 min, 60°C for 1 min, 72°C for 1 min, and 72°C for 10 min. The RT-PCR products were resolved on a 3% agarose gel and visualized using ethidium bromide. The size of unspliced XBP-1 was 137 bp, and that of spliced XBP-1 was 111 bp.
RNA interference with siRNA
When SGC and HGC cells grown in 6-well plates reached approximately 50% confluence, the cells were transfected with 50 nM CHOP-specific siRNA according to the manufacturer’s instructions. After 48 h, the cells were infected with NDV or rL-RVG at an MOI of 10. At 24 h after infection, the cells were harvested and were analyzed by western blotting.
Cell viability assay
Apoptotic and viable cells were analyzed using flow cytometry (FCM) with annexin V/propidium iodide (PI) double staining to detect membrane events according to the manufacturer’s instructions (4A Biotech, Beijing, China). FCM analysis of the labeled cells was performed using a FACSort flow cytometer (FACSCalibur, BD), and the data were analyzed using CellQuest software (BD Biosciences). The cytogram of the four quadrants was used to distinguish normal (annexin V2-/PI-), early apoptotic (annexin V+/PI-), late apoptotic (annexin V+/PI+), and necrotic (annexin V2-/PI+) cells. The sum of the numbers of early and late apoptotic cells is presented as the total amount of apoptosis. All experiments were carried out in triplicate.
Statistical analysis
The data were analyzed using Student’s t-test or one-way analysis of variance (ANOVA) within Statistica (SPSS V17.0) software. Differences with p<0.05 or p<0.01 were considered statistically significant. All experiments were repeated at least three times.
Results
rL-RVG induces an ER stress response in SGC and HGC cells
We tested the peak time of ER stress-related protein expression after infection with 10 MOI rL-RVG. As shown in Figure 1A and 1D, the expression of GRP78 and CHOP began at 12 h after treatment with rL-RVG, peaked at 24 h and was sustained for at least 12 h in SGC cells. The same is shown in Figure 1B and 1E for HGC cells. When cells were stained for two ER stress-specific markers, or GRP78 and CHOP (Figure 2), we observed that GRP78 and CHOP protein expression was markedly upregulated in the rL-RVG- and NDV-infected groups compared with the PBS control group. However, the expression of the two proteins in the rL-RVG-infected group was the strongest among all three groups. Additionally, Figure 1A shows a transient increase in eIF2α phosphorylation by rL-RVG in SGC and HGC cells. The activation of IRE1α was also confirmed by the presence of XBP-1 mRNA splice variants upon rL-RVG treatment, as shown in Figure 1B. The results showed that the spliced XBP-1 mRNA appeared after 12 h of pretreatment, but the effect was transient.
Figure 1.
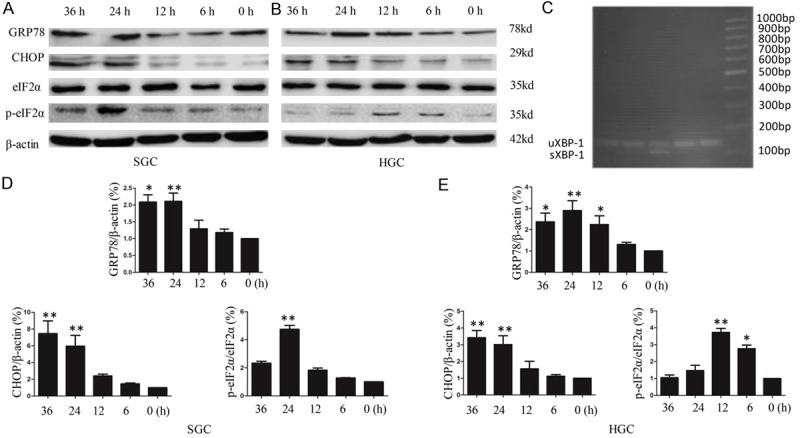
rL-RVG induces the expression of ER stress-related proteins in SGC-7901 and HGC cells in a time-dependent manner. A and D. For SGC; B and E. For HGC. The cells were infected with 10 MOI virus or with vehicle (control) for different amounts of time. Cell lysates were harvested at the same time after infection with virus for different amounts of time, and proteins were detected by western blotting. A and D. The expression of GRP78 and CHOP increased at 12 h after treatment with rL-RVG, peaked at 24 h (**, p<0.01) and was sustained for at least 12 h in SGC cells. The same was observed in B and E. for HGC cells. A transient increase in eIF2α phosphorylation following rL-RVG infection was observed at 24 h in SGC cells (**, p<0.01) and at 12 h in HGC cells (**, p<0.01). C. Effect of rL-RVG on the level of spliced mRNA forms of XBP-1. uXBP1, unspliced XBP-1; sXBP-1, spliced XBP-1. The spliced XBP-1 mRNA appeared with 12 h of pretreatment, but the effect was transient.
Figure 2.
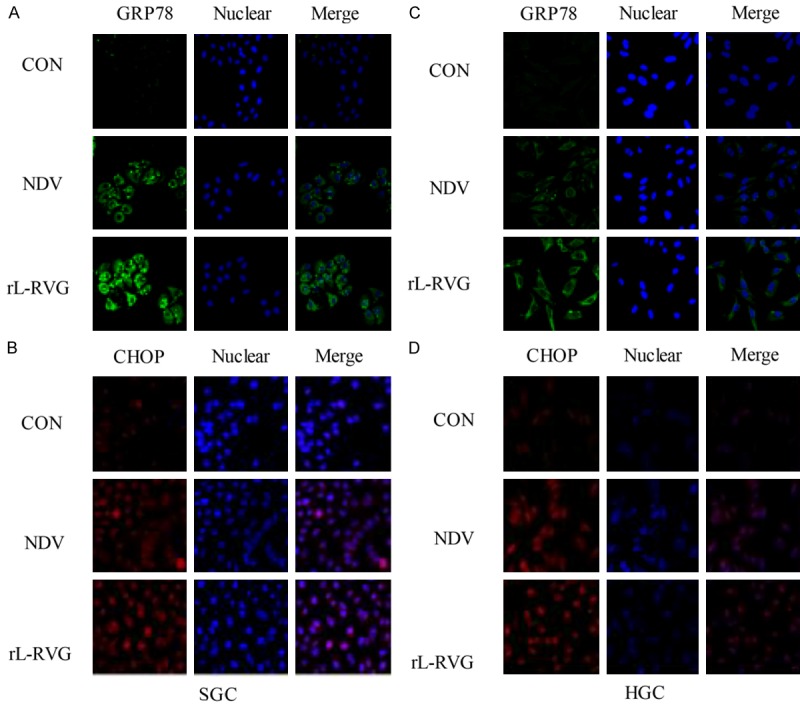
Expression of ER stress-related proteins in infected SGC and HGC cells. The nuclei were stained with Hoechst 33342. The cells had been infected with virus for 24 h. ER stress-related protein expression was monitored by immunofluorescence microscopy (x200 magnification). A and B. For SGC, C and D. For HGC. A and C. GRP78 protein (green) was significantly more strongly expressed in the NDV and rL-RVG groups, and the rL-RVG-infected group exhibited the strongest expression. Similarly, in B and D. CHOP protein expression was strongest in the rL-RVG-infected group.
There are three ER stress sensors, namely, PERK, IRE1 and ATF6, that can activate the UPR response during ER dysfunction. Figure 3A and 3B show that the expression levels of phosphorylated eIF2α, ATF6(f), and XBP-1 proteins were higher in SGC and HGC cells at 24 h after infection with 1 MOI or 10 MOI NDV or rL-RVG compared with the levels in the PBS control group. Based on the above, NDV and rL-RVG can induce ER stress, in which all three branches of the ER stress signaling pathways participate.
Figure 3.
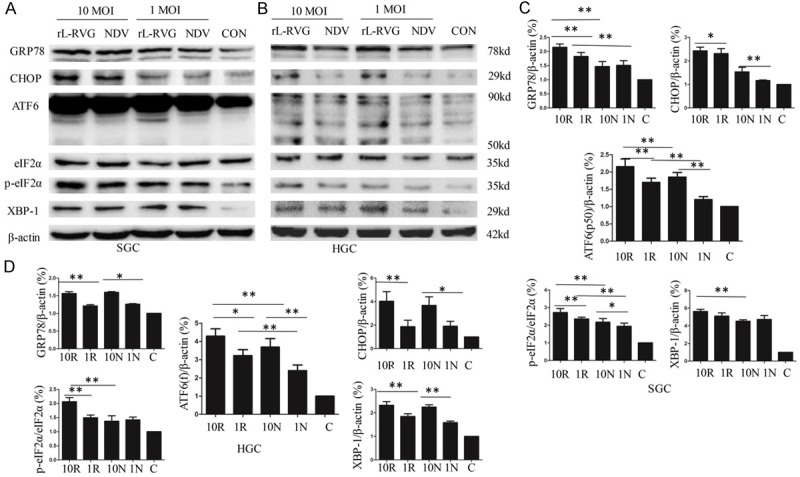
NDV and rL-RVG each induce the expression of ER stress-related proteins in SGC-7901 and HGC cells in a concentration-dependent manner. A and C. For SGC; B and D. For HGC. The cells were infected with 10 MOI or 1 MOI virus for 24 h. NDV and rL-RVG infection increased the amounts of GRP78 and CHOP in SGC and HGC cells at 24 h post-infection, and the rL-RVG-infected group exhibited the strongest expression. Activation of the three branches of ER stress signaling pathway proteins, or XBP-1, ATF6 and p-eIF2α, increased significantly in SGC and HGC cells (*, p<0.05; **, p<0.01).
The ER stress response contributes to the SGC and HGC autophagy and apoptosis induced by rL-RVG
Previous tests and the present experiments showed that autophagy and apoptosis occur in rL-RVG-infected SGC and AGS cells [2]. Recent reports have additionally shown that ER stress may be a potent inducer of autophagy [17-19]. Chemical chaperones such as 4-PBA constitute a group of compounds known to improve ER folding capacity and to facilitate the trafficking of mutant proteins by stabilizing their conformation [20]. To test whether close relationships exist among the ER stress response, autophagy and apoptosis, we investigated the effects of rL-RVG-induced ER stress on the autophagy marker proteins beclin-1 and LC3 and the apoptosis-related protein cleaved caspase-3 by pretreating cells with 4-PBA. As shown in Figure 4A and 4B, the SGC and HGC cells infected with rL-RVG and pretreated with 4-PBA exhibited downregulated expression of beclin-1, LC3-II/I and cleaved caspase-3 compared with the group infected with NDV or rL-RVG alone. As shown in Figure 4C and 4D, FCM analysis indicated that the percentages of annexin V+/PI+ and annexin V+/PI- cells decreased in the combined treatment group compared with the rL-RVG treatment group. These results support the conclusion that rL-RVG induces autophagy and apoptosis via the ER stress response, via the upstream regulators.
Figure 4.

N+4: NDV+4-PBA; R+4: rL-RVG+4-PBA. The contribution of ER stress downregulation to the expression of autophagy- and apoptosis-associated proteins in SGC and HGC cells after infection. A. For SGC; B. For HGC. The expression of the autophagy-associated proteins beclin-1 and LC3-II/I decreased in cells with 4-PBA treatment compared with cells without 4-PBA treatment. The same was observed for apoptosis-associated proteins. C and D. Early apoptotic (annexin V+/PI-) and late apoptotic (annexin V+/PI+) cells. Prior to fluorescence-activated cell sorting analysis of annexin V/PI staining, SGC cells were pretreated with 4-PBA (10 mM) 2 h before exposure to NDV or rL-RVG (10 MOI). Annexin V/PI staining was then performed to assess apoptosis/necrosis. Annexin V staining showed that pretreatment with 10 mM 4-PBA resulted in a significant decrease in apoptosis in the rL-RVG-infected SGC cells (**, p<0.01).
CHOP, a downstream signaling protein in the ER stress response, is one of the bridges between the rL-RVG induced ER stress response and autophagy
The CHOP promoter receives positive input from other components of the UPR and then upregulates the expression of autophagy marker proteins and provokes caspase activation [21]. To investigate the mechanism underlying ER stress response-induced autophagy, the expression of CHOP was silenced using specific siRNA. As shown in Figure 5A, the specific siRNA (FAM-siRNA) was transferred into cells according to the manufacturer’s instructions, and the potency of the siRNA treatment was determined by western blotting. Of the three sequences of siRNA (shown in Table 1 of the supplementary data) that were used to knock down CHOP, siCHOP 3 caused significant silencing (Figure 5B). In particular, CHOP-specific siRNA significantly downregulated the CHOP protein level in SGC cells. The expression of beclin-1 and the conversion of LC3 protein during the autophagy induced by rL-RVG were also reduced by CHOP-specific siRNA. Parallel experiments showed that the cleavage of caspase-3 had decreased as well, as shown in Figure 5C. The results thus indicate that CHOP is one of the pivotal mediators of the ER stress-mediated autophagy and apoptosis induced by rL-RVG.
Figure 5.

si+N: siCHOP+NDV; si+4: siCHOP+rL-RVG. Knockdown of CHOP decreased autophagy- and apoptosis-related protein expression. A. Fluorescently labeled siRNA (FAM-siRNA) was transferred into cells according to the manufacturer’s instructions, and the effects were monitored by immunofluorescence microscopy (x200 magnification). B. I expression of CHOP protein was silenced significantly in the group expressing siRNA 3. C. For SGC; D. For HGC. The expression of the autophagy-associated proteins beclin-1 and LC3-II/I decreased in cells after silencing CHOP compared with expression in cells without treatment. The same was observed for apoptosis-associated proteins.
Table 1.
The sequence interference of CHOP gene
| Gene Name | Sequence | |
|---|---|---|
|
| ||
| Sense (5’-3’) | Antisense (5’-3’) | |
| CHOP (GADD153) | ||
| SiRNA1 (CHOP-HOMO-805) | GAGCUCUGAUUGACCGAAUTT | AUUCGGUCAAUCAGAGCUCTT |
| SiRNA2 (CHOP-HOMO-291) | CCCAUUAUCCUGCAGAUGUTT | ACAUCUGCAGGAUAAUGGGTT |
| SiRNA3 (CHOP-HOMO-650) | CGGAAACAGAGUGGUCAUUTT | AAUGACCACUCUGUUUCCGTT |
The autophagy induced by rL-RVG is mediated by the IRE1/JNK/beclin-1 signaling pathway
Once ER stress is activated, JNK phosphorylates the anti-apoptotic protein bcl-2, which interacts with beclin-1 under physiological conditions; this phosphorylation leads to the disruption of the bcl-2/beclin-1 complex and allows beclin-1 to trigger autophagy [22,23]. To investigate the involvement of the IRE1/JNK/beclin-1 pathway in the relationship between the ER stress response and autophagy, we examined the activity of p-JNK and beclin-1 following exposure to rL-RVG or NDV. The activation of JNK and beclin-1 was specifically determined by western blot analysis. As shown in Figure 6A and 6B, pretreatment with SP600125, a specific inhibitor of JNK, at 2 h before infection with rL-RVG not only prevented the phosphorylation of JNK but also suppressed the phosphorylation of bcl-2 and beclin-1 synchronously. The conversion of LC3-I was significantly decreased in the same sample. As shown in Figure 6C, the numbers of rL-RVG-induced autophagosomes were also markedly decreased after pretreatment with SP600125 according to electron microscopy. This finding suggests that rL-RVG promotes ER stress-induced autophagy through the IRE1/JNK/beclin-1 signaling pathway.
Figure 6.
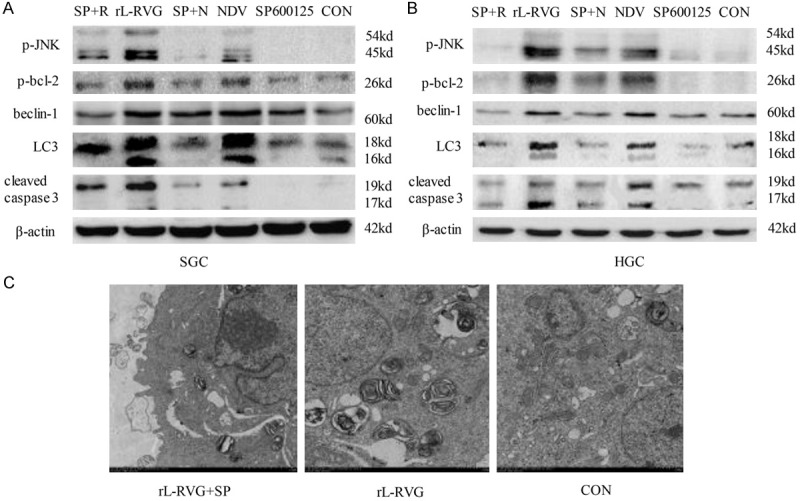
SP+N: SP600125+NDV; SP+R: SP600125+rL-RVG. The autophagy induced by rL-RVG was mediated by the IRE1/JNK/beclin-1 signaling pathway. A. For SGC; B. For HGC. The phosphorylation of JNK and bcl-2 was upregulated in the rL-RVG- or NDV-infected SGC and HGC cells. The expression of the autophagy-associated proteins beclin-1 and LC3-II/I decreased in cells after treatment with a specific inhibitor of JNK, or SP600125, compared with cells without the treatment. The same was observed for apoptosis-associated proteins. C. The ultrastructure of the SGC cells after infection with virus or pretreatment with SP600125. rL-RVG infection increased the number of autophagosomes in SGC cells, but the number of rL-RVG-induced autophagosomes was markedly decreased after pretreatment with SP600125.
Autophagy plays a role in ER stress-mediated apoptosis
Previous experiments found that virus-induced apoptosis in SGC and AGS cells infected with NDV or rL-RVG after pretreatment with 3MA was significantly decreased, so we concluded that autophagy contributes to the SGC and HGC apoptosis induced by rL-RVG and NDV [2]. Following up on these experiments, in the present study, we tested the expression levels of the ER stress-related proteins GRP78, CHOP and p-eIF2α in samples with the same treatment. As shown in Figure 7, when rL-RVG- or NDV-induced autophagy was inhibited with 3MA, the expression of not only apoptosis-related proteins but also ER stress-associated proteins was decreased significantly. Collectively, these results indicate that both rL-RVG-induced autophagy and NDV-induced autophagy are pro-apoptotic mechanisms and that blockage of autophagy reduces rL-RVG- and NDV-induced ER stress and apoptosis.
Figure 7.
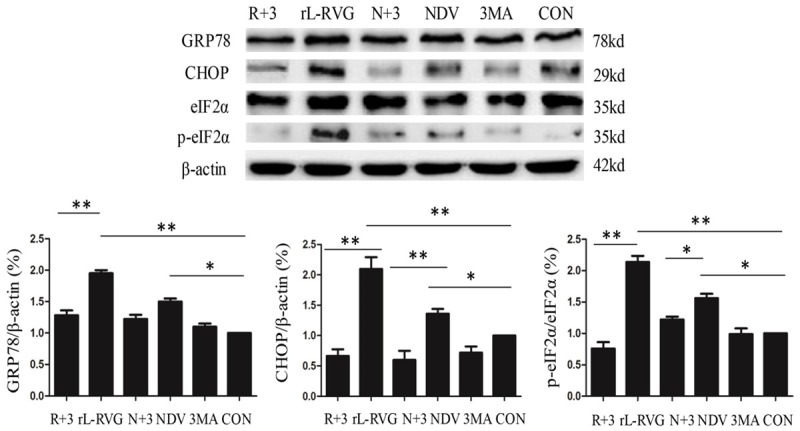
N+3: NDV+3MA; R+3: rL-RVG+3MA. The contribution of autophagy downregulation to the expression of ER stress-related proteins in SGC cells after infection. The expression of the ER stress-related proteins GRP78, CHOP and p-eIF2α decreased in cells after 3MA treatment compared with cells without 3MA treatment (*, p<0.05; **, p<0.01).
Discussion
The oncolytic virus NDV has been used extensively as an oncolytic agent in both preclinical and clinical studies [24,25]. These application prospects benefit from the virus’ selectivity for tumor cells and interferon (IFN)-deficient cells [26]. Previous reports have demonstrated that rL-RVG spreads more easily within cells and has much more antitumor potency [2]. Multiple mechanisms underlying NDV-induced cytotoxicity have been elucidated, including immunomodulatory mechanisms [27-29], autophagy pathways [30,31] and apoptotic pathways [32]. In addition, several reports have proposed possible crosstalk among these mechanisms [31].
Our study clearly demonstrates for the first time that the ER stress response is activated in gastric carcinoma cells exposed to the NDV LaSota strain and rL-RVG. This finding is supported by the following evidence. First, ultrastructural analysis of SGC cells treated with rL-RVG or NDV showed dilatations of ER cisternae [2]. Second, GRP78/BiP is a molecular chaperone in the ER that interacts with the hydrophobic domains of proteins and prevents misfolding during translocation [33]. This chaperone is proposed to be the master initiator of the UPR [34], with depletion of GRP78 resulting in widespread activation of the UPR [35]. When SGC and HGC cells were infected with NDV or rL-RVG in the present study, the expression level of GRP78 protein was significantly increased, with the strongest increase in the rL-RVG group. Third, the UPR cellular responses are downstream of the parallel activation of three branches of signaling pathways named after the transmembrane ER stress sensors IRE1, ATF6, and PERK [36]. In the current study, rL-RVG and NDV each increased PERK and eIF2α phosphorylation and the expression of XBP-1, ATF6(f) and CHOP. RT-PCR analysis also revealed the presence of XBP-1 mRNA splice variants. Fourth, suppression of ER stress by the chemical chaperone 4-PBA reduced rL-RVG- and NDV-induced autophagy and apoptosis. Fifth, knockdown of CHOP using specific siRNA attenuated rL-RVG- and NDV-induced LC3-II conversion and the expression of cleaved caspase-3, suggesting that induction of ER stress led to autophagy and apoptosis. To the best of our knowledge, these data preliminarily indicate that the induction of autophagy and apoptosis by rL-RVG involves ER stress.
ER stress triggers autophagy through PERK/eIF2α, IRE1/JNK or potentially cleavage of eIF2α by caspase-3 [37]. The IRE1/JNK pathway participates in the release of beclin-1, which in turn triggers the formation of autophagosomes and the conversion of LC3-II, promoting autophagy by alleviating beclin-1 inhibition through downregulation of bcl-2 [38]. Herein, western blot analysis revealed that the expression of p-JNK and beclin-1 increased in rL-RVG-infected SGC cells. Pretreatment with SP600125, a specific inhibitor of JNK, suppressed the expression of p-JNK, p-bcl-2, and beclin-1 and the conversion of LC3-I. CHOP is downstream of the PERK/eIF2α signaling pathway, which receives positive input from other components of the UPR, and upregulation of CHOP protein can provoke caspase activation [39]. In the present study, knockdown of CHOP using CHOP-specific siRNA suppressed not only the activation of caspase-3 but also the conversion of LC3-I and beclin-1 protein expression. We propose that the PERK/eIF2α signaling pathway increases the expression of CHOP in SGC and HGC cells, which in turn mediates rL-RVG- and NDV-induced autophagy and apoptosis. Therefore, we demonstrate that rL-RVG-induced ER stress triggers autophagy through the PERK/eIF2α/CHOP and IRE1/JNK/beclin-1 signaling pathways. In other words, ER stress is the upstream regulator that induces autophagy and apoptosis.
Certain reports have shown that excess ER stress can cause autophagy and that inhibition of autophagy augments apoptotic cell death but does not affect ER stress [18]. However, in the present study, when rL-RVG- and NDV-induced autophagy was inhibited with 3MA in SGC and HGC cells, the expression levels of the ER stress-related proteins GRP78, CHOP and p-eIF2α decreased significantly. Combined with previous research, our work suggests that inhibition of rL-RVG-induced autophagy downregulates the virus-induced ER stress and apoptosis. We speculate that there are two possible conclusions. First, several recent observations have suggested that oncolytic activity is dependent on virus replication [40,41]. NDV triggers autophagy in cancer cells and chicken cells to enhance virus replication, and inhibition of autophagy decreases virus production [16,42,43]. Therefore, we propose that cancer cells infected with NDV or rL-RVG experience excessive ER stress, which increases autophagy through the PERK/eIF2α/CHOP and IRE1/JNK/beclin-1 signaling pathways. The autophagy then enhances virus replication, which induces even more ER stress and autophagy. All of these events form a positive feedback loop and finally induce cell death. However, when we inhibited NDV- and rL-RVG-induced autophagy, the above-mentioned positive feedback loop was broken. In particular, the inhibitor of autophagy decreased virus production, which remitted the ER stress. Thus, NDV- and rL-RVG-induced ER stress and apoptosis were decreased. Second, previous reports suggest that inhibition of autophagy at a late stage can effectively augment the cytotoxicity of anticancer therapy. However, inhibiting autophagy at an early stage attenuates the cytotoxicity of anticancer therapy [44,45]. In the present study, rL-RVG-induced ER stress and apoptosis were relieved after treatment with 3MA. The changes in ER stress and apoptosis that occur after inhibition of autophagy depend on the type of anticancer reagent used and the stage in which the autophagy inhibitor is applied.
In conclusion, this study shows that three branches of UPR signaling pathways participate in the effects of NDV and rL-RVG infection in SGC and HGC cells. In addition, rL-RVG can induce autophagy by activating the PERK/eIF2α/CHOP and IRE1/JNK/beclin-1 signaling pathways. Inhibition of autophagy relieves the rL-RVG-induced ER stress and apoptosis, suggesting that autophagy plays an important pro-apoptotic role in virus-induced apoptosis. As shown in Figure 8. Taken together, the results of this study demonstrate that rL-RVG-induced ER stress and autophagy mediate apoptosis and also provide an understanding of their interplay in gastric carcinoma cells. Our results also suggest that rL-RVG may be a powerful candidate for antitumor treatments.
Figure 8.
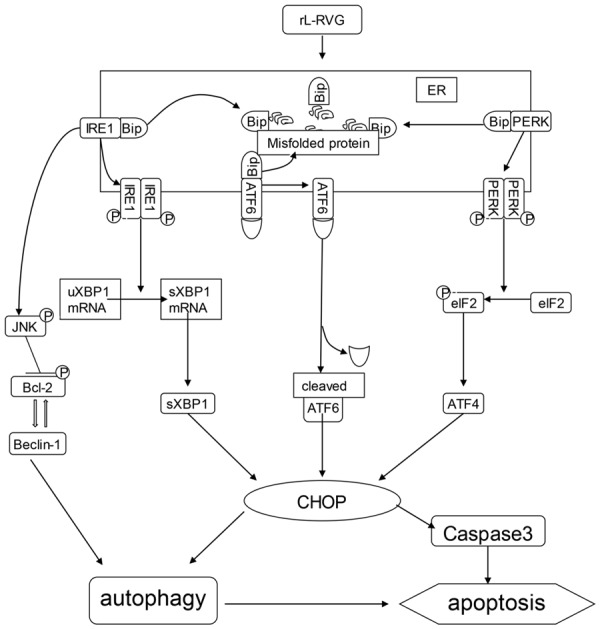
Proposed mechanism of rL-RVG-induced cell death.
Acknowledgements
This research was supported by the Natural Science Foundation of Jiangsu Province (grant no. BK20151333) and the Social Development Fund of Zhenjiang City, China (SH2014046). The authors would like to thank Professor Zhigao Bu and Dr Jinying Ge from the State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Harbin, China, for supplying the recombinant Newcastle disease virus. The authors would also like to thank Ai-hua Gong from Jiangsu University (Jiangsu, China) for kindly providing suggestions regarding the experiments performed.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Bu XF, Wang MB, Zhang ZJ, Zhao YH, Li M, Yan YL. Autophagy is involved in recombinant Newcastle disease virus (rL-RVG)-induced cell death of stomach adenocarcinoma cells in vitro. Int J Oncol. 2015;47:679–689. doi: 10.3892/ijo.2015.3039. [DOI] [PubMed] [Google Scholar]
- 3.Cuddington BP, Mossman KL. Oncolytic bovine herpesvirus type 1 as a broad spectrum cancer therapeutic. Curr Opin Virol. 2015;13:11–16. doi: 10.1016/j.coviro.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 4.Vaha-Koskela MJ, Heikkila JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zamarin D, Palese P. Oncolytic Newcastle disease virus for cancer therapy: old challenges and new directions. Future Microbiol. 2012;7:347–367. doi: 10.2217/fmb.12.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganar K, Das M, Sinha S, Kumar S. Newcastle disease virus: current status and our understanding. Virus Res. 2014;184:71–81. doi: 10.1016/j.virusres.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ge J, Wang X, Tao L, Wen Z, Feng N, Yang S, Xia X, Yang C, Chen H, Bu Z. Newcastle disease virus-vectored rabies vaccine is safe, highly immunogenic, and provides long-lasting protection in dogs and cats. J Virol. 2011;85:8241–8252. doi: 10.1128/JVI.00519-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 9.Koumenis C. ER stress, hypoxia tolerance and tumor progression. Curr Mol Med. 2006;6:55–69. doi: 10.2174/156652406775574604. [DOI] [PubMed] [Google Scholar]
- 10.Jheng JR, Ho JY, Horng JT. ER stress, autophagy, and RNA viruses. Front Microbiol. 2014;5:388. doi: 10.3389/fmicb.2014.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 12.Fung TS, Torres J, Liu DX. The emerging roles of viroporins in ER stress response and autophagy induction during virus infection. Viruses. 2015;7:2834–2857. doi: 10.3390/v7062749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 14.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 16.Meng G, Xia M, Wang D, Chen A, Wang Y, Wang H, Yu D, Wei J. Mitophagy promotes replication of oncolytic Newcastle disease virus by blocking intrinsic apoptosis in lung cancer cells. Oncotarget. 2014;5:6365–6374. doi: 10.18632/oncotarget.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasanain M, Bhattacharjee A, Pandey P, Ashraf R, Singh N, Sharma S, Vishwakarma AL, Datta D, Mitra K, Sarkar J. alpha-Solanine induces ROS-mediated autophagy through activation of endoplasmic reticulum stress and inhibition of Akt/mTOR pathway. Cell Death Dis. 2015;6:e1860. doi: 10.1038/cddis.2015.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu WT, Huang CY, Lu IC, Gean PW. Inhibition of glioma growth by minocycline is mediated through endoplasmic reticulum stress-induced apoptosis and autophagic cell death. Neuro Oncol. 2013;15:1127–1141. doi: 10.1093/neuonc/not073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciechomska IA, Gabrusiewicz K, Szczepankiewicz AA, Kaminska B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine a-induced cell death. Oncogene. 2013;32:1518–1529. doi: 10.1038/onc.2012.174. [DOI] [PubMed] [Google Scholar]
- 20.Welch WJ, Brown CR. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones. 1996;1:109–115. doi: 10.1379/1466-1268(1996)001<0109:iomacc>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen S, Zhang Y, Wang Z, Liu R, Gong X. Bufalin induces the interplay between apoptosis and autophagy in glioma cells through endoplasmic reticulum stress. Int J Biol Sci. 2014;10:212–224. doi: 10.7150/ijbs.8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorence RM, Roberts MS, O’Neil JD, Groene WS, Miller JA, Mueller SN, Bamat MK. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic Newcastle disease virus. Curr Cancer Drug Targets. 2007;7:157–167. doi: 10.2174/156800907780058853. [DOI] [PubMed] [Google Scholar]
- 25.Burke J, Nieva J, Borad MJ, Breitbach CJ. Oncolytic viruses: perspectives on clinical development. Curr Opin Virol. 2015;13:55–60. doi: 10.1016/j.coviro.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Elankumaran S, Chavan V, Qiao D, Shobana R, Moorkanat G, Biswas M, Samal SK. Type I interferon-sensitive recombinant newcastle disease virus for oncolytic virotherapy. J Virol. 2010;84:3835–3844. doi: 10.1128/JVI.01553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng J, Fournier P, Schirrmacher V. Induction of interferon-α and tumor necrosis factor-related apoptosis-inducing ligand in human blood mononuclear cells by hemagglutinin-neuraminidase but not F protein of Newcastle disease virus. Virology. 2002;297:19–30. doi: 10.1006/viro.2002.1413. [DOI] [PubMed] [Google Scholar]
- 28.Jestin V, Cherbonnel M. Interferon-induction in mouse spleen cells by the Newcastle disease virus (NDV) HN protein. Ann Rech Vet. 1991;22:365–372. [PubMed] [Google Scholar]
- 29.Park MS, Shaw ML, Munoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, Garcia-Sastre A, Basler CF. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J Virol. 2003;77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, De Vleeschouwer S, Agostinis P, Graf N, Van Gool SW. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer. 2015;136:E313–325. doi: 10.1002/ijc.29202. [DOI] [PubMed] [Google Scholar]
- 32.Meng S, Zhou Z, Chen F, Kong X, Liu H, Jiang K, Liu W, Hu M, Zhang X, Ding C, Wu Y. Newcastle disease virus induces apoptosis in cisplatin-resistant human lung adenocarcinoma A549 cells in vitro and in vivo. Cancer Lett. 2012;317:56–64. doi: 10.1016/j.canlet.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 33.Naidoo N. ER and aging-Protein folding and the ER stress response. Ageing Res Rev. 2009;8:150–159. doi: 10.1016/j.arr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez A, Ordonez R, Reiter RJ, Gonzalez-Gallego J, Mauriz JL. Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J Pineal Res. 2015;59:292–307. doi: 10.1111/jpi.12264. [DOI] [PubMed] [Google Scholar]
- 35.Eletto D, Maganty A, Eletto D, Dersh D, Makarewich C, Biswas C, Paton JC, Paton AW, Doroudgar S, Glembotski CC, Argon Y. Limitation of individual folding resources in the ER leads to outcomes distinct from the unfolded protein response. J Cell Sci. 2012;125:4865–4875. doi: 10.1242/jcs.108928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanderson TH, Gallaway M, Kumar R. Unfolding the unfolded protein response: unique insights into brain ischemia. Int J Mol Sci. 2015;16:7133–7142. doi: 10.3390/ijms16047133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schleicher SM, Moretti L, Varki V, Lu B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: implications for future therapeutic approaches. Drug Resist Updat. 2010;13:79–86. doi: 10.1016/j.drup.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 38.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 39.Ma Y, Shimizu Y, Mann MJ, Jin Y, Hendershot LM. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones. 2010;15:281–293. doi: 10.1007/s12192-009-0142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dortmans JC, Rottier PJ, Koch G, Peeters BP. The viral replication complex is associated with the virulence of Newcastle disease virus. J Virol. 2010;84:10113–10120. doi: 10.1128/JVI.00097-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yaacov B, Eliahoo E, Lazar I, Ben-Shlomo M, Greenbaum I, Panet A, Zakay-Rones Z. Selective oncolytic effect of an attenuated Newcastle disease virus (NDV-HUJ) in lung tumors. Cancer Gene Ther. 2008;15:795–807. doi: 10.1038/cgt.2008.31. [DOI] [PubMed] [Google Scholar]
- 42.Meng C, Zhou Z, Jiang K, Yu S, Jia L, Wu Y, Liu Y, Meng S, Ding C. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch Virol. 2012;157:1011–1018. doi: 10.1007/s00705-012-1270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun Y, Yu S, Ding N, Meng C, Meng S, Zhang S, Zhan Y, Qiu X, Tan L, Chen H, Song C, Ding C. Autophagy benefits the replication of Newcastle disease virus in chicken cells and tissues. J Virol. 2014;88:525–537. doi: 10.1128/JVI.01849-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 45.Shingu T, Fujiwara K, Bogler O, Akiyama Y, Moritake K, Shinojima N, Tamada Y, Yokoyama T, Kondo S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int J Cancer. 2009;124:1060–1071. doi: 10.1002/ijc.24030. [DOI] [PubMed] [Google Scholar]