

Abstract
Tamoxifen and aromatase inhibitors (AIs) have shown efficacy in prevention of estrogen receptor-positive (ER+) breast cancer; however, there exists no proven prevention strategy for estrogen receptor-negative (ER-) breast cancer. Up to 40% of ER- breast cancers have human epidermal growth factor receptor 2 overexpression (HER2+), suggesting HER2 signaling might be a good target for chemoprevention for certain ER- breast cancers. Here, we tested the feasibility of the HER2-targeting agent lapatinib in prevention and/or early intervention of an ER-/HER2+ early-stage breast disease model. We found that lapatinib treatment forestalled the progression of atypical ductal hyperplasia (ADH)-like acini to ductal carcinoma in situ (DCIS)-like acini in ER-/HER2+ human mammary epithelial cells (HMECs) in 3D culture. Mechanistically, we found that inhibition of HER2/Akt signaling by lapatinib led to downregulation of GLUT4 and a reduced glucose uptake in HER2-overexpressing cells, resulting in decreased proliferation and increased apoptosis of these cells in 3D culture. Additionally, our data suggest that HER2-driven glycolytic metabolic dysregulation in ER-/HER2+ HMECs might promote early-stage breast disease progression, which can be reversed by lapatinib treatment. Furthermore, low-dose lapatinib treatment, starting at the early stages of mammary grand transformation in the MMTV-neu* mouse model, significantly delayed mammary tumor initiation and progression, extended tumor-free survival, which corresponded to effective inhibition of HER2/Akt signaling and downregulation of GLUT4 in vivo. Taken together, our results indicate that lapatinib, through its inhibition of key signaling pathways and tumor-promoting metabolic events, is a promising agent for the prevention/early intervention of ER-/HER2+ breast cancer progression.
Keywords: Lapatinib, ER-/HER2+ breast cancer, cancer prevention, GLUT4
Introduction
Breast cancer is the second most commonly diagnosed cancer and accounts for the second highest number of cancer deaths among American women, according to the American Cancer Society (www.cancer.org). There is an urgent need for effective prevention and early intervention strategies to ultimately reduce breast cancer morbidity and mortality. Although chemo-prevention trials using anti-estrogenic tamoxifen (Tam) and raloxifene have shown an encouraging 42% decrease in estrogen receptor positive (ER+) breast cancer incidence, there is no effective agent for the prevention of ER negative (ER-) breast cancer, which generally have worse clinical outcomes among different subtypes of breast cancer [1-3].
Breast cancer develops through multiple steps. In a simplified scenario, many (although not all) breast cancer develop from normal epithelium, to ductal hyperplasia, to atypical ductal hyperplasia (ADH or atypia), to ductal carcinoma in situ (DCIS), to invasive (infiltrating) ductal carcinoma (IDC), and ultimately, to metastatic breast cancer [4]. HER2 amplification has been identified in about 50% of high-grade DCIS and 25% of advanced-stage breast cancer [5]. The high frequency of HER2 amplification in DCIS suggests that HER2 overexpression may play an important role in the early stage of mammary carcinogenesis. Additionally, HER2 overexpression (HER2+) can be detected in up to 40% of ER- breast cancer, and several lines of evidence have suggested a positive correlation between HER2 overexpression in benign breast lesion and higher risk of breast cancer incidence [6]. Thus, targeting HER2 signaling could be a chemoprevention strategy for certain ER- breast cancer.
Lapatinib, a FDA approved dual tyrosine kinase inhibitor for both epidermal growth factor receptor (EGFR) and HER2 receptor tyrosine kinases, is efficacious in clinical treatment of breast cancer and may have potential in prevention of HER2-overexpressing breast disease progressing [7,8]. Lapatinib offers several advantages over other HER2-targeting agents: orally available and well tolerated, making it applicable for chemoprevention, especially, at a lower dose than that used for treatment in the clinic.
Dysregulation of cellular metabolism has been described as a hallmark of cancer which facilitates tumor progression [9]. Tumor cells are known to have enhanced aerobic glycolysis and increased glucose uptake compared to normal cells [10,11]. Recently, it has been reported that increased aerobic glycolysis occurs in early-stages of breast cancer and contributes to the transition from normal to neoplastic cells [12]. Increased glucose uptake is required for generation of higher energy and production of precursor molecules needed for cell division [13]. Transportation of glucose across cell membrane is one of a rate limiting step in glycolysis, thus glucose transporters (GLUTs) plays an important role in tumor initiation and progression [13,14]. Overexpression of glucose transporters have been observed in various types of cancer including breast cancer [13,15-17].
In this study, we tested the prevention and/or early intervention effect of lapatinib on ER-/HER2+ overexpressing early stage breast disease progression. We investigated the efficacy of lapatinib for preventing/inhibiting the transition from ADH-like to DCIS-like acini of ER-/HER2+ human mammary epithelial cells (HMECs) in 3D cell culture and in MMTV-neu* transgenic mouse models in vivo [18-20]. Excitingly, we found that lapatinib treatment decreased glucose uptake rate by inhibiting GLUT4 expression, leading to decreased cell proliferation in ER-/HER2+ early stage breast disease model. Together, data from our investigations indicate that lapatinib could intervene ER-/HER2+ early stage breast disease progression toward cancer both in vitro and in vivo.
Materials and methods
Cell line and cell culture
MCF10A cell line was obtained from American Type Culture Collection (ATCC) and cultured in MCF10A medium, and the modified variants were obtained, generated, and cultured as previously described [21,22].
Antibodies and reagents
Rabbit monoclonal antibodies against Erk1/2 (4695), p-Erk1/2 (4370), total AKT (4691), p-AKT (4060), EGFR (4267), p-EGFR (3777), c-caspase-3 (9664) and cyclin D2 (3741) were from Cell Signaling Technology. Rabbit polyclonal antibody against HER2/ErbB2 and p-HER2/ErbB2 (2244) were also from Cell Signaling Technology. Mouse monoclonal anti-β-actin antibody (A5441) was from Sigma. Mouse anti-laminin-5 (MAB1947) and rabbit anti-Glut4 antibodies were from EMD Millipore. Rabbit polyclonal antibodies against Ki67 (ab833) was from Abcam and against GLUT1 (sc7903) was from Santa Cruz. The HRP linked secondary antibodies against mouse (NA931) and rabbit (NA934) were from GE Healthcare. Alexa Fluor 488 anti-rabbit (A11008) and Alexa Fluor 594 anti-mouse (A21201) secondary antibodies and ProLong® Gold antifade reagent with DAPI (P36931) were obtained from Life Technologies. Lapatinib was purchased from GlaxoSmithKline and LC Laboratories. Akt inhibitor MK-2206 (S1078) was purchased from Selleckchem.
Generation of GLUT4 knockdown cells
To knockdown GLUT4 in the MCF10A.B2 cells, two GLUT4 siRNA (MISSION siRNA, Sigma) targeting the GLUT4 mRNA were used in the experiments: siGLUT4.1 and siGLUT4.2. PepMute™ siRNA transfection reagent (SignaGen Laboratories) was used for siRNA transfection and the protocol was followed per manufacturer’s instruction.
Cell proliferation assay
Cells were plated at 1000 cells/well in triplicate in 96-well cell culture plates. Four hours before each time points (24, 48 and 72 hr), 20 μl of 5 mg/ml MTT in PBS (pH 7.5) were added into 200 ul of culture media and incubated at 37°C in the dark. After 4 hours of incubation, media with MTT was removed and 100 μl of DMSO were added into each well. After mixing wells, the absorbance was determined at 570 nm and 620 nm with a microtiter plate reader (BioTek).
Glucose uptake assay
2x105 cells/well were cultured in 6-well plate for 24 hours, in triplicate. Before the assay, cells were washed with cold PBS and cultured in 1 ml DMEM glucose free medium (Gibco) for 3 hours. 1 μCi of 2-deoxy-D-glucose [1,2-3H(N)] (Moravek Biochemicals) was directly added into medium and incubated for 1 hour. Tritium signal (3H), which corresponds to glucose uptake, was detected by liquid scintillation analyzer (PerkinElmer). Cell associated radioactivity was detected by hemocytometer and normalized with total cell number.
Measurement of lactate production
Cells were plated at 1000 cells/well in triplicate in 96-well cell culture plates for 24 hours. Before the assay, the medium was replaced with fresh complete medium and incubated for 1 hour. Lactate production was then measured by using Lactate Colorimetric/Fluorometric Assay Kit (BioVision) and the protocol was followed per manufacturer’s instruction.
Measurement of oxygen consumption rate
Cells were plated at 5000 cells/well in triplicate in 96-well cell culture plates for 24 hours. Prior to analysis, cell culture media was replaced with DMEM XF base medium (Seahorse Biosciences) and real-time oxygen consumption rates were analyzed on an XF96 Extracellular Flux Analyzer (Seahorse Biosciences) under basal conditions.
Western blot analysis
Western blot analysis was performed as previously described [23]. Briefly, total cell lysates were collected with immunoprecipitation (IP) lysis Buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerolphosphate, 1 mM sodium orthovanadate, 1 mM PMSF and protease inhibitor cocktail). Protein concentration was measured using BCA protein assay kit (Pierce) and equal concentration of cell lysates were subjected to electrophoresis using SDS-PAGE. Once the protein was run in gel, it was transferred to PVDF membrane (Bio-Rad). Membranes were blocked with 5% milk in phosphate buffer saline with 0.05% Tween (PBS-T) for 30 min, followed by primary antibody incubation at 4°C for overnight. Membranes were washed thrice with PBST (5 min each), incubated with secondary antibody (5% milk in PBS-T) for 60 min and signal was detected by ECL (Amersham) following the manufacture’s instruction.
Three dimensional (3D) cell culture assay
Three dimensional culture assay was performed by following the protocol as previously described [18,24]. 3D culture assay medium (DMEM/F12 supplemented with 2% donor horse serum, 10 µg/mL insulin, 1 ng/mL cholera toxin, 100 µg/mL hydrocortisone, 50 units/mL penicillin, and 50 µg/mL streptomycin) containing 5 ng/mL epidermal growth factor and 2% growth factor-reduced Matrigel (BD Biosciences) was replaced every 4 days. Cell Recovery Solution (BD BioSciences) were used to remove Matrigel and collect cells at the end point of the experiment.
Immunofluourescence staining on 3D culture cells
Cells in 3D cultures were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 in PBS for 10 minutes. Cells were then washed thrice with PBS/Glycine buffer (130 mM, 7 mM, Na2HPO4, 3.5 mM NaH2PO4, and 100 mM glycine) and incubated with IF buffer (130 mM, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 7.7 mM NaN3, 0.1% bovine serum albumin, 0.2% Triton X-100, and 0.05% Tween 20) for 1 hour. Cells were further incubated with primary antibodies which were suspended in IF buffer for overnight at 4°C. Next day, cells were washed thrice with PBS for 5 minutes each and were incubated with Alexa fluorophore conjugated rabbit or mouse secondary antibodies (Life Technologies), counterstained with 4’, 6-diamidino-2-phenylindole (DAPI) and mounted with anti-fade solution (Life Technologies). Imaging was done using Zeiss LSM 710 laser scanning confocal microscope (Carl Zeiss AG).
Animal experiments
All procedures involving mice and experimental protocols (ACUF ID #: 04-02-05134) were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Texas M.D. Anderson Cancer Center. MMTV-neu NDL 2-5 (referred to as MMTV-neu*) transgenic mouse model, which expresses activated neu under the control of the mouse mammary tumor virus (MMTV) promoter and spontaneously develops estrogen receptor negative (ER-) and HER2+ mammary tumors were used [19]. Female MMTV-neu* mice were treated with either vehicle (0.5% hydroxypropyl methylcellulose with tween-80) or lapatinib dose (30 mg/Kg) by oral gavage once daily for 6 days a week. Tumor sizes were measured twice a week using digital caliper. Tumor-free survival was defined as the time from date of first treatment to the first appearance of a palpable mammary tumor at least 100 mm3 in size. For histological analyses non-serial sections thought-out the MFPs were analyzed. Tumor-free survival was monitored.
RNA extraction, RT-PCR (reverse-transcription-polymerase chain reaction) and quantitative real-time RT-PCR
Total RNA from cells were isolated using Trizole reagent (Invitrogen) following the manufacturer’s instruction. 1 µg of total RNA was reverse transcribed into cDNA using iScriptTM cDNA synthesis kit (Bioad) following the manufacturer’s instruction. An equivalent volume (1 µL) of cDNA was used as template for quantitative real-time PCR with iQTM SYBR Green Supermix (Biorad) and the StepOnePlusTM (Applied Biosystem) instrument according to the manufacturer’s instruction. The sequences of the primers are given in the following table (Table 2). The threshold cycles for specific targets were then normalized to the threshold cycles of GAPDH to calculate the relative fold change difference.
Primers
Table 2.
The sequences of the primers
| Gene | Primers |
|---|---|
| CCND2 | F: 5’-ACCTTCCGCAGTGCTCCTA-3’ |
| R: 5’-CCCAGCCAAGAAACGGTCC-3’ | |
| GLUT4 | F: 5’-CTCAGCAGCGAGTGACTGG-3’ |
| R: 5’-AGCCACGTCTCATTGTAGCTC-3’ | |
| GLUT1 | F: 5’-AAGGTGATCGAGGAGTTCTACA-3’ |
| R: 5’-ATGCCCCCAACAGAAAAGATG-3’ | |
| GAPDH | F: 5’-TGGTATCGTGGAAGGACTCATGAC-3’ |
| R: 5’-ATGCCAGTGAGCTTCCCGTTCAGC-3’ |
cDNA microarray and analysis
Unbiased platform, whole genome 44K long oligo array (Agilent), was applied to gene profiling of MCF10A.B2 cells compared to MCF10A.Vec and parental cells in collaboration with the cDNA microarray core facility in MD Anderson Cancer Center (MDACC). To identify the ErbB2/HER2 regulated genes in 10A cells, R software and limma package were used to identify differentially expressed genes using a 1.5-fold change (FC) threshold and an adjusted p value cutoff at 0.01. Ingenuity Pathway Analysis (IPA) software (http://www.ingenuity.com) was used to perform the functional annotation and pathway analysis of the differentially expressed genes.
Reverse phase protein array and analysis
Reverse phase protein array (RPPA) was performed by RPPA-Functional Proteomics Core facility at MDACC. Sample preparation and data analysis was performed as described previously [23,25].
Statistical analysis
Quantitative results were analyzed either by one-way ANOVA (multiple groups) or t test (2 groups). Differences with P < 0.05 (2-sided) were considered statistically significant. *, P < 0.05, **, P < 0.01 and ***, P < 0.001. For patient samples, Wilcoxon rank-sums test was used. Tumor-free survival analyses were performed using the Kaplan-Meier Wilcoxon test. Error bars represent mean ± standard error of mean (SEM).
Results
HER2 overexpression activated multiple cancer signaling events in ER- human mammary epithelial cells in 3D cell culture
We have stably overexpressed HER2 in the MCF10A non-transformed, ER- HMECs using retroviral expression vector and generated 10A.B2 cells with the empty vector transfected MCF10A cells (10A.Vec) as the control cell line [21]. When cultured under 3D conditions, the 10A.Vec cells formed a well-organized acini structure with hollow-lumen, as formed by MCF10A parental (10A.Pr) cells, which mimics the morphology of human mammary gland. However, 10A.B2 cells had the multi-acinar structures in 3D culture and exhibit two distinct morphologies: 80% of the acini were enlarged, atypical acini with multilayered epithelium and incomplete luminal filling which resemble ADH stage breast lesions; the remaining 20% of the structures were large, multi-lobular, and non-invasive acini with filled lumens sharing structural properties of DCIS, as we previously reported [20,21,24].
Here, we examined the gene expression profile of 10A.Vec and 10A.B2 cells grown in 3D culture by cDNA microarray analysis using Agilent 4X 44K whole human genome microarray platform. Compared to 10A.Vec cells, around 2000 genes were significantly (P < 0.01) differentially expressed in 10A.B2 cells. We performed functional annotation of differentially expressed genes between 10A.Vec and 10A.B2 cells using the Ingenuity Pathway Analysis (IPA), and found that these genes were involved in various canonical pathways, including angiogenesis, molecular mechanism of cancer, cyclins and cell cycle regulations, Wnt/β-catenin signaling, and PI3/AKT signaling etc. (Figure 1A). Compared to 10A.Vec cells, some of the upregulated genes in 10A.B2 cells are involved in cell cycle regulation, as shown by IPA analysis (Table 1). According to microarray data, Cyclin D2 (CCND2) was one of a top candidate gene involved in cell cycle regulation and was upregulated in 10A.B2 cells compared to 10A.vec cells and 10A.Pr cells, which was further validated using qRT-PCR (Figure 1B). Meanwhile, RPPA (reverse phase protein array) of protein lysates from these MCF10A stable lines revealed the signaling events activated by ErbB2/HER2 overexpression in 10A.B2 cells at the protein level compared to 10A.Vec cells (Figure 1C). Consistent with previous report, p-Akt (S473) was dramatically upregulated in 10A.B2 cells compared to 10A.Vec cells and 10A.parental cells without change in total Akt level (Figure 1C and 1D) [7]. Both cDNA microarray and RPPA data demonstrated that HER2-overexpression activated multiple signaling pathways important for early transformation in ER- human mammary epithelial cells.
Figure 1.
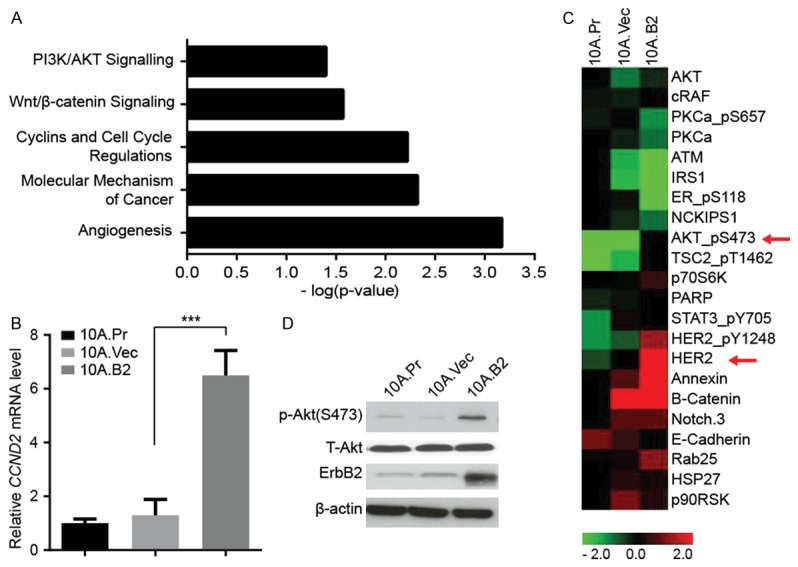
HER2 overexpression activated multiple cancer signaling events in ER- human mammary epithelial cells in 3D cell culture. A. Significantly altered canonical pathways associated with 10A.B2 cells compared with 10A.Vec cells after IPA analysis. B. Quantitative reverse transcription PCR (qRT-PCR) validation of CCND2 expression in mRNA level in indicated cells. ***, P < 0.001. C. Heat map of Reverse Phase Protein Array (RPPA) analysis of 10A.P, 10A.Vec and 10A.B2 cells. D. Western blotting analysis of p-Akt (S473), T-Akt and ErbB2 expression in protein level in indicated cells.
Table 1.
List of genes involved in cell cycle regulation and are upregulated in 10A.B2 cells compared with 10A.Vec cells, as shown by IPA analysis
| ID | Symbol | Description | Fold Change |
|---|---|---|---|
| A_23_P139881 | CCND2 | Cyclin D2 | 34.7 |
| A_24_P284420 | ERBB2 | PI3K/AKT Signaling | 9.79 |
| A_23_P138507 | CDK1 | Cyclin-Dependent Kinase 1 | 7.2 |
| A_24_P227141 | ELF5 | E74-Like Factor 5 | 3.15 |
| A_23_P16523 | GDF15 | Growth Differentiation Factor 5 | 2.86 |
| A_23_P401055 | SOX2 | SRY (Sex Determining Region Y)- Box 2 | 2.72 |
| A_24_P397107 | CDC25A | Cell Division Cycle 25A | 2.54 |
| A_23_P26810 | TP53 | Tumor Protein 53 | 2.38 |
| A_23_P64873 | DCN | Decorin | 2.25 |
| A_24_P81841 | CDKN1B | Cyclin-Dependent Kinase Inhibitor 1B | 2.08 |
Lapatinib inhibit PI3K-Akt pathway and prevented the transition of 10A.B2 cells acini from atypia to DCIS in 3D model
To determine whether inhibiting HER2 signaling in ER-/HER2+ HMEC (10A.B2 cells) could prevent transition of ADH-like to DCIS-like acini in 3D model, we treated 10A.B2 cell acini with the EGFR/HER2 inhibitor lapatinib at various doses (0.1, 0.5, 1, 1.5, and 2 μM, respectively) starting from day 6 of 3D culture and monitored acini morphology daily until day 12. We found that low concentrations of lapatinib (0.5 or 1 μM) reversed the abnormal acini growth of 10A.B2 cells (Figure 2A). Notably, higher doses of lapatinib at 1.5 and 2 μM had a cytotoxic effect on the acinar growths (data not shown). Thus, we treated 10A.B2 cell acini with an effective low dose (0.5 μM) lapatinib starting from day 6 for prevention study, and found the number of DCIS-like acini had largely inhibited after lapatinib treatment compared to vehicle group, and the majority of the DCIS-like acini were converted to normal or ADH-like acini (Figure 2B). We also performed immunofluorescence staining in 10A.Vec and vehicle- or lapatinib-treated 10A.B2 acini with acini polarity marker Laminin V, apoptosis marker cleaved Caspase-3, and proliferation marker Ki-67. We found that lapatinib treatment (0.5 μM) reversed the abnormal acini structure of 10A.B2 to well-organized and singular acini structure similar to that of 10A.Vec cells (Figure 2B). Lapatinib (0.5 μM) treatment also reduced proliferation and increased apoptosis of 10A.B2 cells when compared to vehicle-treated cells (Figure 2B).
Figure 2.
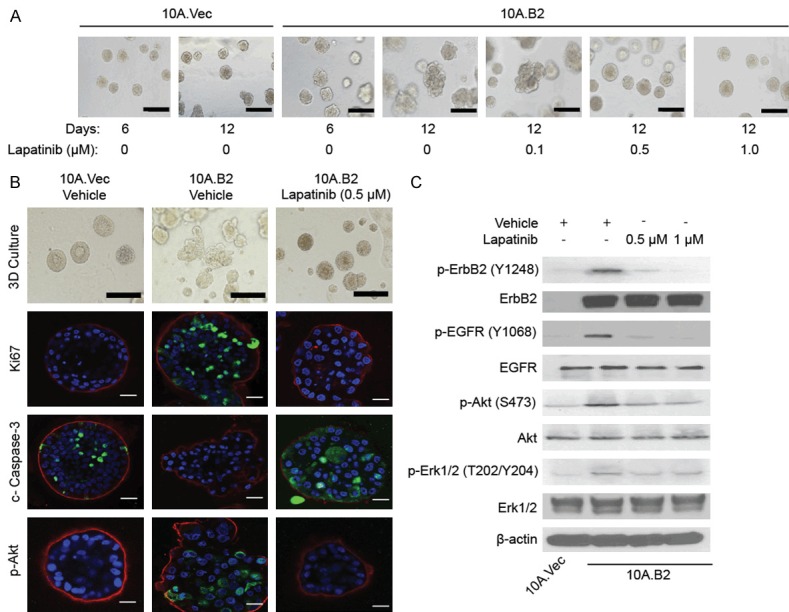
Lapatinib inhibited PI3K-Akt pathway and prevented the transition of 10A.B2 cell acini from atypia to DCIS in 3D model. A. Phase contrast microscopy images of 10A.Vec and 10A.B2 cells treated with Vehicle or Lapatinib at indicated concentrations in 3D culture. Scale bar represents 200 µm. B. Immunofluorescence staining analysis of Ki-67, cleaved caspase-3 and p-Akt in 10A.Vec, 10A.B2 and 10A.B2 cells treated with lapatinib (0.5 µM) for prevention study in 3D culture on day 12. Ki-67, cleaved caspase-3, and p-Akt are shown in green. Laminin V is shown in red. DAPI is shown in blue. Scale bar represents 50 µm. Light microscopy photos (top panel) were used as reference. Scale bar represents 200 µm in light microscopy images. C. Western blotting analysis of p-ErbB2, p-EGFR, p-Akt and p-Erk1/2 expression in protein level in indicated cells with Vehicle or Lapatinib treatment.
Western blot analyses of protein lysates extracted from 3D cultures confirmed that lapatinib treatment inhibited the phosphorylation of both EGFR and HER2 in MCF10A HMEC, as well as the phosphorylation of HER2 downstream signaling molecules, Akt and Erk1/2 (Figure 2C). Taken together, the above data suggested that lapatinib treatment could inhibit HER2/EGFR downstream signaling, and prevent transition of ADH-like to DCIS-like acini in 3D cultured ER-/HER2+ HMEC (10A.B2 cells), which largely dependent on the increased proliferation and impaired apoptosis by HER2 overexpression.
Lapatinib treatment reversed the cancer-promoting metabolic program of 10A.B2 cells
The increased proliferation of 10A.B2 cells requires metabolic reprograming to provide energy and substrates for biosynthesis. HER2 and its downstream signaling molecules, including the PI3K-Akt pathway and Ras pathway, are intimately involved in regulating cancer metabolism and play a critical role in cancer initiation and progression [26,27]. However, the role of HER2 overexpression induced metabolic alterations in the early stages of breast disease remain elusive, and whether lapatinib reverses HER2-induced metabolic alterations is not clear. To this end, we first performed glucose uptake assay and found that 10A.B2 cells consumed significantly more glucose than control 10A.Vec cells, which could be significantly inhibited by lapatinib treatment (1 μM for 4 hrs.), suggesting that HER2 overexpression might increase glucose uptake via its kinase activity (Figure 3A). Secondly, we measured the oxygen consumption rate and found that 10A.B2 cells consumed less oxygen than 10A.Vec cells, while lapatinib treatment (1 µM, 4 hr) did not have significant effect on the oxygen consumption in both 10A.Vec and 10A.B2 cells (Figure 3B). Thirdly, we compared lactate production between 10A.Vec and 10A.B2 cells and found that 10A.B2 cells produced more lactate than 10A.Vec cells, and lapatinib treatment (1 µM, 4 hr) significantly inhibited the lactate production in 10A.B2 cells (Figure 3C). Together, these data indicated that HER2 overexpression/activation in the MCF10A ER- HMECs induce glycolytic metabolic alterations (i.e. increased glucose uptake and lactate production), which may contribute to the increased proliferation of ER- HMECs, which could facilitate early breast disease progression. On the other hand, lapatinib treatment could reverse the HER2 kinase-driven metabolic reprogramming in 10A.B2 cells.
Figure 3.

Lapatinib treatment reversed the cancer-promoting metabolic program of 10A.B2 cells. A. Glucose uptake assay of 10A.Vec and 10A.B2 cells treated with Vehicle or Lapatinib (1 µM, 4 hr). B. Oxygen consumption assay of 10A.Vec and 10A.B2 cells treated with Vehicle or Lapatinib (1 µM, 4 hr). C. Lactate production assay of 10A.Vec and 10A.B2 cells treated with Vehicle or Lapatinib (1 µM, 4 hr). n.s., not significant, *, P < 0.05, **, P < 0.01 and ***, P < 0.001.
Lapatinib treatment inhibited HER2 induced GLUT4 expression in 10A.B2 cells
Among the reprogramed metabolic parameters measured, lapatinib is most effective in inhibition of the elevated glucose uptake in 10A.B2 cells (Figure 3). To dissect the underlying mechanism of lapatinib-induced inhibition of the elevated glucose uptake in 10A.B2 cells, we first examined the expression of major glucose transporters in 10A.B2 cells. We found that HER2 overexpression increased GLUT1 and GLUT4 gene expression at both mRNA and protein levels (Figure 4A and 4B). Notably, GLUT4 protein upregulation in 10A.B2 cells is more dramatic than GLUT1 protein upregulation; and lapatinib treatment (0.5 μM and 1 μM) of 10A.B2 cells inhibited GLUT4 expression more dramaticaly than GLUT1 expression (Figure 4A and 4B).
Figure 4.
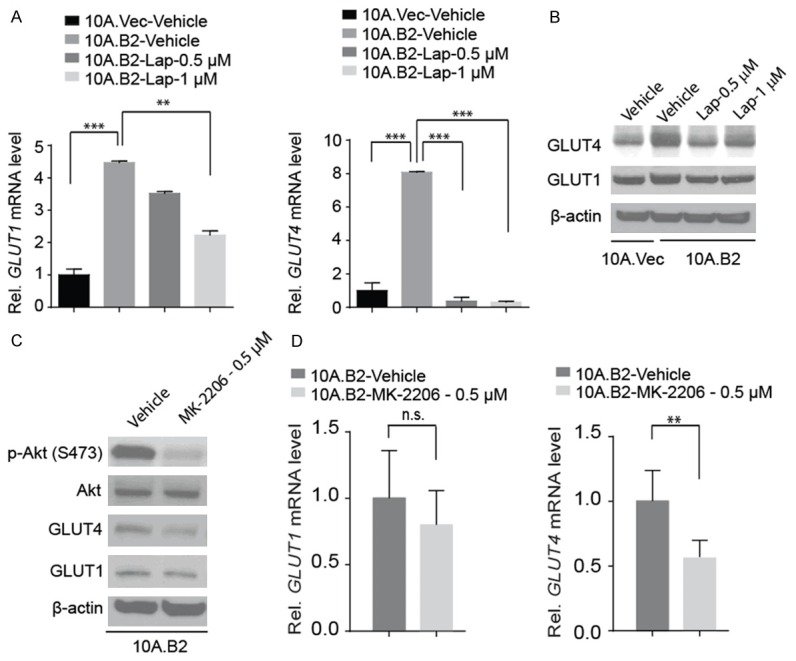
Lapatinib treatment inhibited HER2 induced GLUT4 expression in 10A.B2 cells. A. qRT-PCR analysis of GLUT1 (left panel) and GLUT4 (right panel) mRNA expression in indicated cell lines with Vehicle or Lapatinib treatment. **, P < 0.01 and ***, P < 0.001. B. Western blotting analysis of GLUT1 and GLUT4 expression in protein level in indicated cell lines with Vehicle or Lapatinib treatment. C. Western blot analysis of p-Akt, GLUT4 and GLUT1 in 10A.B2 cells with Vehicle or MK-2206 treatment. D. qRT-PCR analysis of GLUT1 (left panel) and GLUT4 (right panel) mRNA expression in indicated cell lines with Vehicle or MK-2206 treatment. n.s., not significant, and **, P < 0.01.
Akt-mediated the expression of GLUT genes has been previously described [28-30]. Therefore, we treated 10A.B2 cells with Akt inhibitor MK-2206 (0.5 µM) (Figure 4C), and found that it could inhibit HER2-induced GLUT4 expression at both protein (Figure 4C) and mRNA levels (Figure 4D) but MK-2206 treatment did not significantly change GLUT1 expression (Figure 4C and 4D). These data indicated that lapatinib treatment inhibited GLUT4 expression in 10A.B2 cells through inhibition of Akt activation.
GLUT4 is a key executor of HER2-driven ADH-like acini progression to DCIS-like acini
To test whether GLUT4 plays a critical role in HER2-driven ADH-like acini progression to DCIS-like acini of HMEC cells, we knocked down GLUT4 in 10A.B2 cells with two distinct siRNAs targeting GLUT4 (10A.B2 siGLUT4). GLUT4 knockdown was validated in both mRNA level and protein level (Figure 5A and 5B), and GLUT4 knockdown resulted in decreased proliferation rate of 10A.B2 cells in 2D culture (Figure 5C). To investigate whether GLUT4 knockdown could intervene ADH-like acini progression to DCIS-like acini of 10A.B2 cells, we grew 10A.B2 siGLUT4 cells in 3D culture and compared their acini morphology with that of the control cells (10A.B2 Ctrl). We found that the number of DCIS-like acini had largely decreased after GLUT4 knockdown, and majority of the acini were normal or ADH-like acini (Figure 5D). GLUT4 knockdown inhibited the abnormal multi-acini structure of 10A.B2 to well-organized and complete single-acinar structure as shown by Laminin V staining (Figure 5D). Moreover, staining of Ki-67 and cleaved caspase-3 showed that GLUT4 knockdown decreased 10A.B2 cell proliferation but had no significant effect on 10A.B2 apoptosis in 3D culture compared to that of control cells (Figure 5D). These data indicated that GLUT4 plays a critical role in HER2-driven ADH-like acini progression to DCIS-like acini of 10A.B2 cells, and thus downregulation of GLUT4 by lapatinib plays a key role in preventing ADH-like acini progression to DCIS-like acini of 10A.B2 cells by lapatinib.
Figure 5.
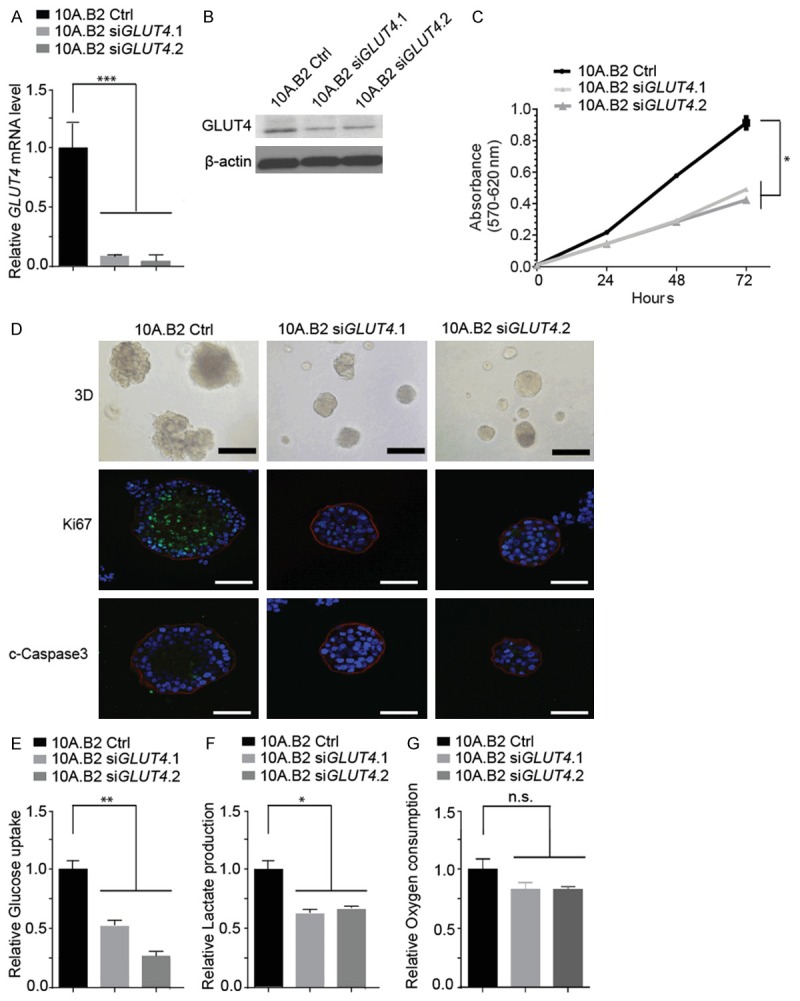
GLUT4 is a key executor of HER2-driven ADH-like acini progression to DCIS-like acini. A. qRT-PCR analysis of GLUT4 mRNA expression in 10A.B2 cells transfected with control siRNA (Ctrl) or GLUT4 siRNA (siGLUT4) four days after transfection. B. Western blotting analysis of GLUT4 protein level in indicated cells as in A. C. Proliferation assay for indicated cells as in A measured by MTT assay at 0, 24, 48 and 72 hours. D. Immunofluorescence staining analysis (on day 12 of 3D culture) of Ki-67 and cleaved caspase-3 in 10A.B2 cells transfected with siCtrl or siGLUT4 on day 0. Ki-67 and cleaved caspase-3 are shown in green. Laminin V is shown in red. DAPI is shown in blue. Scale bar represents 50 µm. Light microscopy photos (top panel) are used as reference. Scale bar represents 100 µm for light microscopy images. E. Glucose uptake assay of indicated cell lines. F. Lactate production assay of indicated cell lines. G. Oxygen consumption assay of indicated cell lines. n.s., not significant, *, P < 0.05, **, P < 0.01 and ***, P < 0.001.
Next, we examined the effect of GLUT4 knockdown on metabolic functions of ER-/HER2+ 10A.B2 cells. Indeed, GLUT4 knockdown reduced the glucose uptake in 10A.B2 siGLUT4 cells compared to 10A.B2 Ctrl cells (Figure 5E). Meanwhile, 10A.B2 siGLUT4 cells had reduced lactate production compared to the 10A.B2 Ctrl cells (Figure 5F). On the other hand, oxygen consumptions were not significantly different between the 10A.B2 siGLUT4 cells and 10A.B2 Ctrl cells (Figure 5G). Together, these data suggested that GLUT4-induced glucose uptake could play an important role in ER-/HER2+ 10A.B2 cells’ metabolic dysregulation that plays critical role in early transformation partly by increasing cell proliferation.
Lapatinib prevented early stage breast lesion progression in MMTV-neu* mouse model
Next, we investigated whether lapatinib may prevent and intervene ER-/HER2+ mammary tumor initiation and progression by inhibiting proliferation via downregulation of GLUT4, in addition to targeting HER2 signaling, in vivo using MMTV-neu* transgenic mouse model [19]. We previously reported that, MMTV-neu* mice developed hyperplastic lesions between 8-11 weeks of age, mammary intraepithelial neoplasia (MIN) between 11-19 weeks of age, and invasive ductal carcinoma (IDC) lesions between 19 to 26 weeks of age [24].
To determine the effect of lapatinib on the initiation and progression of ER- mammary tumors in vivo, we began treating female MMTV-neu* mice with low dose of lapatinib (30 mg/kg) from 10 weeks of age (mostly having hyperplastic lesions) and continued the treatment after mice developed mammary tumor and till they were euthanized between 21 and 29 weeks of age (end point). The lowest effective dose of 30 mg/kg/day for six days per week was adopted for the prevention study and vehicle was used as controls. All vehicle-treated mice developed mammary tumors by 85 days into the treatment, while three out of nine mice treated with lapatinib remained free of palpable tumor until 105 days (p=0.0042) (Figure 6A). We then performed IHC staining to examine whether lapatinib treatment inhibited proliferation and induced apoptosis, downregulated GLUT4, in addition to targeting HER2 signaling in mammary tumors of MMTV-neu* mice. Indeed, the level of p-ErbB2, Ki-67, p-Akt, Cyclin D2 and GLUT4 were significantly inhibited in lapatinib treatment group compared to vehicle treated group (Figure 6B and 6C), whereas apoptosis (cleaved caspase-3 staining) in mammary tumor of MMTV-neu* mice was increased by lapatinib treatment (Figure 6B and 6C). All these data suggested that lapatinib treatment inhibited HER2 signaling and downregulated GLUT4, which contribute to decreased cell proliferation and increased cell death, and consequently, a significant delay of mammary tumor progression in the MMTV-neu* ER-/HER2+ mouse model. Therefore, low doses of lapatinib could be adapted for the prevention of ER-negative and HER2 overexpressing breast cancer in the clinic.
Figure 6.
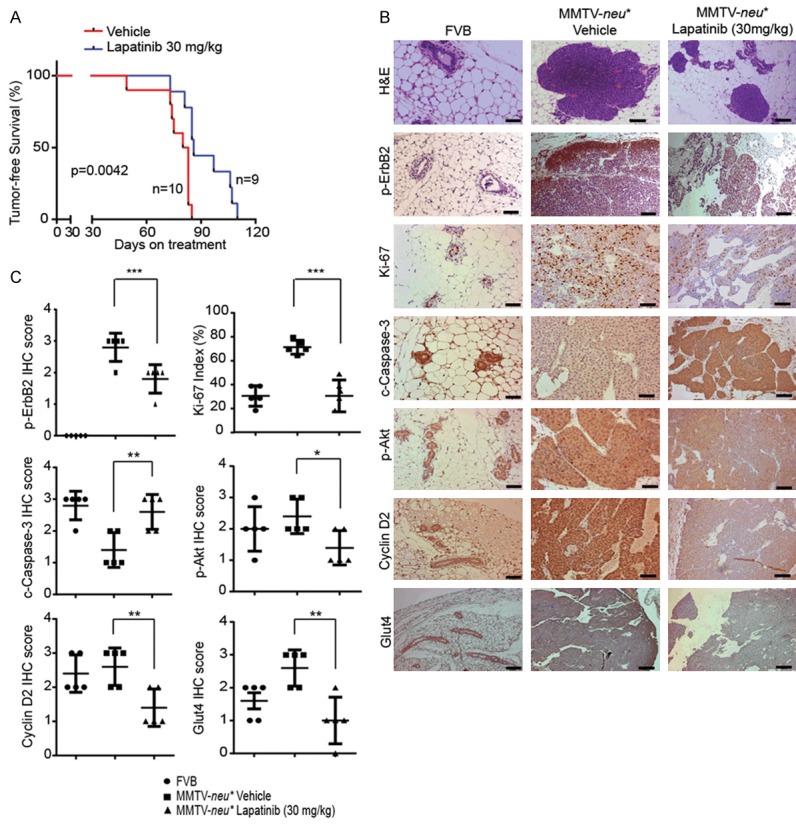
Lapatinib prevented early stage breast lesion progression in MMTV-neu* mouse model. A. Kaplan-Meier Survival analysis of MMTV-neu* mice treated with Vehicle and 30 mg/kg Lapatinib for prevention. B. Representative H&E and IHC images of pErbB2 (Y1248), Ki-67, cleaved caspase-3, p-Akt (S473), Cyclin D2 and GLUT4 staining in the mammary gland from FVB control mice and MMTV-neu* mice treated with Vehicle or 30 mg/kg lapatinib. H&E images (for both Vehicle and Lapatinib treatment) were from samples collected at 21 weeks and IHC staining was done on samples collected at end point. Scale bar represents 50 µm. C. Quantitative analysis of IHC staining intensity scores of various markers in vehicle and lapatinib treated MMTV-neu* mice along with WT FVB mice as shown in B. *, P < 0.05, **, P < 0.01 and ***, P < 0.001.
Discussion
Currently, Tamoxifen is used for preventing ER+ breast disease progression to breast cancer. ER expression profiling is used as a biomarker to predict the response to Tamoxifen and prognosis of ER+ breast cancer patients. For ER- breast cancer patients, there is still a lack of effective prevention agents in the clinical setting [31-36]. The high frequency of HER2 amplification in ~ 40% of ER- breast cancers suggests that HER2 amplification in early stage of breast diseases may play an important role in early stage breast cancer development in these patients. However, the HER2-activated signaling networks in early stage breast cancer progression remain elusive. Our paired 10A.Vec and 10A.B2 cells in 3D cell culture provide a good in vitro model for the rapid identification of HER2-activated signaling networks in early stage breast cancer which may also serve as biomarkers to assess the cancer risk. We have mapped the global signaling events activated by HER2 overexpression using RPPA array and performed profiling on HER2 overexpression induced transcriptome by cDNA microarray in this system (Figure 1). These unbiased approaches revealed HER2-mediated activation of multiple signaling pathways that may contribute to abnormal acini growth in 3D culture. The findings also provide insights on developing strategies to effectively target HER2 for prevention of ER-/HER2+ early stage breast disease progression to cancer.
Among HER2-targeting agents, lapatinib (Tykerb, GlaxoSmithKline) is a small molecule that enters the cell and blocks the kinase function of HER2 and EGFR by blocking the ATP binding site on their tyrosine kinase domains. FDA approved lapatinib on March 13, 2007 for use in patients with advanced metastatic breast cancer in conjunction with capecitabine (XelodaTM) (http://www.cancer.gov/about-cancer/treatment/drugs/fda-lapatinib#Anchor-HER6789). Specifically, it is used for the treatment of advanced HER2+ cancers that has failed to respond to other frontline regimens [37,38]. Here, we tested the effect of lapatinib to intervene ADH-like acini progression to DCIS-like acini of 10A.B2 cells in 3D culture and showed that lapatinib treatment inhibited the abnormal multi-acini structure of 10A.B2 to well-organized and complete single-acinar structure (Figure 2B). Cancer cells rely on high glycolytic flux in order to generate sufficient energy and biosynthesis substrates for supporting proliferation, and indeed, we found that 10A.B2 cells have a high glycolytic activities, especially high glucose uptake, that can be effectively inhibited by lapatinib. We then examined the expression of GLUTs which are the high affinity isoforms for glucose transport in our model system [13], and found that 10A.B2 cells expressed high levels of GLUT1 and GLUT4, which are significantly downregulated by lapatinib. Taking advantage that 10A.B2 cells in 3D culture are amendable for rapid dissection of molecular mechanisms, we discovered that lapatinib treatment downregulated GLUT4 expression in 10A.B2 cells via inhibition of HER2/Akt signaling and downregulation of GLUT4 expression is effective in preventing ADH-like acini progression to DCIS-like acini of 10A.B2 cells in 3D culture (Figures 3, 4 and 5). The findings indicate that metabolic dysregulation in ER-/HER2+ HMECs plays a crucial role in early stage breast disease progression and inhibiting tumor-prone metabolic events enables lapatinib prevention function. We further validated these findings by treating the MMTV-neu* mouse model with low dose of lapatinib at early stage before developing ER-/HER2+ mammary tumors, which significantly delayed tumor initiation and progression corresponding to significant inhibitions of HER2/Akt phosphorylation and GLUT4 expression (Figure 6). Understanding the mechanistic link between cellular metabolism and cancer cell growth control have provided additional treatments options for human cancer [11] . The same principle may also apply to cancer prevention. Our above new findings suggest that targeting tumor-prone metabolic events by modulating cellular metabolism, e.g., calorie restrictions, especially when combined with low dose of lapatinib, may have better effect on prevention of ER-/HER2+ breast cancers.
Lapatinib treatment decreased GLUT4 expression much more dramatically than GLUT1 expression. Thus, GLUT4 is a critical mediator of lapatinib-induced reversal of HER2-driven metabolic dysregulation. GLUT4 gene expression can be regulated by multiple mechanisms [39]. It was reported that insulin-dependent GLUT4 regulation played a central role in glucose homeostasis as insulin activated PI3K and Akt, resulting in GLUT4 translocation to the membrane in muscle cells and adipose tissue [39,40]. In addition to Akt, PKCλ/ζ [41], AMPK [42], and CaMK2 [43] have been found to also promote GLUT4 translocation to the cell membrane. Recently, various studies showed that breast cancer cells expressed high levels of GLUT4, and knocking down GLUT4 in breast cancer cell lines impaired glucose uptake and reduced lactate production, which was also observed in our early breast disease model [44,45] (Figure 3A and 3C). Therefore, blocking GLUT4 or its upstream activating signals may empower cancer treatment and prevention.
In summary, in this study, we demonstrated that overexpression of HER2 activates multiple signaling events in ER-/HER2+ 10A.B2 HMECs of an early stage breast disease model. We found that lapatinib effectively intervened ADH-like acini progression to DCIS-like acini of 10A.B2 cells in 3D culture by, at least partly, decreasing glucose uptake via downregulation of GLUT4 expression after inhibiting HER2/Akt signaling. Furthermore, low dose lapatinib treatment starting from early stages of mammary gland transformation delayed tumor initiation and progression which are associated with inhibition of HER2/Akt signaling and downregulation of GLUT4 expression. The data presented in this study indicated that lapatinib is a promising agent for the prevention/intervention of ER-/HER2+ breast cancer by inhibiting key signal events in early stage breast disease and by reversing cancer-promoting metabolic events, which warrant further clinical follow-ups in the future.
Acknowledgements
This work was supported by Susan G. Komen Breast Cancer Foundation promise grant KG091020 (D.Y.), NIH grants P30-CA 16672 (MDACC), PO1-CA099031 project 4 (D.Y.), RO1-CA112567-06 (D.Y.), RO1-CA184836 (D.Y.), T34 GM008718 (J.B.), DOD Pre-doctoral Fellowship W81XWH-10-1-0238 (J.X.), and China Medical University Research Fund (D.Y.). Dr. D. Yu is the Hubert L. & Olive Stringer Distinguished Chair in Basic Science at MD Anderson Cancer Center.
Disclosure of conflict of interest
None.
References
- 1.Jordan VC. Tamoxifen for breast cancer prevention. Proc Soc Exp Biol Med. 1995;208:144–149. doi: 10.3181/00379727-208-43846c. [DOI] [PubMed] [Google Scholar]
- 2.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 3.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER Jr, Wade JL 3rd, Robidoux A, Margolese RG, James J, Lippman SM, Runowicz CD, Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan VC, Wolmark N. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295:2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 4.Lakhani SR. The transition from hyperplasia to invasive carcinoma of the breast. J Pathol. 1999;187:272–278. doi: 10.1002/(SICI)1096-9896(199902)187:3<272::AID-PATH265>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 5.Latta EK, Tjan S, Parkes RK, O’Malley FP. The role of HER2/neu overexpression/amplification in the progression of ductal carcinoma in situ to invasive carcinoma of the breast. Mod Pathol. 2002;15:1318–1325. doi: 10.1097/01.MP.0000038462.62634.B1. [DOI] [PubMed] [Google Scholar]
- 6.Stark A, Hulka BS, Joens S, Novotny D, Thor AD, Wold LE, Schell MJ, Melton LJ 3rd, Liu ET, Conway K. HER-2/neu amplification in benign breast disease and the risk of subsequent breast cancer. J. Clin. Oncol. 2000;18:267–274. doi: 10.1200/JCO.2000.18.2.267. [DOI] [PubMed] [Google Scholar]
- 7.Strecker TE, Shen Q, Zhang Y, Hill JL, Li Y, Wang C, Kim HT, Gilmer TM, Sexton KR, Hilsenbeck SG, Osborne CK, Brown PH. Effect of lapatinib on the development of estrogen receptor-negative mammary tumors in mice. J Natl Cancer Inst. 2009;101:107–113. doi: 10.1093/jnci/djn436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 11.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dupuy F, Griss T, Blagih J, Bridon G, Avizonis D, Ling C, Dong Z, Siwak DR, Annis MG, Mills GB, Muller WJ, Siegel PM, Jones RG. LKB1 is a central regulator of tumor initiation and pro-growth metabolism in ErbB2-mediated breast cancer. Cancer Metab. 2013;1:18. doi: 10.1186/2049-3002-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202:654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 14.Hatanaka M. Transport of sugars in tumor cell membranes. Biochim Biophys Acta. 1974;355:77–104. doi: 10.1016/0304-419x(74)90008-0. [DOI] [PubMed] [Google Scholar]
- 15.Brown RS, Wahl RL. Overexpression of Glut-1 glucose transporter in human breast cancer. An immunohistochemical study. Cancer. 1993;72:2979–2985. doi: 10.1002/1097-0142(19931115)72:10<2979::aid-cncr2820721020>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 16.Younes M, Lechago LV, Somoano JR, Mosharaf M, Lechago J. Immunohistochemical detection of Glut3 in human tumors and normal tissues. Anticancer Res. 1997;17:2747–2750. [PubMed] [Google Scholar]
- 17.Medina RA, Owen GI. Glucose transporters: expression, regulation and cancer. Biol Res. 2002;35:9–26. doi: 10.4067/s0716-97602002000100004. [DOI] [PubMed] [Google Scholar]
- 18.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 19.Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999;18:2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu J, Guo H, Treekitkarnmongkol W, Li P, Zhang J, Shi B, Ling C, Zhou X, Chen T, Chiao PJ, Feng X, Seewaldt VL, Muller WJ, Sahin A, Hung MC, Yu D. 14-3-3zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer Cell. 2009;16:195–207. doi: 10.1016/j.ccr.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li SH, Hawthorne VS, Neal CL, Sanghera S, Xu J, Yang J, Guo H, Steeg PS, Yu D. Upregulation of neutrophil gelatinase-associated lipocalin by ErbB2 through nuclear factor-kappaB activation. Cancer Res. 2009;69:9163–9168. doi: 10.1158/0008-5472.CAN-09-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, Acharya S, Sahin O, Zhang Q, Saito Y, Yao J, Wang H, Li P, Zhang L, Lowery FJ, Kuo WL, Xiao Y, Ensor J, Sahin AA, Zhang XH, Hung MC, Zhang JD, Yu D. 14-3-3zeta turns TGF-beta’s function from tumor suppressor to metastasis promoter in breast cancer by contextual changes of Smad partners from p53 to Gli2. Cancer Cell. 2015;27:177–192. doi: 10.1016/j.ccell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain S, Wang X, Chang CC, Ibarra-Drendall C, Wang H, Zhang Q, Brady SW, Li P, Zhao H, Dobbs J, Kyrish M, Tkaczyk TS, Ambrose A, Sistrunk C, Arun BK, Richards-Kortum R, Jia W, Seewaldt VL, Yu D. Src Inhibition Blocks c-Myc Translation and Glucose Metabolism to Prevent the Development of Breast Cancer. Cancer Res. 2015;75:4863–4875. doi: 10.1158/0008-5472.CAN-14-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Huang WC, Zhang L, Zhang C, Lowery FJ, Ding Z, Guo H, Wang H, Huang S, Sahin AA, Aldape KD, Steeg PS, Yu D. SRC family kinases as novel therapeutic targets to treat breast cancer brain metastases. Cancer Res. 2013;73:5764–5774. doi: 10.1158/0008-5472.CAN-12-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 27.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 28.Hernandez R, Teruel T, Lorenzo M. Akt mediates insulin induction of glucose uptake and up-regulation of GLUT4 gene expression in brown adipocytes. FEBS Lett. 2001;494:225–231. doi: 10.1016/s0014-5793(01)02353-5. [DOI] [PubMed] [Google Scholar]
- 29.Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- 30.Hajduch E, Alessi DR, Hemmings BA, Hundal HS. Constitutive activation of protein kinase B alpha by membrane targeting promotes glucose and system A amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes. 1998;47:1006–1013. doi: 10.2337/diabetes.47.7.1006. [DOI] [PubMed] [Google Scholar]
- 31.Anderson E, Clarke RB. Steroid receptors and cell cycle in normal mammary epithelium. J Mammary Gland Biol Neoplasia. 2004;9:3–13. doi: 10.1023/B:JOMG.0000023584.01750.16. [DOI] [PubMed] [Google Scholar]
- 32.Anderson E, Clarke RB, Howell A. Estrogen responsiveness and control of normal human breast proliferation. J Mammary Gland Biol Neoplasia. 1998;3:23–35. doi: 10.1023/a:1018718117113. [DOI] [PubMed] [Google Scholar]
- 33.Clarke RB, Anderson E, Howell A. Steroid receptors in human breast cancer. Trends Endocrinol Metab. 2004;15:316–323. doi: 10.1016/j.tem.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 34.Lee S, Mohsin SK, Mao S, Hilsenbeck SG, Medina D, Allred DC. Hormones, receptors, and growth in hyperplastic enlarged lobular units: early potential precursors of breast cancer. Breast Cancer Res. 2006;8:R6. doi: 10.1186/bcr1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen OW, Hoyer PE, van Deurs B. Frequency and distribution of estrogen receptor-positive cells in normal, nonlactating human breast tissue. Cancer Res. 1987;47:5748–5751. [PubMed] [Google Scholar]
- 36.Shoker BS, Jarvis C, Sibson DR, Walker C, Sloane JP. Oestrogen receptor expression in the normal and pre-cancerous breast. J Pathol. 1999;188:237–244. doi: 10.1002/(SICI)1096-9896(199907)188:3<237::AID-PATH343>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 37.Moy B, Kirkpatrick P, Kar S, Goss P. Lapatinib. Nat Rev Drug Discov. 2007;6:431–432. doi: 10.1038/nrd2332. [DOI] [PubMed] [Google Scholar]
- 38.Yu D, Hung MC. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene. 2000;19:6115–6121. doi: 10.1038/sj.onc.1203972. [DOI] [PubMed] [Google Scholar]
- 39.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR, Klip A. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol. 1999;19:4008–4018. doi: 10.1128/mcb.19.6.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farese RV, Sajan MP, Standaert ML. Insulin-sensitive protein kinases (atypical protein kinase C and protein kinase B/Akt): actions and defects in obesity and type II diabetes. Exp Biol Med (Maywood) 2005;230:593–605. doi: 10.1177/153537020523000901. [DOI] [PubMed] [Google Scholar]
- 42.Richter EA, Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev. 2013;93:993–1017. doi: 10.1152/physrev.00038.2012. [DOI] [PubMed] [Google Scholar]
- 43.Ojuka EO, Goyaram V, Smith JA. The role of CaMKII in regulating GLUT4 expression in skeletal muscle. Am J Physiol Endocrinol Metab. 2012;303:E322–331. doi: 10.1152/ajpendo.00091.2012. [DOI] [PubMed] [Google Scholar]
- 44.Garrido P, Osorio FG, Moran J, Cabello E, Alonso A, Freije JM, Gonzalez C. Loss of GLUT4 induces metabolic reprogramming and impairs viability of breast cancer cells. J Cell Physiol. 2015;230:191–198. doi: 10.1002/jcp.24698. [DOI] [PubMed] [Google Scholar]
- 45.Medina RA, Meneses AM, Vera JC, Guzman C, Nualart F, Astuya A, Garcia MA, Kato S, Carvajal A, Pinto M, Owen GI. Estrogen and progesterone up-regulate glucose transporter expression in ZR-75-1 human breast cancer cells. Endocrinology. 2003;144:4527–4535. doi: 10.1210/en.2003-0294. [DOI] [PubMed] [Google Scholar]