

Abstract
Amounting evidence has demonstrated that phenethyl isothiocyanate (PEITC) is a strong inducer of reactive oxygen species (ROS) and functions as a selective killer to various human cancer cells. However, it remains obscure whether PEITC has potential selective lethality to malignant glioma cells. Thus in this study, we performed multiple analysis such as MTT assay, Hoechst 33258 staining, flow cytometry, foci formation, RT-PCR, Western blot, and transfection to explore the selective lethality of PEITC to malignant glioma cells and the underlying mechanisms. We found that PEITC induced a selective apoptosis and suppressed tumorigenicity and migration of malignant glioma cells. Furthermore, we found PEITC significantly induced GSH depletion, ROS production, caspase-9 and caspase-3 activation, and miR-135a upregulation in malignant glioma cells but not in normal cells. Moreover, PEITC activated the miR-135a-mitochondria dependent apoptosis pathway as demonstrated by downregulation of STAT6, SMAD5 and Bcl-xl while upregulation of Bax expression and Cytochrome-C release in malignant glioma cell lines but not in the immortalized human normal glial HEB cells. Correspondingly, the above PEITC-induced activation of the ROS-MiR-135a-Mitochondria dependent apoptosis pathways in malignant glioma was attenuated by pre-transfection with miR-135a inhibitor, pre-treatment with multidrug resistance-associated protein 1 (MRP1) inhibitor Sch B, or combination with glutathione (GSH). These results revealed that PEITC selectively induced apoptosis of malignant glioma cells through MRP1-mediated export of GSH to activate ROS-MiR-135a-Mitochondria dependent apoptosis pathway, suggesting a potential application of PEITC for treating glioma.
Keywords: Phenethyl isothiocyanate, miR-135a, MRP1, selective lethality, glioma
Introduction
Glioblastoma multiforme (GBM) is the most common primary central nervous system (CNS) tumor (account for approximately 80%) [1,2]. Despite recent advances in the diagnosis, surgery, chemotherapy and radiation therapy, the prognosis for GBM remains very poor with the median survival time of only 12-15 months [3]. In clinical, temozolomide (TMZ) is the first line chemotherapy drug for GBM [4]. However, due to blood brain barrier (BBB), low O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation rate [5] and multiple drug resistance of glioblastoma, the efficacy of TMZ-based radiochemotherapy is low. Thus, it is urgent to develop novel and effective treatment modalities including new chemotherapy drugs for the management of glioblastoma.
Phenethyl isothiocyanate (PEITC) is released from glucosinolates by cutting or chewing activated enzyme myrosinase [6] and known to be one of the major bioactive compounds present in cruciferous vegetables such as watercress, broccoli and Brussels sprouts [7]. Previous studies have revealed PEITC has broad spectrum and remarkable anti-cancer effects by inducing apoptosis [8-11] and reversing chemotherapy-drug resistance [8,12-15]. PEITC has been shown to selectively kill malignant cancer cells but not the corresponding normal cells [13,15-17] through potent induction of reactive oxygen species (ROS) in malignant cancer cells but not in normal cells [18]. However, this “ROS-based cancer therapy” has been recently questioned [19]. Thus, the mechanisms of the selective lethality of PEITC to cancer cells remain to be determined in suitable models.
MicroRNAs (miRNAs) are endogenous small non-coding RNAs that act as crucial gene regulators at post-transcriptional level, and play an important role in the initiation, progression and prognostic of various human cancers [20]. Emerging evidences have revealed miRNAs play pivotal roles in the response and resistance of anti-cancer agents [21,22]. Moreover, various studies have revealed anti-cancer agents trigger cancer cells apoptosis through inducing ROS, which in turn regulate a wide range of miRNAs [23-25], revealing new roles of miRNA in cancer therapy responses. MiR-135a is one of the ROS-regulated miRNAs [26] and has been demonstrated to function as a selective killer of malignant glioma through mitochondria-dependent pathways [27]. A previous study showed that PEITC, a strong ROS inducer and selective killer of malignant cancer cells, could induce apoptosis of glioma cells through the extrinsic- and intrinsic-apoptosis signaling pathways [28]. Thus, we hypothesize whether PEITC may function as a selective killer to malignant glioma cells through ROS production to activate miR-135a-mitochondria dependent apoptosis pathway. Moreover, various studies have demonstrated that multidrug resistance-associated protein 1(MRP1) is overexpressed in glioblastoma and plays a pivotal role in PEITC-induced ROS production through depleting GSH in cancer cells. Thus, it is also possible that MRP1 is involved in the selective lethality of PEITC to malignant glioma cells via ROS-MiR-135a-Mitochondria dependent apoptosis pathways.
In the current study, using immortalized human normal glial cell line (HEB) and malignant glioma cell lines (U87, U251, T98G) as models, we explored the potential mechanisms of PEITC as a selective killer to malignant glioma cells. Our results demonstrated that PEITC induced selective lethality and suppressed tumorigenicity and migration of malignant glioma cells through MRP1-mediated ROS production thereby activating miR-135a-mitochondria dependent apoptosis pathways, suggesting PEITC is a potential efficient agent for the treatment of glioma.
Materials and methods
Cell culture
Human normal glial cell line (HEB) and malignant glioma U87, U251 and T98G cell lines were kindly provided by Dr. Wu (Department of Pharmacology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, China). Malignant glioma U343 and HS683 cell lines were kindly provided by Dr. Gong (Department of Pharmacy, Xiangya Hospital, Central South University, Changsha, Hunan, China). Murine hippocampal HT22 cells, Non-cancerous human keratinocytes HaCaT cells, Human embryonic kidney HEK293 cells and Human Umbilical Vein Endothelial Cells (HUVEC) were obtained from the American Type Culture Collection (Manassas VA, USA). All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum at 37°C in an incubator with 5% CO2.
MTT and apoptosis assays
PEITC was purchased from Sigma-Aldrich (St. Louis, MO). The cytotoxic effects of PEITC on HEB, U87, U251, T98G, U343, HS683, HT22, HaCaT, HEK293, or HUVEC cells were evaluated by methyl thiazolyl tetrazolium (MTT) assay (sigma, St.Louis, MO, USA) according to the manufacturer’s instruction. Apoptosis was evaluated by the apoptotic morphology, Hoechst 33258 Staining, flow cytometry, and Caspases’ activity. Apoptotic morphology was observed by microscope (Nikon, Japan) and Hoechst 33258 Staining (Beyotime, Beijing, China). Cells with condensed and fragmented nuclei were considered and calculated as the apoptotic cells. For flow cytometry analysis, cells were collected and stained with Annexin V-FITC Apoptosis Detection Kit (Beyotime, C1063) and analyzed by a flow cytometer (Beckman Coulter, Brea, CA, USA). Caspases’ activity was determined by Caspase-3 and 9 Activity Assay Kit (Beyotime, C116 and C1158). Caspases inhibitor Z-VAD-FMK (Beyotime, C1202) was dissolved in dimethyl sulfoxide (DMSO) and diluted to suitable concentration for experiments (sigma, St.Louis, MO, USA).
Foci-formation assay
Four hundred viable malignant glioma cells (U87, U251 and U343) were placed in 6-well plate in triplicate for 24 h, then continually maintained in complete medium with or without 20 μM PEITC for 14 days. Foci were fixed with 4% polyoxymethylene and stained with 0.1% crystal violet (Beyotime, C0121). The stained foci were washed by PBS 3 times and then detected by microscope. The foci were further dissolved in 500 μl DMSO and quantified by spectrophotometer at 540 nm.
Cell scratch assay
Malignant glioma cells (U87, U251 and T98G) were cultured in 6-well plate. After cells approached almost 100% confluence, the supernatant was absorbed and the cells were scratched with a 20 μl tips. Then the cells were washed with PBS and added medium with 20 μM PEITC. The scratched area was photographed with a microscope at 0 h and 24 h, respectively.
Cellular GSH measurement
The GSH assay is based on the chemical conjugation of GSH with 5, 59-Dithiobis (2-nitrobenzoic acid) [DTNB] (Beyotime, S0052). Cell extracts were prepared by sonication and deproteination using the conditions recommended by the manufacturer. Total GSH was detected by measuring the product of glutathionylated DTNB by spectrophotometer at 412 nm.
Cellular ROS detection
The intracellular ROS was detected by Reactive Oxygen Species Assay Kit (Beyotime, S0033) based on 2’,7’-dichlorofluoresceindiacetate (DCFH-DA).DCF fluorescence was detected using fluorescence microscope and photographs were taken by inverted fluorescence microscope (Nikon, Japan). The intensity of fluorescence was measured with a spectrofluorimeter (BioTek, USA) at excitation/emission wavelengths of λ=488/525 nm.
RT-qPCR and RT-PCR
Total RNAs were isolated by TRIzol reagent (Invitrogen, Grand Island, NY, USA). The concentration of RNA was determined by measuring the absorbance at 260 nm (BioSpec-nano). Reverse transcription was performed using SuperScript III reverse transcriptase (Invitrogen). Q-PCR was performed with Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) and detected with the LightCycler 480 instrument (Roche, Basel, Switzerland). Comparative CT method (2-ΔΔCT) was adopted to evaluate the relative quantitation. The levels of U6 and β-actin were used to normalize miRNA and mRNA level, respectively. Primer sequence used were synthesized by Sangon Biotech (Shanghai, China) with the following sequence: hMRP1-forward-5’-GGTGCTTCCCACGGAGG-3’, reverse-5’-TCAACCACAAAACTGCAGCC-3’, hBeta-actin-forward-5’-AGCGGGAAATCGTGCGTG-3’, reverse-5’-CAGGGTACATGGTGGTGCC-3’. Primers of stem-loop method for miR-135a detection were designed and synthesized by RiboBio (Guangzhou, China).
Cell transfection
MiR-135a inhibitor and miR-135a inhibitor negative control were designed and synthesized by RiboBio (Guangzhou, China). Cells were grown to 70%-90% confluence in 6-well plates before transfection. Cells were transfected with these oligonucleotides by using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer’s instructions.
Western blot analysis
Cells were lysed in the ice-cold RIPA lysis buffer (Beyotime, P0013B). The protein concentration of the lysate was determined by BCA kits (Beyotime, P0010S). Protein samples were separated by electrophoresis on SDS-PAGE gel and transferred onto a Hybond ECL transfer membrane (Amersham Pharmacia, Piscataway, NJ, USA). After blocking, the membrane was incubated with primary antibodies for STAT6, SMAD5, Bcl-xl, Bax, Cyto-C, MRP1, or β-actin (Santa Cruz, CA) followed by exposure to horseradish peroxidase (HRP)-conjugated secondary anti-mouse or rabbit antibodies (Santa Cruz, CA). To visualize protein bands, chemiluminescence (ECL) system (BIO-RAD, USA) was used.
Statistical analyses
Statistical analyses of the data were conducted using GraphPad Prism 5 software. All data are expressed as Mean±SD (n=3) except special indication. Difference was considered to be statistically significant at P<0.05 or fold change>2.
Results
PEITC has selective lethality to malignant glioma cells
To determine whether PEITC had selective lethality to malignant glioma cell lines, MTT assay was performed on a series of cell lines including the non-cancerous cell lines HEB, HT22, HACAT, HEK293 and HUVEC, and the malignant glioma cell lines U87, U251, T98G, U343 and HS683. Cells were plated in 96-well plates at a density of 5×103 and treated with various concentrations (0, 10, 15, 20, 25, 30, 35, 40 μM) of PEITC for 24 h and 48 h. PEITC induced more loss of cell viability in the malignant glioma cell lines than in the non-cancerous cell lines in a dose- and time-dependent manner (Figure 1A). The IC50 values of PEITC treatment for 24 h and 48 h in the above cell lines were calculated by GraphPad Prism 5 and the results showed that the IC50 values of PEITC treatment for 24 h and 48 h in malignant glioma cell lines were significantly lower than that in the non-cancerous cell lines (Figure 1B). These results demonstrated that the malignant glioma cell lines were more sensitivity to PEITC than the non-cancerous cell lines, suggesting that PEITC has selective lethality to malignant glioma cells.
Figure 1.
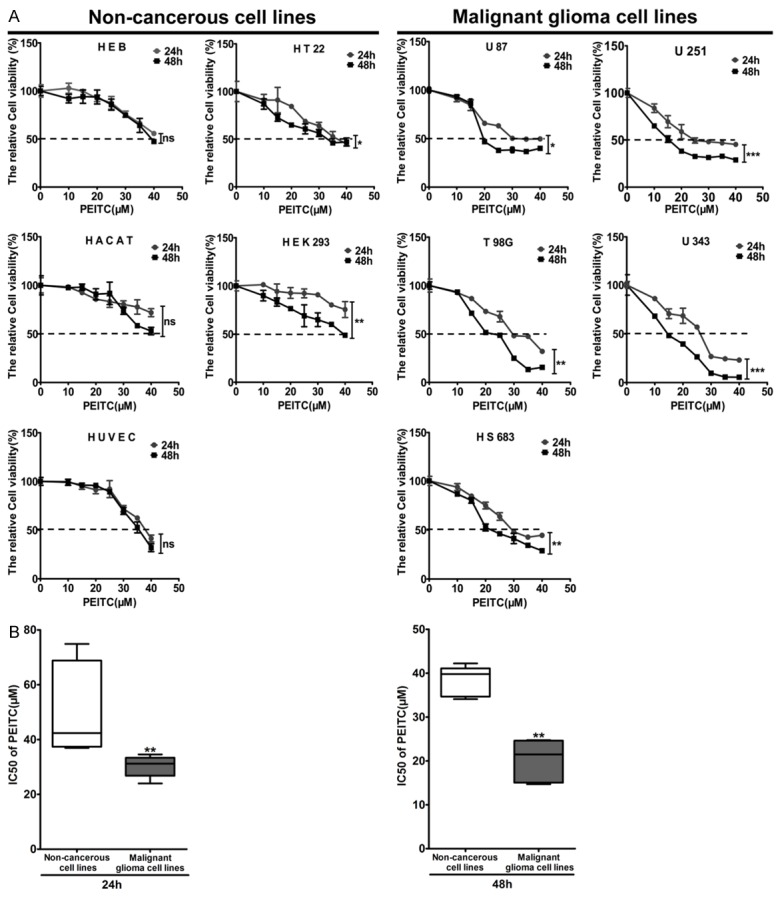
PEITC caused more cytotoxicity in the malignant glioma cell lines than in the non-cancerous cell lines. Non-cancerous HEB, HT22, HACAT, HEK293 and HUVEC cells, and the malignant glioma U87, U251, T98G, U343 and HS683 cells were treated with various concentrations (0, 10, 15, 20, 25, 30, 35, 40 μM) of PEITC for 24 h and 48 h. A. The relative cell viability was determined by MTT assay. Data are presented as mean ± SD in triplicate. *P<0.05, **P<0.01, ***P<0.001, ns: no significant. B. The IC50s of cell lines treated with PEITC for 24 h and 48 h were compared in the malignant glioma cell lines and the non-cancerous cell lines, respectively. **P<0.01.
PEITC selectively induces mitochondria-dependent apoptosis of malignant glioma cells
Carefully analysis of the data of MTT assay revealed that 20 μM of PEITC for 24 h could cause significant cytotoxicity on malignant glioma cells, but had no effect on the non-cancerous cell lines. Accordingly, we chose PEITC at the dose of 20 μM and treatment for 24 h for further experiments. Since the immortalized human normal glial cell line (HEB) and malignant glioma cell lines U87, U251 and T98G have been well documented as the cell models for identifying miR-135a functioning as the selective killer of malignant glioma [27], we consequently selected these cell lines for exploring the potential mechanism of PEITC to selectively kill malignant glioma cells.
After treatment with 20 μM PEITC for 24 h, significant apoptotic morphological changes such as smaller, round and blunt in size of malignant glioma cells (U87, U251 and T98G) were observed, but there was no such cell morphological alteration in HEB cells (Figure 2A). Hoechst 33258 staining (Figure 2B) and Annexin V/PI flow-cytometry assay (Figure 2C) showed PEITC selectively triggered apoptosis in malignant glioma cells but not in HEB cells. Caspases are in charge of cell apoptosis and caspase-9 is directly activated in mitochondrial pathway and caspase-3 is a terminal executioner in apoptosis [27]. Next, cells were treated with or without PEITC (20 μM) for 24 h and the activities of caspase-9 and caspase-3 were measured by the corresponding activity assay kits. PEITC (20 μM) selectively activated caspase-9 and caspase-3 in malignant glioma cells, but not in HEB cells (Figure 2D and 2E). Consistent with the above results, caspases inhibitor Z-VAD-FMK significantly reduced the loss of cell viability of malignant glioma cell lines by PEITC (Figure 2F). These results implied that mitochondria-dependent apoptosis pathway plays a pivotal role in PEITC-mediated selective lethality to malignant glioma cells.
Figure 2.

PEITC the selectively killed malignant glioma cell lines through mitochondria-dependent apoptosis pathway. The indicated cells were treated with 20 μM of PEITC for 24 h. (A) Cell morphological changes were determined under phase contrast microscope at 20×. (B) Cells were stained with Hoechst 33258 detected and calculated by fluoresent photomicrographs at 10×. Mean ± SD, n=3, ***P<0.001 VS control. (C) Cells were labeled with AnnexinV/PI and detected by flow cytometry. Right lower and upper quadrant showed apoptotic cells and left upper quadrant showed necrosis cells. (D, E) The activity of caspase-9 (D) and caspase-3 (E) was measured by the corresponding caspase activity assay kits, respectively. Mean ± SD, n=3, **P<0.01, ***P<0.001 VS control. (F) Cell viability after treatment with 20 μM PEITC alone or in combination with caspases inhibitor Z-VAD-FMK (10 μM) was determined by MTT assay. Mean ± SD, n=3, *P<0.05, **P<0.01, ***P<0.001 VS control, #P<0.05, ##P<0.01, ###P<0.001 VS 20 μM PEITC.
PEITC suppresses tumorigenicity and migration of malignant glioma cells in vitro
The potent induction of apoptosis in malignant glioma cell lines by PEITC led us to explore its ability to suppress tumorigenicity of malignant glioma cell lines in vitro with foci-formation assay. As shown in Figure 3A, PEITC (20 μM) treated groups displayed much fewer and smaller foci of malignant glioma cell lines (U87, U251 and U343) as compared with control. When detecting the foci formation ability by microscope, we found the pseudopodia of the malignant glioma cells were damaged by PEITC, suggesting PEITC may suppress migration ability of malignant glioma cells. Indeed, further cell scratch assays showed 20 μM PEITC for 24 h significantly suppressed migration ability (Figure 3B) of the malignant glioma cell lines (U87, U251 and T98G). Thus, PEITC suppressed the foci formation and migration of malignant glioma cells in vitro.
Figure 3.
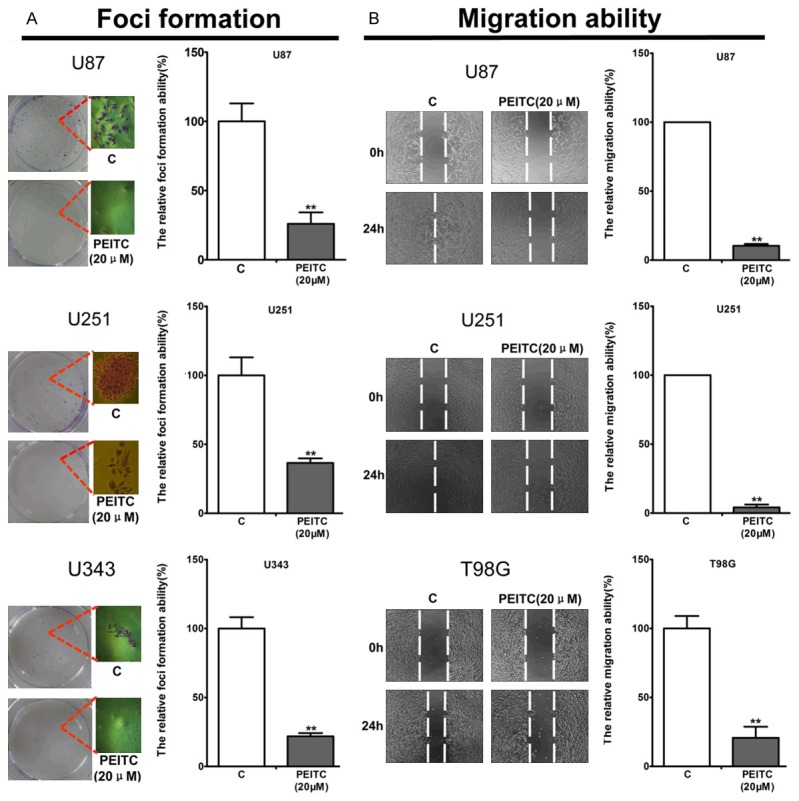
PEITC suppressed tumorigenicity and migration ability of malignant glioma cell lines in vitro. Cells were treated with or without 20 μM PEITC. A. Foci formation of U87, U251 and U343 cells was determined as described in materials and methods. B. The scratched areas of U87, U251 and T98G were determined as described in material and methods. Mean ± SD, n=3, **P<0.01 VS control.
Cellular GSH depletion and ROS production are critical factors for PEITC mediated selective lethal to malignant glioma cell lines
Previous studies have revealed the cellular GSH and ROS level are important factors in determining the selective lethality of PEITC to cancer cells [13,16,29]. To investigate whether cellular GSH and ROS are key factors for PEITC to selectively kill malignant glioma cells, we detected GSH and ROS levels in HEB and malignant glioma cells (U87, U251 and T98G) in the presence or absence of PEITC. ROS staining by DCFH-DA probe demonstrated no significant difference of cellular GSH and ROS level among these cell lines under normal growth conditions (data not shown). However, treatment of 20 μM PEITC for 5 h induced more ROS production in malignant glioma cells than in HEB (Figure 4A). Further quantitative analysis by microplate fluorometer showed that PEITC for 5 h significantly induced the production of ROS in malignant glioma cells but not in HEB cells (Figure 4B). Moreover, treatment of these cells with different concentrations of PEITC for 5h showed that PEITC induced ROS production in malignant glioma cells in a dose-dependent manner, but almost no change in HEB within 20 μM PEITC (Figure 4C). In addition, treatment of these cells with 20 μM PEITC for different time demonstrated that PEITC induced ROS production in malignant glioma cells but no HEB cells in a time-dependent way (Figure 4D). Correspondingly, these cells were treated with different concentrations of PEITC for different time, and the cellular GSH levels were assessed by GSH assay. As shown in Figure 4E and 4F, PEITC depleted cellular GSH in a dose- and time- dependent manner, but almost no change in HEB within 20 μM PEITC. These results revealed cellular GSH depletion and ROS production were critical factors for PEITC functioning as a selective killer to malignant glioma cell lines, but the “basis ROS cancer therapy” determining PEITC selective lethality may not adapt in Wu’s cell model, suggesting there are other additional mechanisms that mediate the selective lethal of PEITC in malignant glioma cell lines.
Figure 4.
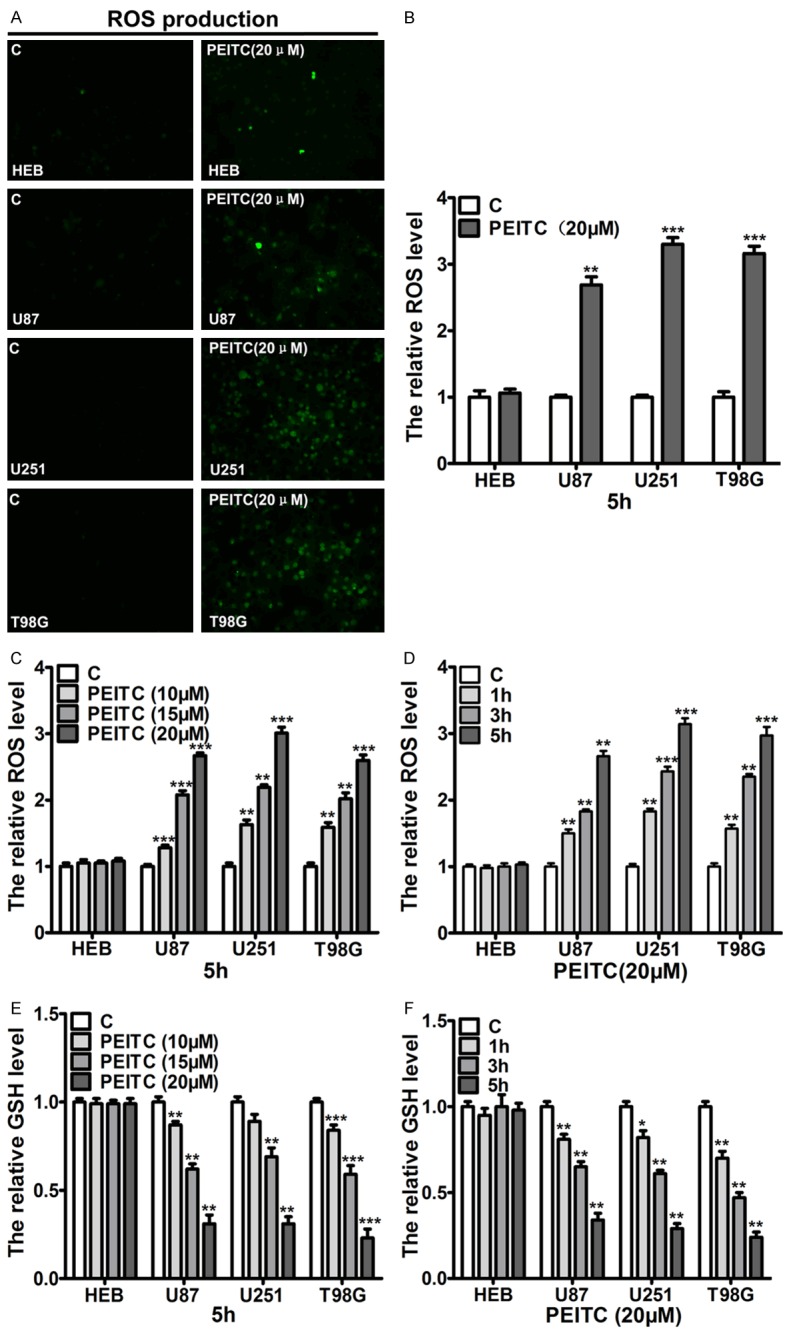
Cellular GSH depletion and ROS production played a pivotal role in PEITC selectively killing the malignant glioma cells. Cells were treated with indicated concentrations of PEITC for different time periods. A. The cellular ROS production induced by 20 μM PEITC for 5 h in HEB and U87, U251 and T98G cells was detected by inverted fluorescence microscope. B. The cellular ROS production induced by 20 μM PEITC for 5 h in HEB and U87, U251 and T98G cell was assessed by microplate fluorometer. C. HEB, U87, U251 and T98G cells were treated the indicated concentrations of PEITC for 5 h, cellular ROS levels were measured by microplate fluorometer. D. HEB, U87, U251 and T98G cells were treated with 20 μM PEITC for the indicated time, cellular ROS levels were measured by microplate fluorometer. E. HEB, U87, U251 and T98G cells were treated the indicated concentrations of PEITC for 5 h, cellular GSH was measured by spectrophotometer. F. HEB, U87, U251 and T98G cells were treated 20 μM PEITC for the indicated time, cellular GSH was measured by spectrophotometer. Mean ± SD, n=3, *P<0.05, **P<0.01, ***P<0.001 VS control.
Cellular ROS-induced MiR-135a upregulation plays a pivotal role in the selective lethality of PEITC to malignant glioma cells
In previous studies, Wu et al. revealed miR-135a, which is abundant in human normal glial cells but is reduced with glioma grading, functions as a selective killer to malignant glioma cells through mitochondria-dependent apoptosis pathway [27]. Further, Shin et al. found chemicals-induced miR-135a-3p could trigger prostate cancer cells apoptosis [26]. PEITC has been demonstrated as a strong ROS inducer and mainly activates mitochondria-dependent apoptosis pathway to eliminate cancer cells. These data suggest that PEITC-induced ROS production may upregulate miR-135a to selectively trigger apoptosis in malignant glioma cells. To this end, we firstly detected the relative expression level of miR-135a in non-cancerous cell lines and malignant glioma cell lines. The results showed miR-135a was abundant in non-cancerous cell lines with overexpression in HEB cells but was low in malignant glioma cell lines (Figure 5A). In average, the relative expression level of miR-135a in non-cancerous cell lines was significant higher than in malignant glioma cell lines (Figure 5B). Further, we detected miR-135a levels in HEB and malignant glioma cells (U87, U251 and T98G) after treatment with 20 μM PEITC or 20 μM PEITC in combination with 7.5 mM GSH for 24 h. The results showed, in malignant glioma cell lines but not in HEB cells, PEITC significantly upregulated miR-135a, which was however completely reversed by 7.5 mM GSH, a ROS scavenger (Figure 5C). Further experiments showed 50 nm miR-135a inhibitor and 7.5 mM GSH partly and completely reversed PEITC-induced caspase-9 and caspase-3 activation in malignant glioma cells, but whichever treatments had no effects on caspase-9 and caspase-3 activation in HEB (Figure 5D and 5E). In consistent, Western blot analyses showed PEITC significantly down-regulated STAT6 and SMAD5, which are miR-135a targets and play a pivotal role in mitochondria-dependent apoptosis pathways [27]. Moreover, PEITC treatment significantly resulted in Bcl-xl down-regulation, Bax up-regulation and Cyto-chrome C releasing. Pre-transfection with 50 nM miR-135a inhibitor or combination with 7.5 mM GSH partly and completely reversed the PEITC-induced effects on these proteins associated with miR-135a-mitochondria dependent apoptosis pathway, respectively. Interestingly, the proteins associated with miR-135a-mitochondria dependent apoptosis pathway were not significantly altered by these treatments in HEB cells (Figure 5F). In addition, the reduced cell viability of malignant glioma cells that was induced by PEITC was partly and completely prevented by pre-transfection with 50 nm miR-135a inhibitor and 7.5 mM GSH, respectively (Figure 5G). These results revealed ROS-miR-135a-mitochondria dependent apoptosis pathway plays a pivotal role in PEITC-mediated selective lethality to malignant glioma cells.
Figure 5.
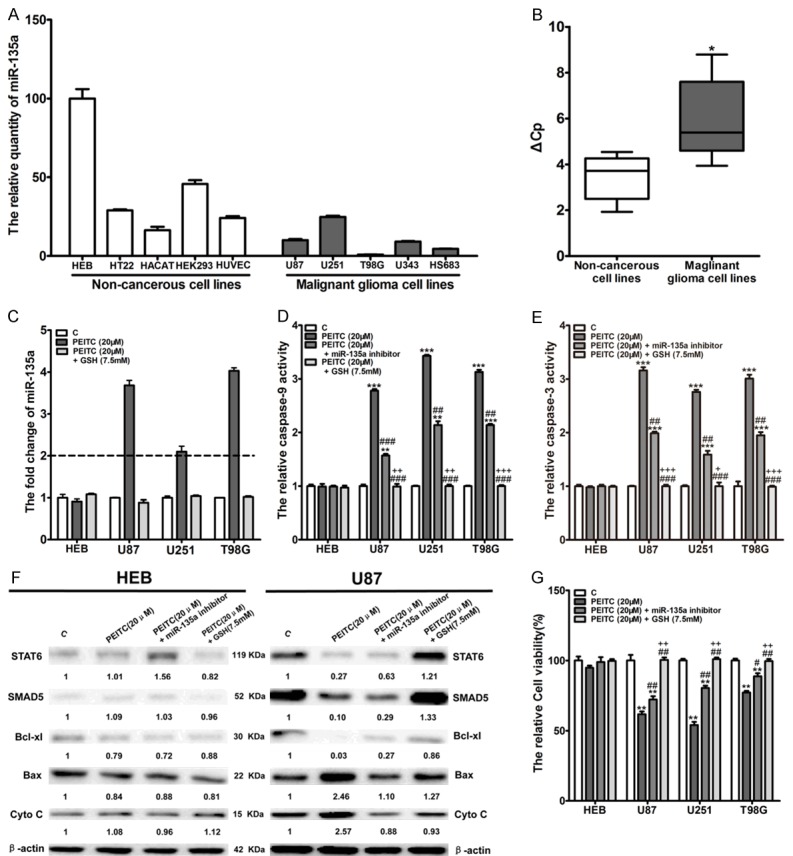
MiR-135a upregulation-induced by ROS contributed to PEITC-mediated selective lethality to malignant glioma cells. (A) The relative expression of miR-135a was determined by RT-qPCR with U6 as internal control. HEB shows the highest level of endogenous miR-135a. (n=3). (B) The relative abundance of miR-135a in non-cancerous cell lines versus malignant glioma cell lines (*P<0.05). (C) Cells were treated with 20 μM of PEITC alone or in combination with 7.5 mM GSH for 24 h. The relative expression of miR-135a was determined by RT-qPCR with U6 as internal control. n=3, fold change>2 regarded as significant difference. (D, E) Cells were treated with 20 μM of PEITC alone or in combination with 7.5 mM GSH or pre-transfection with 50 nM miR-135a inhibitor for 24 h. The activation of caspase-9 (D) and caspase-3 (E) was measured by the corresponding caspase activate assay kit, respectively (Mean ± SD, n=3, **P<0.01, ***P<0.001 VS control group, ##P<0.01, ###P<0.001 VS 20 μM PEITC group, +P<0.05, ++P<0.01, +++P<0.001 VS pre-transfection with 50 nm miR-135a inhibitor group). (F) Cells were treated with 20 μM of PEITC alone or in combination with 7.5 mM GSH or pre-transfection with 50 nM miR-135a inhibitor for 24 h. The proteins associated with miR-135a-mitochondria dependent apoptosis pathway in HEB and U87 affected were determined by western blot. (G) Cells were treated with 20 μM of PEITC alone or in combination with 7.5 mM GSH or pre-transfection with 50 nM miR-135a inhibitor for 24 h. Cell viability was determined by MTT assay. (Mean ± SD, n=3, **P<0.01 VS control group, #P<0.05, ##P<0.01 VS 20 μM PEITC group, ++P<0.01 VS pre-transfection with 50 nm miR-135a inhibitor group).
MRP1 plays a pivotal role in PEITC-mediated selective activation of the ROS-miR-135a-mitochondria dependent apoptosis pathway in malignant glioma cells
PEITC is lipophilic and can enter into cells through passive diffusion across lipid bilayer of HEB and malignant glioma cells [30]. After entering into cells, PEITC would conjugate with GSH to PEITC-GSH conjugate, which would be quickly exported out with the identified specialty transporters such as MRP1, MRP2 and BCRP [18], which are overexpressed in cancer cells [31,32], leading to ROS production. It has been proposed that keeping the PEITC-GSH conjugates longer retention in cells through inhibiting these identified specialty transporters would release GSH again to keep ROS and anti-ROS system balance [18]. Since MRP1 is overexpressed in glioblastoma [33,34], it is possible that MRP1 may mediate the difference of ROS-response induced by PEITC in HEB and malignant glioma cells, thereby leading to selective activiation of ROS-miR-135a-mitochondria dependent apoptosis pathway in U87, U251 and T98G. To this end, we firstly determined MRP1 expression through RT-PCR and Western blot, and found a higher expression of MRP1 in malignant glioma cell lines (U87, U251 and T98G) than HEB both in mRNA and protein levels (Figure 6A and 6B). Schisandrin B (Sch B), a specific inhibitor of MRP1 [35], was used to explore whether MRP1 mediates ROS-miR-135a-mitochondria dependent apoptosis pathway activation in malignant glioma cells. Through MTT assay, we found Sch B showed no apparent cytotoxicity to HEB, U87, U251 and T98G within 150 μM (data not shown). With Carboxyfluorescein diacetate (CFDA), a specific substrate of MRP1, we found pre-incubation with 75 μM Sch B for 2 h was the optimal condition for Sch B to inhibit MRP1 in malignant glioma (data not shown). Further experiments showed the cellular GSH and ROS changes in malignant glioma (U87, U251 and T98G) induced by 20 μM PEITC were significantly attenuated by pre-incubation with 75 μM Sch B for 2 h (Figure 6C and 6D). Similarly, the increase of miR-135a expression and the activation of caspase-9 and caspase-3 in malignant glioma (U87, U251 and T98G) by 20 μM PEITC were significantly reduced by pre-incubation with 75 μM Sch B for 2 h (Figure 6E-G). In addition, PEITC-mediated downregulation of STAT6, SMAD5 and Bcl-xl protein levels, and upregulation of Bax protein level and Cyto-chrome C release in malignant glioma cells was attenuated by pre-incubation with 75 μM Sch B for 2 h (Figure 6H). Finally, pre-incubation with 75 μM Sch B for 2 h significantly reduced the loss of cell viability of malignant glioma cells by PEITC (Figure 6I). It was noted that pre-incubation with 75 μM Sch B for 2 h did not lead to alteration of the molecules of the ROS-miR-135a-mitcohondria dependent apoptosis pathways in HEB cells treated with PEITC, implying MRP1 plays a pivotal role in PEITC-mediated selective lethality to malignant glioma cells.
Figure 6.
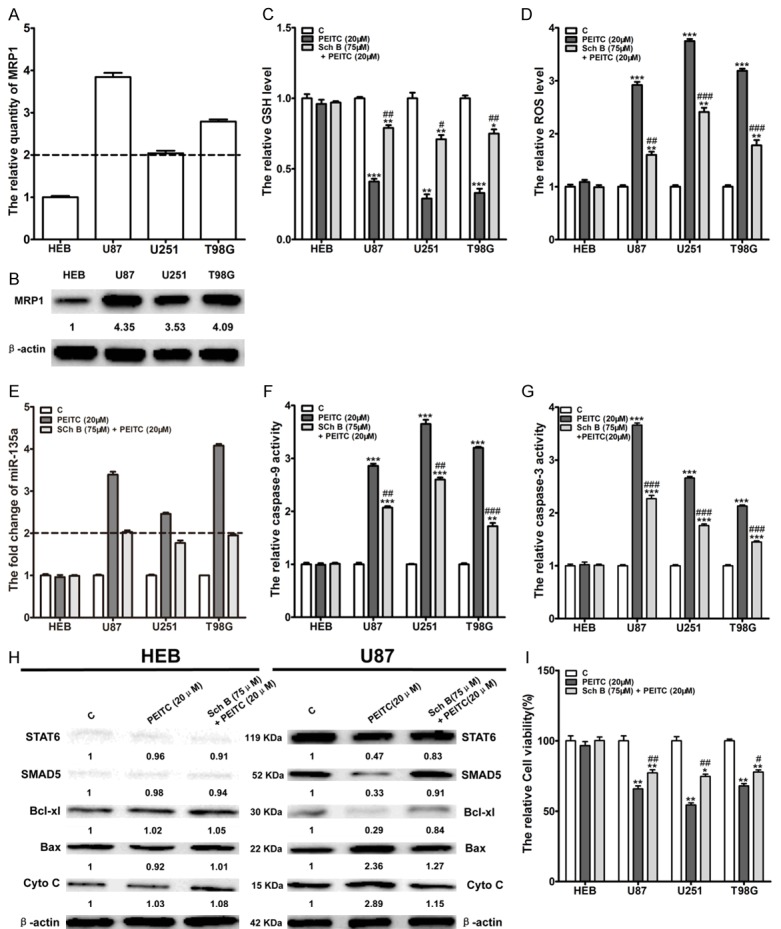
MRP1 played a pivotal role in the activation of ROS-miR-135a-mitochondria dependent apoptosis pathway in malignant glioma cell lines by PEITC. (A) The mRNA levels of MRP1 in HEB, U87, U251 and T98G cells were detected by RT-PCR. (B) The protein levels of MRP1 in HEB, U87, U251 and T98G cells were detected by Western blot. Mean ± SD, n=3, fold>2 regarded as significant difference. (C, D) Cells were pre-incubated with 75 μM Sch B for 2 h, followed by treatment with or without 20 μM PEITC for 5 h. Cellular GSH (C) and ROS (D) were measured by spectrophotometer. Mean ± SD, n=3,*P<0.05, **P<0.01, ***P<0.001 VS control group, #P<0.05, ##P<0.01, ###P<0.001 VS 20 μM PEITC group. (E, F, G) Cells were pre-incubated with 75 μM Sch B for 2 h, followed by treatment with or without 20 μM PEITC for 24 h. MiR-135a levels were determined by RT-qPCR (E) the activity of caspase-9 (F) and caspase-3 (G) were determined by caspase activation assay kits. Mean ± SD, n=3, **P<0.01, ***P<0.001 VS control group, ##P<0.01, ###P<0.001 VS 20 μM PEITC group, fold change>2 regarded as significant difference. (H) HEB and U87 cells were pre-incubated with 75 μM Sch B for 2 h, followed by treatment with or without 20 μM PEITC for 24 h. The proteins associated with miR-135a-mitochondria dependent apoptosis were determined through Western blot. (I) HEB and U87 cells were pre-incubated with 75 μM Sch B for 2 h, followed by treatment with or without 20 μM PEITC for 24 h. The cell viability was determined by MTT assay. Mean ± SD, n=3, *P<0.05, **P<0.01 VS control group, #P<0.05, ##P<0.01 VS 20 μM PEITC group.
Discussion
In our present study, we found that PEITC had selective lethality to malignant glioma cells. We further demonstrated PEITC induced apoptosis and suppressed tumorigenicity and migration in malignant glioma cells. Mechanistically, our findings demonstrated that PEITC exerted its selective lethality functions through MRP-1 mediated GSH depletion leading to ROS production, thereby activating miR-135a-mitochondria dependent apoptosis pathway in malignant glioma (Figure 7). These results suggest that PEITC is an effective agent for treating glioma.
Figure 7.
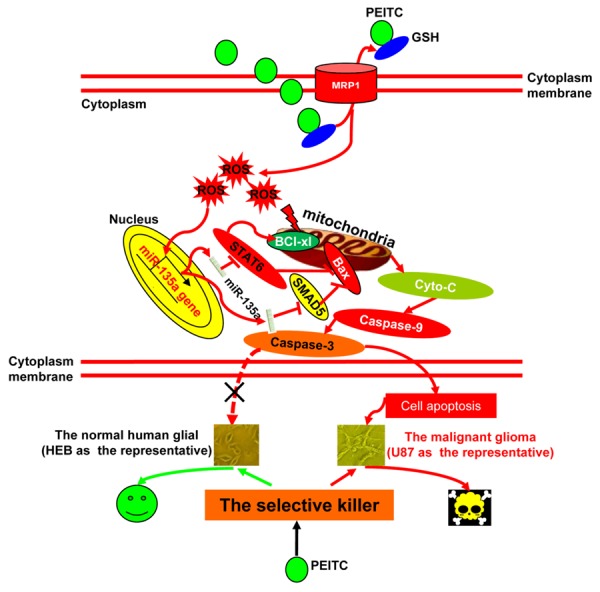
Schematic representation of the signaling pathway mediating PEITC selective lethality to malignant glioma cells.
In recent years, PEITC has been shown as a selective killer of malignant cells such as ovarian cancer cells and leukemic cells depending on the cellular basis ROS, which are higher in malignant cancer cells than in the corresponding normal cells [13,16]. Herein, we are the first to report PEITC functions as a selective killer to malignant glioma cells. However, we did not find cellular basis ROS difference between HEB and malignant glioma cells. Further experiments revealed that PEITC induced ROS production, accompanied with upregulation of miR-135a and activation of mitochondria-dependent apoptosis pathways in malignant glioma cells, but no significant ROS and miR-135a changes were observed in HEB treated within 20 μM PEITC. The regulation of miRNAs by anti-cancer agent-induced ROS production has been demonstrated in several studies, such as the regulation of miR-27a by Betulinic acid, Curcumin and Celastrol in the stimulation of apoptosis in breast cancer, colon cancer and bladder cancer cells [23,24,36] and re-activation of miR-491 by Arsenic trioxide (As2O3) in attenuating the invasion potential of human liver cancer cells [37]. MiR-135a is abundant in normal glial cell but reduced with glioma grading, suggesting it may be silenced in glioblastoma through hyper-methylation, which is an important epigenetic event during tumorigenesis. ROS has been demonstrated to regulate DNA Methyltransferases (DNMTs) leading to hyper-methylation silenced tumor suppressors in cancer cells [38,39]. PEITC is a strong ROS inducer and functions as an epigenetic regulator in cancer cells [40,41]. It is thus possible that the PEITC-induced ROS-mediated DNA demethylation may be the potential mechanism of miR-135a upregulation in malignant glioma cells, which is needed to be further studied.
No significant difference of cellular basis GSH and ROS level between HEB and malignant glioma cells (U87, U251 and T98G) was observed, however, malignant glioma cells but not HEB displayed significant ROS increase after treatment with 20 μM PEITC for 5 h. It remains unclear regarding the mechanism of the different ROS-response between HEB and malignant glioma cells after treatment with the same concentration of PEITC. In our knowledge, the lipophilic PEITC can enter into cells through passive diffusion [30], suggesting there might be no rate difference in crossing the lipid bilayers of HEB and malignant glioma cells. Once inside cell, PEITC conjugates with GSH to form the PEITC-GSH complex, which is rapidly exported out cells by specific transporters such as MRP1, MRP2 and BCRP, leading to ROS production [18]. As MRP1, MRP2 and BCRP are overexpressed in cancer cells, the different expression of these specific transporters may cause the difference rates of exporting out PEITC-GSH conjugates and consequently result in the different ROS-response induced by PEITC in HEB and malignant glioma cells. MRP1 is overexpressed in glioblastoma [33,34]. We found MRP1 played a pivotal role in the different ROS-response and selectively killing malignant glioma cells through activating miR-135a-mitochondria dependent apoptosis pathways by PEITC. GSH is an important anti-ROS molecule inside cells and plays an important role in drug resistance of cancer cells to chemotherapy drugs such as cisplatin, doxorubicin and vincristine. The mechanism of the enhanced cytotoxicity by increased export of PEITC-GSH conjugates from cells remains elusive. Unlike the normal chemotherapy drugs such as cisplatin, doxorubicin and vincristine, which depend on enough intracellular concentration of drugs to inhibit specific drug-targets [42-44], PEITC has no specific drug targets and mainly depends on GSH depletion to cause ROS production thereby triggering cancer cell death. Thus, more PEITC-GSH conjugates export out of cells will induce more ROS production to cause more cytotoxic. Consequently, if PEITC-GSH conjugates are kept longer retention in cancer cells, it will release free GSH to keep cellular ROS balance, causing less cytotoxicity [18]. In supporting the above hypothesis, we found that pharmacologic inhibition of MRP1 by Sch B significantly attenuated PEITC-induced activation of ROS-miR-135a-mitochondria-dependent apoptosis pathway and loss of cell viability, suggesting PEITC is a promising agent for treating glioma. The lipophilic and small molecule nature of PEITC suggests that it could easily cross the blood brain barrier and accumulate enough concentration in glioblastoma in theory, but the suitable given dose and parameters of pharmacokinetics require further investigation in animal models and clinical trials.
In conclusion, our studies revealed PEITC had selective lethality to malignant glioma cells through activiation of the ROS-MiR-135a-Mitochondria dependent apoptosis pathways via MRP1-mediated export of GSH, suggesting PEITC is a potential effective agent for treating glioblastoma.
Acknowledgements
This work was supported by grants from Key program, National Natural Science Foundation of China (No. 81373477, No. 81570533).
Disclosure of conflict of interest
None.
References
- 1.Wang L, Ye X, Cai X, Su J, Ma R, Yin X, Zhou X, Li H, Wang Z. Curcumin suppresses cell growth and invasion and induces apoptosis by down-regulation of Skp2 pathway in glioma cells. Oncotarget. 2015;6:18027–37. doi: 10.18632/oncotarget.4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J, Yang J, Chen Y, Mao Q, Li S, Xiong W, Lin Y, Chen J, Ge J. Genetic Variants of VEGF (rs201963 and rs3025039) and KDR (rs7667298, rs2305948, and rs1870377) Are Associated with Glioma Risk in a Han Chinese Population: a Case-Control Study. Mol Neurobiol. 2016;53:2610–8. doi: 10.1007/s12035-015-9240-0. [DOI] [PubMed] [Google Scholar]
- 3.Yin D, Ogawa S, Kawamata N, Leiter A, Ham M, Li D, Doan NB, Said JW, Black KL, Phillip Koeffler H. miR-34a functions as a tumor suppressor modulating EGFR in glioblastoma multiforme. Oncogene. 2013;32:1155–1163. doi: 10.1038/onc.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pojo M, Goncalves CS, Xavier-Magalhaes A, Oliveira AI, Goncalves T, Correia S, Rodrigues AJ, Costa S, Pinto L, Pinto AA, Lopes JM, Reis RM, Rocha M, Sousa N, Costa BM. A transcriptomic signature mediated by HOXA9 promotes human glioblastoma initiation, aggressiveness and resistance to temozolomide. Oncotarget. 2015;6:7657–7674. doi: 10.18632/oncotarget.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eom KY, Cho BJ, Choi EJ, Kim JH, Chie EK, Wu HG, Kim IH, Paek SH, Kim JS, Kim IA. The Effect of Chemoradiotherapy with SRC Tyrosine Kinase Inhibitor, PP2 and Temozolomide on Malignant Glioma Cells in vitro and in vivo. Cancer Res Treat. 2016;48:687–97. doi: 10.4143/crt.2014.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Getahun SM, Chung FL. Conversion of glucosinolates to isothiocyanates in humans after ingestion of cooked watercress. Cancer Epidemiol Biomarkers Prev. 1999;8:447–451. [PubMed] [Google Scholar]
- 7.Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol Biomarkers Prev. 1998;7:1091–1100. [PubMed] [Google Scholar]
- 8.Tang K, Lin Y, Li LM. The role of phenethyl isothiocyanate on bladder cancer ADM resistance reversal and its molecular mechanism. Anat Rec (Hoboken) 2013;296:899–906. doi: 10.1002/ar.22677. [DOI] [PubMed] [Google Scholar]
- 9.Satyan KS, Swamy N, Dizon DS, Singh R, Granai CO, Brard L. Phenethyl isothiocyanate (PEITC) inhibits growth of ovarian cancer cells by inducing apoptosis: role of caspase and MAPK activation. Gynecol Oncol. 2006;103:261–270. doi: 10.1016/j.ygyno.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Xiao D, Singh SV. p66Shc is indispensable for phenethyl isothiocyanate-induced apoptosis in human prostate cancer cells. Cancer Res. 2010;70:3150–3158. doi: 10.1158/0008-5472.CAN-09-4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakubikova J, Bao Y, Sedlak J. Isothiocyanates induce cell cycle arrest, apoptosis and mitochondrial potential depolarization in HL-60 and multidrug-resistant cell lines. Anticancer Res. 2005;25:3375–3386. [PubMed] [Google Scholar]
- 12.Wu WJ, Zhang Y, Zeng ZL, Li XB, Hu KS, Luo HY, Yang J, Huang P, Xu RH. beta-phenylethyl isothiocyanate reverses platinum resistance by a GSH-dependent mechanism in cancer cells with epithelial-mesenchymal transition phenotype. Biochem Pharmacol. 2013;85:486–496. doi: 10.1016/j.bcp.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 13.Trachootham D, Zhang H, Zhang W, Feng L, Du M, Zhou Y, Chen Z, Pelicano H, Plunkett W, Wierda WG, Keating MJ, Huang P. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood. 2008;112:1912–1922. doi: 10.1182/blood-2008-04-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang T, Song X, Liu YF, Wang WY. PEITC reverse multi-drug resistance of human gastric cancer SGC7901/DDP cell line. Cell Biol Int. 2014;38:502–510. doi: 10.1002/cbin.10169. [DOI] [PubMed] [Google Scholar]
- 15.Denis I, Cellerin L, Gregoire M, Blanquart C. Cisplatin in combination with Phenethyl Isothiocyanate (PEITC), a potential new therapeutic strategy for malignant pleural mesothelioma. Oncotarget. 2014;5:11641–11652. doi: 10.18632/oncotarget.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 18.Gupta P, Wright SE, Kim SH, Srivastava SK. Phenethyl isothiocyanate: A comprehensive review of anti-cancer mechanisms. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2014;1846:405–424. doi: 10.1016/j.bbcan.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu C, Hu W, Wu H, Hu X. No evident dose-response relationship between cellular ROS level and its cytotoxicity--a paradoxical issue in ROS-based cancer therapy. Sci Rep. 2014;4:5029. doi: 10.1038/srep05029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohtsuka M, Ling H, Doki Y, Mori M, Calin GA. MicroRNA Processing and Human Cancer. J Clin Med. 2015;4:1651–1667. doi: 10.3390/jcm4081651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen SM, Chou WC, Hu LY, Hsiung CN, Chu HW, Huang YL, Hsu HM, Yu JC, Shen CY. The Effect of MicroRNA-124 Overexpression on Anti-Tumor Drug Sensitivity. PLoS One. 2015;10:e0128472. doi: 10.1371/journal.pone.0128472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.To KK, Leung WW, Ng SS. Exploiting a novel miR-519c-HuR-ABCG2 regulatory pathway to overcome chemoresistance in colorectal Cancer. Exp Cell Res. 2015;338:222–31. doi: 10.1016/j.yexcr.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Jutooru I, Lei P, Kim K, Lee SO, Brents LK, Prather PL, Safe S. Betulinic acid targets YY1 and ErbB2 through cannabinoid receptor-dependent disruption of microRNA-27a:ZBTB10 in breast cancer. Mol Cancer Ther. 2012;11:1421–1431. doi: 10.1158/1535-7163.MCT-12-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gandhy SU, Kim K, Larsen L, Rosengren RJ, Safe S. Curcumin and synthetic analogs induce reactive oxygen species and decreases specificity protein (Sp) transcription factors by targeting microRNAs. BMC Cancer. 2012;12:564. doi: 10.1186/1471-2407-12-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chintharlapalli S, Papineni S, Lei P, Pathi S, Safe S. Betulinic acid inhibits colon cancer cell and tumor growth and induces proteasome-dependent and -independent downregulation of specificity proteins (Sp) transcription factors. BMC Cancer. 2011;11:371. doi: 10.1186/1471-2407-11-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin EA, Sohn EJ, Won G, Choi JU, Jeong M, Kim B, Kim MJ, Kim SH. Upregulation of microRNA135a-3p and death receptor 5 plays a critical role in Tanshinone I sensitized prostate cancer cells to TRAIL induced apoptosis. Oncotarget. 2014;5:5624–5636. doi: 10.18632/oncotarget.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu S, Lin Y, Xu D, Chen J, Shu M, Zhou Y, Zhu W, Su X, Qiu P, Yan G. MiR-135a functions as a selective killer of malignant glioma. Oncogene. 2012;31:3866–3874. doi: 10.1038/onc.2011.551. [DOI] [PubMed] [Google Scholar]
- 28.Chou YC, Chang MY, Wang MJ, Harnod T, Hung CH, Lee HT, Shen CC, Chung JG. PEITC induces apoptosis of Human Brain Glioblastoma GBM8401 Cells through the extrinsic- and intrinsic -signaling pathways. Neurochem Int. 2015;81:32–40. doi: 10.1016/j.neuint.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Syed Alwi SS, Cavell BE, Donlevy A, Packham G. Differential induction of apoptosis in human breast cancer cell lines by phenethyl isothiocyanate, a glutathione depleting agent. Cell Stress Chaperones. 2012;17:529–538. doi: 10.1007/s12192-012-0329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji Y, Morris ME. Transport of dietary phenethyl isothiocyanate is mediated by multidrug resistance protein 2 but not P-glycoprotein. Biochemical Pharmacology. 2005;70:640–647. doi: 10.1016/j.bcp.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 31.Guan Y, Mizoguchi M, Yoshimoto K, Hata N, Shono T, Suzuki SO, Araki Y, Kuga D, Nakamizo A, Amano T, Ma X, Hayashi K, Sasaki T. MiRNA-196 is upregulated in glioblastoma but not in anaplastic astrocytoma and has prognostic significance. Clin Cancer Res. 2010;16:4289–4297. doi: 10.1158/1078-0432.CCR-10-0207. [DOI] [PubMed] [Google Scholar]
- 32.Molnar J, Engi H, Hohmann J, Molnar P, Deli J, Wesolowska O, Michalak K, Wang Q. Reversal of multidrug resitance by natural substances from plants. Curr Top Med Chem. 2010;10:1757–1768. doi: 10.2174/156802610792928103. [DOI] [PubMed] [Google Scholar]
- 33.Calatozzolo C, Pollo B, Botturi A, Dinapoli L, Carosi M, Salmaggi A, Maschio M. Multidrug resistance proteins expression in glioma patients with epilepsy. J Neurooncol. 2012;110:129–135. doi: 10.1007/s11060-012-0946-9. [DOI] [PubMed] [Google Scholar]
- 34.Tivnan A, Zakaria Z, O’Leary C, Kogel D, Pokorny JL, Sarkaria JN, Prehn JH. Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front Neurosci. 2015;9:218. doi: 10.3389/fnins.2015.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun M, Xu X, Lu Q, Pan Q, Hu X. Schisandrin B: a dual inhibitor of P-glycoprotein and multidrug resistance-associated protein 1. Cancer Lett. 2007;246:300–307. doi: 10.1016/j.canlet.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Chadalapaka G, Jutooru I, Safe S. Celastrol decreases specificity proteins (Sp) and fibroblast growth factor receptor-3 (FGFR3) in bladder cancer cells. Carcinogenesis. 2012;33:886–894. doi: 10.1093/carcin/bgs102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Jiang F, Mu J, Ye X, Si L, Ning S, Li Z, Li Y. Arsenic trioxide attenuates the invasion potential of human liver cancer cells through the demethylation-activated microRNA-491. Toxicol Lett. 2014;227:75–83. doi: 10.1016/j.toxlet.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Afanas'ev I. Mechanisms of superoxide signaling in epigenetic processes: relation to aging and cancer. Aging Dis. 2015;6:216–227. doi: 10.14336/AD.2014.0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015;16:13–19. doi: 10.2174/1389450116666150113121054. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Chakravarty S, Dey M. Phenethylisothiocyanate alters site- and promoter-specific histone tail modifications in cancer cells. PLoS One. 2013;8:e64535. doi: 10.1371/journal.pone.0064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang LG, Chiao JW. Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (review) Int J Oncol. 2010;37:533–539. doi: 10.3892/ijo_00000702. [DOI] [PubMed] [Google Scholar]
- 42.Rose MC, Kostyanovskaya E, Huang RS. Pharmacogenomics of cisplatin sensitivity in non-small cell lung cancer. Genomics Proteomics Bioinformatics. 2014;12:198–209. doi: 10.1016/j.gpb.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei L, Surma M, Gough G, Shi S, Lambert-Cheatham N, Chang J, Shi J. Dissecting the Mechanisms of Doxorubicin and Oxidative Stress-Induced Cytotoxicity: The Involvement of Actin Cytoskeleton and ROCK1. PLoS One. 2015;10:e0131763. doi: 10.1371/journal.pone.0131763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bijman MN, van Nieuw Amerongen GP, Laurens N, van Hinsbergh VW, Boven E. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol Cancer Ther. 2006;5:2348–2357. doi: 10.1158/1535-7163.MCT-06-0242. [DOI] [PubMed] [Google Scholar]