

Abstract
Insulin resistance (IR) is an important mechanism of pathogenesis of endometrial cancer (EC) and explains the pathogenic mechanism of high risk factors including Obesity BMI (body mass index), Type 2 Diabetes Mellitus, PCOS and so on. Relieving IR or inhibiting the function of insulin could be one of the potential therapeutic strategies for EC, which is a PI3K-driven disease. PI3K/Akt are the central mediators for insulin/IGF1 signaling, however, the involvement of HIPPO pathway co-activators, YAP and TAZ, in insulin resistance remains to be elucidated. In the present study, we analyzed the clinical and biological data of EC patients from TCGA and observed a correlation between insulin resistance and EC. By comparing the expression level of IRS1/2 in obese vs non-obese patients, we found that the most important insulin resistance relative (IRR) genes are the contributing factors to IR. Interestingly, IRS1/2 was correlated positively with YAP/TAZ in EC patients. Knockdown of YAP/TAZ by specific siRNA inhibited the phosphorylation of IRS1 while increased the phosphorylation of IGFR1, the inhibitor of insulin signaling. Treating EC with siYAP/TAZ, YAP inhibitor Verteporfin or metformin alone only partially inhibited the function of insulin and IGF1. However, combination of siYAP/TAZ with metformin could completely inhibit the effects of insulin. Thus, our study demonstrated a novel function of YAP and TAZ in the insulin resistance via IRS1/2 in endometrial cancer. Our study also provided the rationale for the potential therapeutic treatment of EC with the combination of inhibiting YAP/TAZ and metformin.
Keywords: YAP/TAZ, insulin signaling, IRS1/2, endometrial cancer
Introduction
Endometrial cancer (EC) is the most common female genital malignant tumor in developed countries. In United States, from 2006 to 2010, both the 5-year rate changes of incidence and mortality of EC including all races ranked the 4th site among cancers with rising average annual percentage changes while ovarian cancer and cervical cancer had falling average annual percentage changes. EC incidence is going to surpass colorectal cancer to take the 3rd leading cancer site among U.S. women by 2030 [1]. This indicates it is urgent to explore the etiological and developmental mechanisms of EC as well as cost-effective therapeutic strategies.
The growing incidence of EC is closely related to multiple high risk factors. Obesity, which is defined as a body mass index (BMI ≥ 30 (weight (kg)/height (m)2)), is clearly linked with an increased risk of several types of cancer including EC [2-6]. Waist circumference and waist: hip ratio was also strongly correlated with EC even after adjustment for BMI [7]. A direct association between Type 2 Diabetes Mellitus and a higher risk of cancer mortality independent of obesity has been revealed [8-10]. Third, epidemiological studies have illustrated that postmenopausal women are at an increased risk of cancers from exogenous estrogen replacement without progesterone [11,12], while polycystic ovarian syndrome (PCOS) in premenopausal women is very closely related to increased EC risk [13,14]. Women with PCOS were predisposed to develop EC compared to women without PCOS [15]. Other factors include leptin and adiponectin [16-18], the A:L (Adiponectin: Leptin) ratio [16,18], oral contraceptive drug use and so on. Among these high risk factors, obesity, PCOS, diabetes mellitus and the A:L ratio are all closely associated with insulin resistance (IR), which has been regarded as one of important mechanisms that contribute to the occurrence of Endometrial cancer [19].
Endometrial cancer is a PI3K-driven disease and PI3K)/Akt pathway was aberrant in more than 90% of ECs [20-23]. In our previous work, we have investigated the Yes-associated protein (YAP) and its homolog, the transcriptional co-activator TAZ (aka WWTR1), which are two key downstream effectors in the cascade of Hippo signaling and has emerged as a major contributor to cancer pathophysiology [24-26], regulated the PI3K/Akt signaling. PI3K/Akt signaling is the main pathway cascade of insulin/IGF1 signaling. So, we tried to investigate the possible interaction between YAP/TAZ and insulin resistance in EC. In this study, firstly, we investigated the characters of insulin resistance in EC patients of TCGA and the close relationship of IRS1/2 with YAP/TAZ in patients, and then we primarily investigated the regulation of IRS1 by YAP/TAZ. Finally, we tested the effects of combining siYAP/TAZ with metformin in EC cell proliferation.
Methods
Reagents and materials
Antibodies for WB were obtained from Cell Signaling Technology; IGF1 (25 ng/ml) was from Upstate Biotechnology (Lake Placid, N.Y.) and insulin (10 ng/ml) was from Gibco-BRL. ON-TARGETplus SMARTpool siRNA libraries were obtained from Dharmacon for human YAP1 (L-012200-00) and human WWTR1 (L-016083-00). Lipofectamine® RNAiMAX Transfection Reagent was from Invitrogen (13778150). Verteporfin were ordered from Sigma. PrestoBlue® Cell Viability Reagent was from Invitrogen (A-13262).
TCGA data of patients
All clinical data, reverse-phase protein arrays (RPPA) data and mRNA (RNAseq) data of patients with endometrial cancer were downloaded from the TCGA Data portal (http://tcga-data.nci.nih.gov/tcga/findArchives.htm). The relative statistic analyses were performed by R statistical software and integrity pathway analysis (IPA) (http://www.ingenuity.com).
Cell culture and transient transfection
KLE and EFE184 were obtained from the Characterized Cell Line Core Facility of the MD Anderson Cancer Center and routinely propagated as a monolayer culture with, respectively, DMEM F12, RPMI1640, all supplemented with 5% heat-inactivated fetal bovine serum (FBS). Cells were cultured at 37°C in a humidified incubator containing 5% CO2. For siRNA knock down studies, cells were reverse transfected with siRNA with RNAiMAX reagent (Invitrogen) using 4-6 × 103 cells/well in a 96-well plate or 2 × 104 cells/well of a 6-well plate for the KLE and EFE184 cell lines.
Sulforhodamine B assay
Cells were seeded in 96-well plates and then treated as described. The inhibition effects on cell growth were determined using sulforhodamine B (SRB) as described previously [27-29]. Adherent cells were fixed with cold (4°C) 10% trichloroacetic acid (TCA) in a 96-well microplate for 1 h at 4°C and then washed with deionized water and air dried at room temperature. Next, 0.4% (w/v) SRB in 1% acetic acid solution was added into each well and incubated at room temperature for 30 min; then the cells were washed with 1% acetic acid and air dried at room temperature. Bound SRB was solubilized with 100-200 μl of 10 mM unbuffered Tris-base solution (pH 10.5). Absorbance was read at 540-560 nm without reference wavelength. Each experiment was performed at least three times in triplicate determinations.
Presto blue cell viability
The cell viability was determined by PrestoBlue cell viability reagent (Invitrogen) following the manufacturer’s instruction. At least 4 hours before the end of treatment time, presto blue reagent was added and incubated for total of 72 hours and fluorescence measured (540 nm excitation/590 nm emissions). Vehicle treated control cell were considered as 100% viable against which treated cells were compared. Experiments were performed in triplicate. Data was expressed as mean ± SD of triplicate experiments and was plotted using Graph Pad Prism Software.
Western blots
Cells were washed twice in ice-cold phosphate-buffered saline (PBS) and then lysed in RIPA Lysis Buffer (Santa Cruz Biotechnology). The resulting suspension was centrifuged for 10 min, 14,000 rpm at 4°C. The supernatant was then collected, and the protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo Scientific). Cell lysates were incubated with 6 × SDS sample buffer for 5 min at 100°C and then were run on SDS-PAGE gels, transferred to polyvinylidene fluoride membranes, and probed with the appropriate primary and secondary antibodies. Protein bands were detected by enhanced chemiluminescence (ECL, GE Healthcare). The Image J software was used for densitometry of bands.
Phospho-RTK array
The human phospho-RTK array kit was a product of R&D Systems, Inc. Cells were transfected with siYAP and siTAZ for 48 h and then washed with PBS, solubilized in lysis buffer, and then 300 Ag of total protein were processed according to the manufacturer’s protocols. Briefly, arrays were incubated with whole-cell lysates overnight at 4 degree with shaking and washed with the supplied washing buffer. Arrays were then incubated with anti-phosphotyrosine-HRP antibodies for 2 h at room temperature on a rocking platform shaker before incubation with a chemiluminescent reagent and finally film exposure.
Statistical analysis
All statistical analysis was conducted via R statistical software and GraphPad Prism version 6.0. Differences between groups of experiments were examined for statistical significance using t-test and one-way or two-way analysis of variance (ANOVA). Differences were accepted as statistically significant at P < 0.05. In all figures, ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.5, ns non-significant. Error bars represent Mean ± SD of triplicates.
Results
BMI is a high risk factor of EC and closely associated with IR
We used public TCGA (http://cancergenome.nih.gov/) data repositories as our primary source of samples. In total, we selected 361 EC patients with clinical information, RPPA and RNA-seq data. Some of clinical parameters were summarized in Tables 1, 2 and 3. Patients were between 50-70 years old. 57.4% patients were obese (BMI ≥ 30). 56.2% patients were grade 3 but 70.4% patients were at early stages (stage I and II), which might explain that BMI did not impact the survival rate (data not shown). All patients were grouped into two groups according to BMI < 30 and BMI ≥ 30. For grade, there was a trend that grade 1 and 2 patients had lower BMI while grade 3 had higher BMI (P = 0.0629) (Figure 1A). For stage, stage I and II patients had lower BMI while stage III and IV patients had higher BMI (P = 0.0157) (Figure 1B). We also grouped the patients as described above and performed the integrity pathway analysis (IPA) to predict the top 50 canonical pathways. The Insulin Receptor Signaling, Endometrial Cancer Signaling, IGF-1 Signaling and other signalings associated with PI3K/Akt/MAPK pathway were in the list of the top 50 canonical pathways. Interestingly, the expression profile of IRS1/2 between obesity and non-obesity patients were totally opposite when we focused on the Insulin signaling pathway and IGF-1 signaling pathway (Figure 1C). We also performed the correlation analysis between BMI and 779 insulin resistant relative (IRR) genes, which were selected from the Genecards website (http://www.genecards.org), and the mRNA expression data of 779 IRR genes were from TCGA. The expression of 5% (45/779) IRR genes was closely associated with BMI with P < 0.05, and the low expression of IRS1 was associated with low BMI with P = 0.01 (Figure 1D).
Table 1.
The clinic characters of 361 EC patients
| Age | < 50 | 50-60 | 60-70 | > 70 |
| 33 (9.1%) | 111 (30.7%) | 128 (35.5%) | 89 (24.7%) | |
| BMI | < 25 | 25-30 | ≥ 30 | |
| 70 (19.4%) | 84 (23.3%) | 207 (57.4%) | ||
| Grade | 1 | 2 | 3 | |
| 70 (19.4%) | 88 (24.4%) | 203 (56.2%) | ||
| Stage | I | II | III | IV |
| 222 (61.5%) | 31 (8.9%) | 86 (23.8%) | 22 (5.8%) | |
| Survival year (31 death) | < 2 | 2-5 | > 5 | |
| 20 (64.5%) | 9 (29%) | 2 (6.5%) |
Table 2.
The clinic characters of 31 dead patients from 361 EC patients
| Age | < 50 | 50-60 | 60-70 | > 70 |
| 1 (3.2%) | 8 (25.8%) | 12 (38.7%) | 10 (32.3%) | |
| BMI | < 25 | 25-30 | ≥ 30 | |
| 7 (22.5%) | 7 (22.5%) | 17 (55%) | ||
| Grade | 1 | 2 | 3 | |
| 0 | 6 (19.4%) | 25 (80.6%) | ||
| Stage | I | II | III | IV |
| 10 (32.3%) | 2 (6.5%) | 12 (38.7%) | 7 (22.5%) |
Table 3.
The IPA analysis of 779 IRR genes from 12 pairs of EC patients and normal women
| Rank | Gene | T value | DF | P-value |
|---|---|---|---|---|
| 1 | PTS | 3.96 | 11 | 0.0023 |
| 2 | DBI | 3.86 | 11 | 0.0027 |
| 3 | NFKB1 | -3.60 | 11 | 0.0042 |
| 4 | IRS1 | -3.47 | 11 | 0.0053 |
| 5 | MAPK3 | -3.46 | 11 | 0.0053 |
| 6 | AQP7 | -3.46 | 11 | 0.0054 |
| 7 | IL6R | -3.44 | 11 | 0.0056 |
| 8 | GAPDH | 3.43 | 11 | 0.0057 |
| 9 | ESRRA | 3.43 | 11 | 0.0057 |
| 10 | PIK3CG | -3.43 | 11 | 0.0057 |
| 11 | HPRT1 | 3.42 | 11 | 0.0058 |
| 12 | AKT1 | 3.40 | 11 | 0.0059 |
| 13 | PRKAR1A | -3.39 | 11 | 0.0061 |
| 14 | CALR | 3.38 | 11 | 0.0061 |
| 15 | TCF7L2 | -3.38 | 11 | 0.0061 |
| 16 | SOD3 | -3.29 | 11 | 0.0072 |
| 17 | GCK | -3.29 | 11 | 0.0073 |
| 18 | ATM | -3.28 | 11 | 0.0073 |
| 19 | COX6A1 | 3.27 | 11 | 0.0074 |
| 20 | PDGFC | -3.26 | 11 | 0.0076 |
Figure 1.
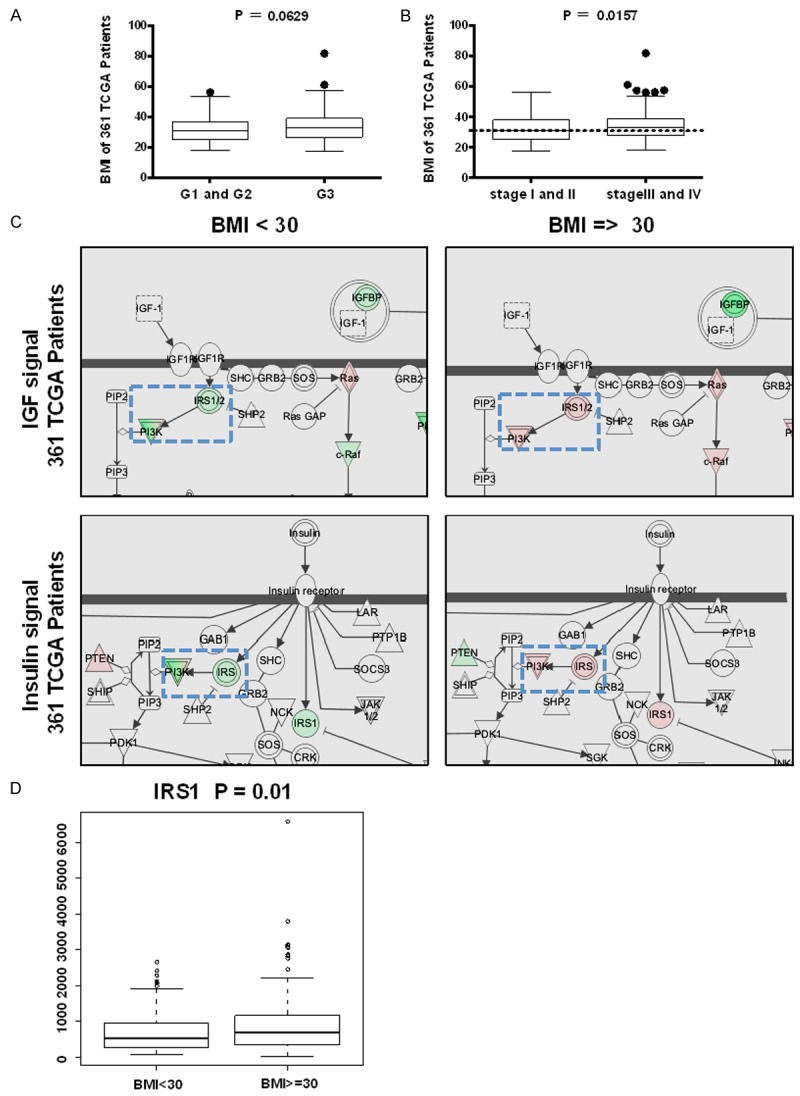
Correlation between BMI and EC grade. Correlation analysis was performed by R language program with 361 EC patients. The clinic data including BMI, Grade, Stage information and RPPA data of 361 EC patients was from the TCGA data on CBio website. Obesityes is a high risk factor of endometrial cancer and associated with insulin resistance closely. A. Grade 1 and 2 (G1 and 2) patients had lower BMI than Grade3 (G3), Unpaired t test (two tailed), P = 0.0629. B. Stage I and II patients had lower BMI than stage III and IV, Unpaired t test (two tailed), P = 0.0157. C. The IGF1 and Insulin signaling network in EC patients with BMI < 30 or EC patients with BMI ≥ 30. In blue frames with dotted line, the patients with BMI < 30 had lower IRS1/2 and PI3K level (light green color), and the patients with BMI ≥ 30 had higher IRS1/2 and PI3K level (light red color). D. The correlation between IRS1 and BMI in 309 of 361 EC patients with mRNA data of Insulin resistant related (IRR) genes. Patients with BMI < 30 had lower IRS1 mRNA level, P = 0.01.
IRS1/2 is the most important IRR gene associated with EC
We performed the upstream prediction with RPPA data of 361 EC as shown in Figure 2. Among 43 proteins that exhibited significant difference between obesity and non-obesity patients, 35% of them (15/43) belonged to IRR genes with red color in the table (Figure 2A). In this analysis, CTNNB1, P38MAPK, TNF were predicted as the dominant regulators, and CDH1, NFKB1, CDH2, YAP1, IRS1, IGFBP2, JUN, FN1, MYC, CCND1, EGFR, GATA3, BCL2, CCNE1 were predicted as the dominant targets (Figure 2B). Among these 17 dominant target proteins, 47% of them (8/17), including P38MAPK, TNF, NFKB1, IRS1, IGFBP2, JUN, CCND1, EGFR belonged to IRR genes. These data suggested that the expression of IRR gene might be related to the development of EC.
Figure 2.
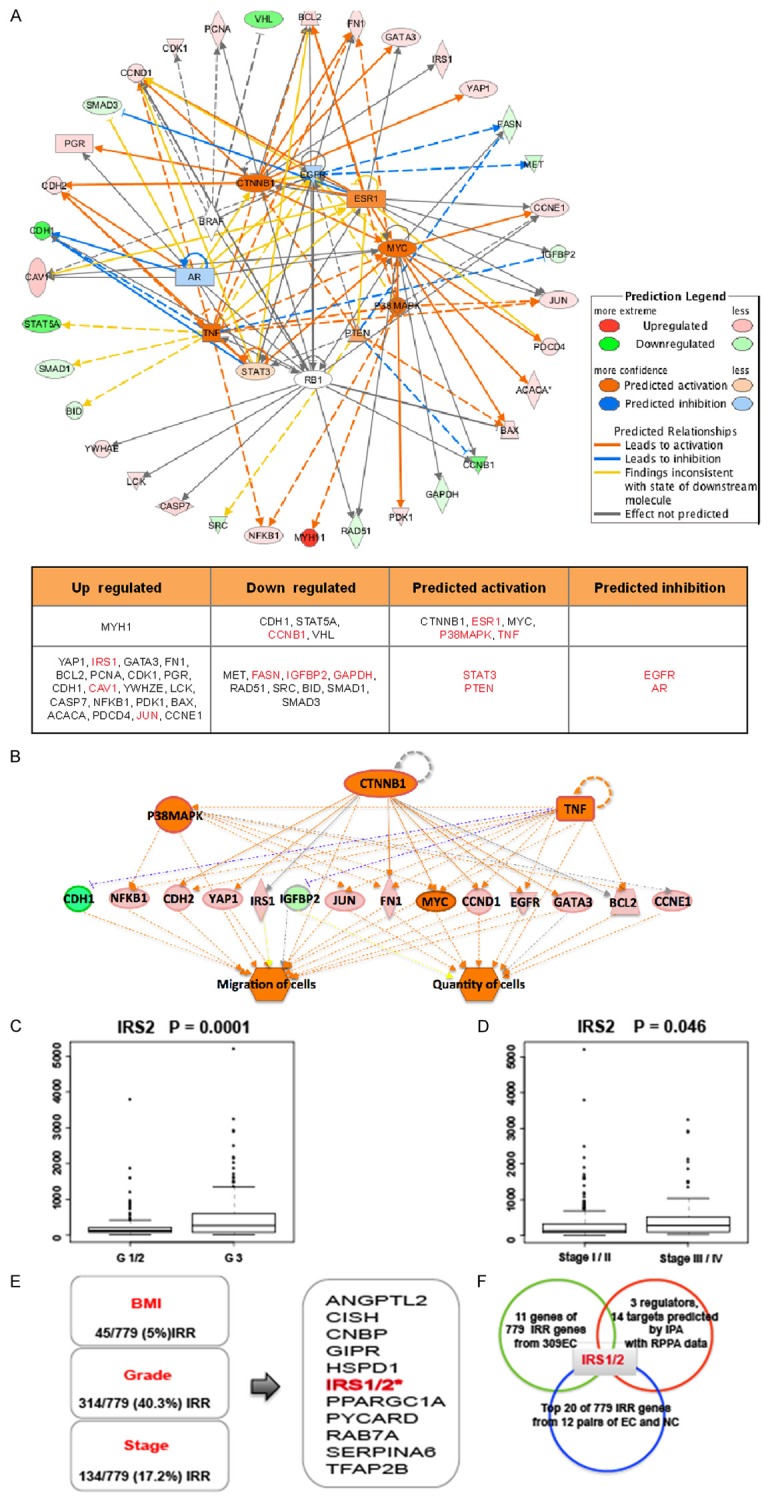
IRS1/2 are the most important IRR genes associated wth the development of endometrial cancer. A. Upstream analysis with RPPA data of 361 EC patients through IPA software. Red color indicated up-regulation; green color indicated down-regulation. Orange color was predicted as more activation and blue color was predicted as more inhibition. Most up-regulated genes belong to the IRR genes. B. Regulator effects analysis with RPPA data of 361 EC patients through IPA software. CTNNB1, P38MAPK, TNF were predicted as the upstream regulators. CDH1, NFKB1, CDH2, YAP1, IRS1, IGFBP2, JUN, FN1, MYC, CCND1, EGFR, GATA3, BCL2, CCNE1 were predicted as the downstream targets. C. Distribution of IRS2 mRNA level according to the grade of EC. D. Distribution of IRS2 mRNA level according to the stage of EC. E. 11 molecules including IRS1/2 are related with occurrence and development of EC closely. The left panel showed the characters of 309 EC patients having IRR genes data, 5% (45/779) IRR genes is associated with the distribution of BMI with P < 0.05, 40.3% (314/779) IRR genes is associated with the degree of grade of EC with P < 0.05. 17.2% (134/779) IRR genes is associated with the degree of stage of EC with P < 0.05. the right frame shows the 11 common IRR genes from three frames at the left side. F. IRS1/2 was the key molecule in EC. 11 common IRR genes in 309 EC patients, 17 regulated molecules in 361 EC patients and top 20 IRR genes in 12 pairs of tumor and control samples were analyzed.
Since we found that 5% (45/779) of IRR genes were correlated with BMI, 40.3% (314/779) of IRR genes were correlated with the grade of EC, and 17.2% (134/779) of IRR genes were correlated with the stage of EC, we analyzed these IRR gene information and focused on 11 important IRR genes including IRS. We found that IRS1 expression was significantly associated with BMI, while IRS2 was associated with grade and stage of EC (Figure 2C-E).
Furthermore, we compared the mRNA level of IRR genes between 12 pairs of EC patients and normal women. We also observed that IRS1 was one of the top 20 IRR genes associated with the development of EC (p < 0.05) (Table 3). Together, above data showed that IRS1/2 were the most important IRR genes in EC development (Figure 2F).
Correlation between IRS1/2 and YAP/TAZ in EC
Our previous study has shown that YAP/TAZ regulate the activity of PI3K/Akt, which are the central signaling molecules for insulin/IGF1 signaling. Hence, we searched the CBio website to determine if there existed a correlation between YAP/TAZ and IRS2 expression. Indeed, the altered YAP1 had the high IRS1 and IRS2 mRNA with the according P = 4.692e-4 and 2.1692-5. The altered TAZ (WWTR1) had high IRS1 with P = 2.623e-4 (Figure 3A-C). With the log odd ratios, we also observed the significance of co-occurrence between YAP1/TAZ and IRS1/2 (Figure 3D).
Figure 3.

YAP and TAZ are associated with IR21/2 closely in endometrial cancer. Correlation of IRS1/2 with YAP and TAZ in EC (Data from CBio website). A. Tumors with altered YAP1 had the high IRS1 mRNA level, P = 0.0004692. B. Tumors with altered YAP1 had the high IRS2 mRNA level, P = 0.00002169. C. Tumors with altered WWTR1 had the high IRS1 mRNA level, P = 0.0002623. D. The co-occurrence possibilities of YAP1, WWTR1, IRS1, and IRS2 are listed in the table. YAP1 co-occurred with IRS1 and IRS2 with P = 0.002 and P = 0.003 separately.
Regulation of IRS1 by YAP/TAZ
We used two cell lines to conduct cell-based assays to validate our clinical observation that there exists an association between YAP/TAZ and IRS1. We confirmed that siYAP/TAZ decreased the phosphorylation of IRS1 but increased the phosphorylation of IGFR1. First, YAP1 and TAZ were successfully knocked down separately and simultaneously in KLE and EFE184 (Figure 4A). In these knockdown cells, we found that 12 hrs of siYAP1/TAZ treatment was enough to decrease the protein level (Figure 4B). After 30 minutes of IGF1 stimulation, total IRS1 increased significantly with P < 0.05 and the IGF1-induced up-regulation of IRS1 level reached peak at 60 minutes. However, treating cells with siYAP/TAZ delayed the function of IGF1 at 30 minutes (Figure 4C, 4D), which indicates that YAP/TAZ levels in cells inhibited the effects of IGF1, similar to what we observed with the effect of YAP/TAZ on cell viability. In addition, we also observed a strong effect of siYAP/TAZ on the level of phospho-IRS1. siYAP/TAZ decreased the phospho-IRS1 level significantly in the presence and absence of IGF1 treatment (Figure 4C). Unexpectedly, we found that siYAP/TAZ increased the phospho-IGFR1 level. This increase in phospho-IGFR1 level might represent a negative feedback due to the decrease of phospho-IRS1. Furthermore, we performed the phospho-RTK array to validate the increase of phospho-IGFR1 induced by siYAP/TAZ (Figure 4E). Indeed, siYAP or siTAZ alone increased the phospho-IGFR1 (data not shown), however, the combined siYAP/TAZ showed stronger effect.
Figure 4.
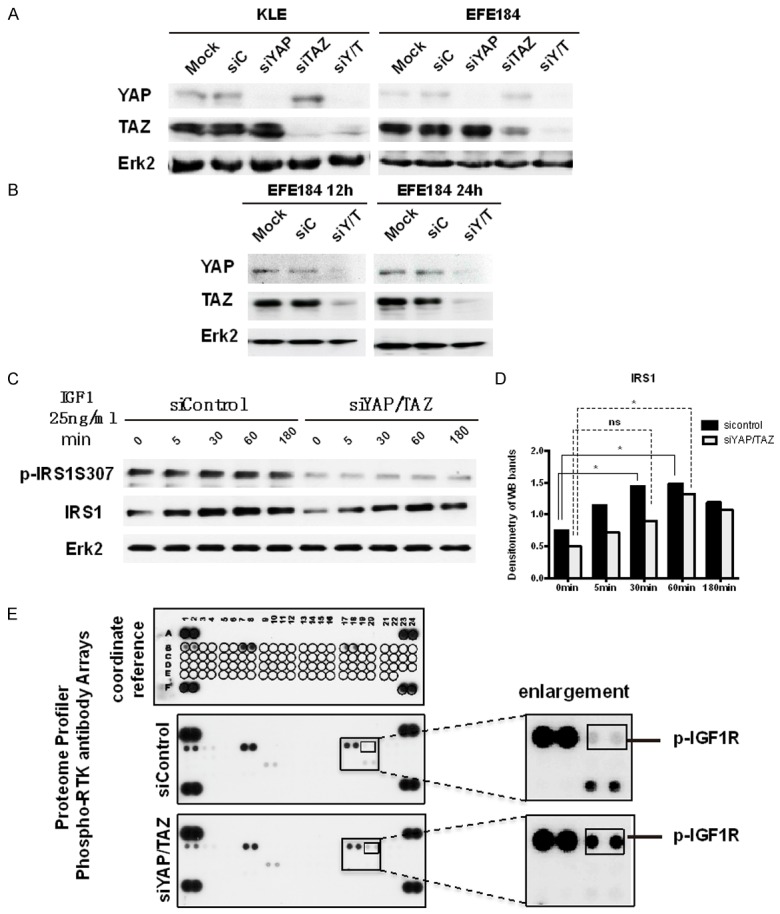
siYAP/TAZ decreased the phosphorylation of IRS1 but increased the phosphorylation of IGFR1. A. KLE cells and EFE184 cells were transfected with mock, siControl, siYAP, siTAZ and siYAP/TAZ (siY/T). At 48 h after transfection, cells were collected for Western blot with the indicated antibodies. Erk2 was used as a loading control. B. EFE184 cells were transfected with mock, siControl, siY/T. At 12 h and 24 h after transfection, cells were collected for Western blot with the indicated antibodies. Erk2 was used as a loading control. C. In KLE cells, YAP and TAZ were knocked down by siRNA, after 24 h, IGF1 25 ng/ml was administrated to cells for 5 min, 30 min, 60 min, 180 min. And then, cells were collected and processed to western blotting with according antibodies. It shows siYAP/TAZ decreased the IRS1 expression level. D. The bar graph is the densitometry of figure c, which indicates siYAP/TAZ decreased the IRS1 expression level significantly. Statistical significance was calculated using one way ANOVA and Dunnett’s multiple comparisons test compared with 0 min. *P < 0.5, ns: non-significant. E. KLE cells were transfected with siYAP/TAZ. After 24 h, cells were processed to Proteome profiler phosphor-RTK antibody arrays (R&D Systems Catalog Number ARY001B). Every antibody was repeated as two dots. siYAP/TAZ increased the p-IGF1R expression, which is shown in two smallest frames.
To further investigate the regulatory role of YAP/TAZ on insulin signaling, we investigated the effect of siYAP/TAZ insulin-induced cell proliferation. We found that siYAP/TAZ could partially attenuate the function of insulin and IGF1 on cell proliferation (Figure 5A). We also used Verteporfin, a YAP inhibitor that could decrease the protein level of YAP and TAZ [30], and observed the abrogated function of insulin and IGF1 (Figure 5B).
Figure 5.
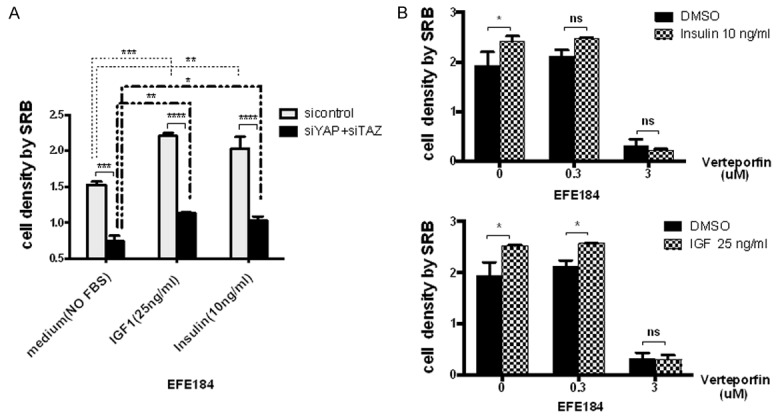
siYAP/TAZ attenuated the effects of IGF and Insulin on cell proliferation. A. EFE184 cells were transfected with siYAP and siTAZ. After 24 h, cells were administrated with IGF1 (25 ng/ml) and Insulin (10 ng/ml). After 48 h, cells were processed to SRB assay to test the cell proliferation. IGF1 and Insulin promoted the cell growth while siYAP/TAZ removed the enhancement of cell proliferation stimulated by IGF1 and Insulin. B. EFE184 cells were subjected to the Verteporfin with 0.3 uM and 3 uM for 24 h, and then cells were subjected to IGF and Insulin too. After 48 h, cells were processed to SRB assay as well. Verteporfin inhibited the effects of IGF and Insulin accordingly.
siYAP/TAZ enhanced the effect of metformin on cell growth
Metformin inhibited the proliferation of both KLE and EFE184 cells in a dose dependent manner (Figure 6A, 6B). 1 uM metformin inhibited the growth of KLE cells significantly, while 2 uM metformin started to inhibit the growth of EFE184 cells. When cells were treated with insulin, we observed that 2 uM was not enough to abrogate the effects of insulin in EFE 184 cells while it was enough to inhibit the cell growth without insulin. However, 4 uM metformin abrogated the effect of insulin (Figure 6C). siYAP/TAZ (siY/T) alone was not efficient enough to inhibit the effects of insulin as shown in the first group of bars. Similarly, merformin treatment alone at 1 uM or 2 uM did not inhibit the function of insulin as shown in the secondary and third groups of bars. siYAP/TAZ (siY/T) plus 1 uM metformin were also not sufficient to block the function of insulin completely as shown in the secondary group of bars. However, we observed that 2 uM or higher concentration of metformin plus siY/T could completely abolish the effects of insulin as shown in the third and forth groups of bars (Figure 6D), suggesting that siYAP/TAZ might sensitized cells to the effect of metformin on cell growth.
Figure 6.

Combining siYAP/TAZ with metformin treatment completely blocked the effects of insulin on cell proliferation. A. Cell proliferation was inhibited by metformin in a dose dependent manner. KLE cells were treated with metformin at 0.5 μM, 1 μM, 2 μM and 4 μM. After 48 h treatment, cell viability was measured by SRB. Statistical significance was calculated using two way ANOVA and Sidak’s multiple comparisons test, ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.5, ns means non-significant. Error bars represent Mean ± SD of triplicates. B. EFE184 were treated in a same way like KLE. Cell viability was measured by SRB. C. EFE184 were treated metformin and insulin with the according dose described in the figure. After 48 h treatment, Cell viability was measured by SRB. D. EFE184 cells were transfected with siYAP and siTAZ. After 24 h, cells were administrated with Insulin (10 ng/ml) and metformin with the dose as shown in the figure. After 48 h, cells were processed to SRB assay to test the cell proliferation.
Discussion
Insulin resistance is traditionally defined as an abnormal metabolic condition, which results in a requirement for increased levels of insulin to achieve a given level of metabolic activity [31]. However, insulin increased cell proliferation and reduced apoptosis in cancer cells [32,33], It was reported that fasting insulin levels of women were positively associated with EC, hyperinsulinemia and C-peptide levels were risk factors of EC independent of estradiol [34,35]. It was also reported that Insulin resistance was positively correlated with the aggressiveness of EC and local tumor dissemination [34,36,37]. Insulin resistance also increased the risk of breast, and ovarian cancers besides endometrial cancers [38-40]. Therefore, strategies to relieve insulin resistance might have a favorable effect on the incidence of endometrial cancer.
As we acknowledged, insulin resistance contributes to obesity, which is also one of the strongest risk factors for EC, accounting for about 40% of EC incidence in developed countries [41]. As the relationship between IRR genes contributing to insulin resistance and BMI in endometrial cancer has not been elucidated so far. We utilized data from TCGA database, which provides detailed information on medical conditions as well as data about proteins and mRNA expression of patients to do pertinent analyses via IPA platform. The large size of this database allowed us to evaluate whether BMI and IRR genes were differentially associated with risk dependent on various clinical parameters of the endometrial cancer, including stage and grade of the tumors. In addition, we were able to evaluate the correlation of IRR genes with BMI and to find the differences in canonical pathways between obesity and non-obesity patients, and even to predict the important regulators and targets involving in the occurrence of EC. By virtue of these analyses, it is possible to find some of the most important molecules that could play critical roles in development of EC. Our findings supported both obesity and insulin resistance as two of the strongest risk factors of EC, which are consistent with previous studies. Considering comprehensively, IRS1/2 became one of our interested targets due to its significant difference of expression in every comparison, which has been described in Figure 2.
In the cascade of insulin signaling, IRS proteins play a central role. Upon insulin binding its cognate receptor at the cell surface, insulin signaling begins to transmit and causes the insulin receptor (IR) autophosphorylation and activation. The activated IR phosphorylates several scaffold molecules including the Insulin Receptor Substrate (IRS) family of proteins. Phosphorylation of IRS-1 (or IRS-2) serves to recruit downstream effectors leading to activation of the MAPK cascade, which promotes transcription, and phosphoinositide 3-kinase (PI3K) cascade, which promotes increased protein and glycogen synthesis through the phosphorylation of the mammalian target of rapamycin (mTOR) and Glycogen Synthase Kinase-3b [42-45]. To date, four members of this family of scaffold molecule sharing a similar structure have been identified in mammalian cells (IRS-1 to IRS-4). Different IRS proteins functions dominantly at different tissue, for example, IRS-1 functions primarily in the skeletal muscle and fat, while IRS-2 acts mainly in liver. IRS1 plays a key role in cancer cell proliferation and mediates the resistance to anticancer drugs, while IRS2 acts mainly in cancer cell motility and metastasis. From this, we selected IRS1 to be followed in the further work and expectedly we observed insulin increased cell proliferation in our study.
Insulin signaling deficiency inhibited the proliferation and metastasis of cancer cells in vitro and in vivo [46,47]. Currently, there are no anticancer agents that target IRS1/2, which promote us to look for the effective ways inhibiting the activity of IRS1/2. Since we found the knock down of YAP/TAZ inhibited PI3K/Akt signaling in EC, we were wondering whether hippo pathway might interfere with the insulin signaling, in other word, hippo pathway might be implicated in the related diseases of insulin resistance, which has not yet been presented explicitly. The information provided by CBio database that there were positive correlation between IRS1/2 and YAP/TAZ gave us more evidences supporting our supposition that the function of YAP/TAZ could contribute to the development of insulin resistance. And our findings from immune blotting and cell viability assay also provided further evidences that low YAP/TAZ level inhibited the function of insulin including the stimulation on cell proliferation and phosphorylation of IRS1 Ser307.
Insulin and IGF-1 share 40%-50% homology whilst IRα and IGF-1 receptor also share 84% homology [48]. It is possible that insulin and IGF-1 signaling pathways might cross talk with each other, which contributes to EC development. An elevated circulating IGF-1 level is an established risk factor for different cancers [49]. In the endometrium, estrogen could increase the level of IGF-1 expression and insulin could increase the bioactivity of IGF-1 in the endometrium [50], however, progesterone inhibits IGF-1 expression and activity through increasing the synthesis of IGFBP-1 [33], which can explain that EC is a estrogen driven cancer and insulin resistance could be a stronger risk factor of EC. Unexpectedly, we found the knock down of YAP/TAZ caused the increase of p-IGFR1 from phospho-RTK array, indeed we have the same results from immune blotting assay (data not shown here). We think the effects of siYAP/TAZ on p-IGFR1 is an outcome of negative feedback rather than cause, because we found the siYAP/TAZ abrogated the effects of IGF1 on the cell proliferation at the same time. Moreover, Verteporfin, which is an inhibitor of YAP and also inhibited the function of TAZ we proved in our previous work (data not shown), removed the function of insulin and IGF1, which was consistent with the results of siYAP/TAZ. It indicated that low YAP and TAZ level inhibited the insulin and IGF1 signaling, in other words, hippo pathway not only controls the size of organs or cell growth but also contributes to the regulation of insulin signaling in case insulin resistance occurs.
Metformin is a biguanide that is a widely prescribed oral medication and has been used as front-line therapy for type 2 diabetes. Metformin had been proved to be able to decrease the incidence of cancer and cancer-related mortality in diabetic patients [51]. It was reported that AMPK inhibits cancer cell growth by inhibiting fatty acid synthesis and mTOR-induced protein translation [52,53]. Metformin has the ability to activate AMPK pathway and inhibits gluconeogenesis to reduce the blood glucose levels and consequently causes to the decrease of circulating insulin concentrations [54], which may be one explanation for metformin’s reduction of cancer risk and mortality due to relieving hyperinsulinemia. In our study, we observed the inhibition of cell viability induced by metformin and the antagonism of metformin to insulin. We also found siYAP/TAZ has a similar effects of insulin with metformin. siYAP/TAZ with 1 uM metformin did not remove the effects of insulin completely, while with 2 uM metformin, siYAP/TAZ totally got rid of the influence of insulin and had the strongest inhibition of cell proliferation. We did not see any evidence that siYAP/TAZ did not impact the effect of metformin, at least it suggested YAP/TAZ might not be the only downstream target of metformin, but it is not reasonable to exclude the possibility that metformin might affect YAP/TAZ. It indicates somehow metformin might be more effective in patients with low YAP/TAZ level than patients with high YAP/TAZ level if we tend to use metformin as a new adjuvant drug.
Altogether, we demonstrated a novel function of YAP and TAZ in the insulin resistance via IRS1/2 in endometrial cancer. Our study also provided the rationale for the potential therapeutic treatment of EC with the combination of inhibiting YAP/TAZ and metformin in future work. At the same time, we need to be aware of the selection of the population of patients upon using metformin. The patients with low YAP/TAZ might benefit from metformin most.
Acknowledgements
We thank the National Natural Science Fundation of China (81001149) and the Shanghai Outstanding Youth Training Plan of China (grant no. XYQ2011062), which provided supports to Chao Wang. We also thank Dr. Gordon B Mills in MD Anderson cancer center for his supports including partial funding and guidance.
Disclosure of conflict of interest
None.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Kushi LH, Doyle C, McCullough M, Rock CL, Demark-Wahnefried W, Bandera EV, Gapstur S, Patel AV, Andrews K, Gansler T American Cancer Society 2010 Nutrition and Physical Activity Guidelines Advisory Committee. American Cancer Society Guidelines on nutrition and physical activity for cancer prevention: reducing the risk of cancer with healthy food choices and physical activity. CA Cancer J Clin. 2012;62:30–67. doi: 10.3322/caac.20140. [DOI] [PubMed] [Google Scholar]
- 3.Delahanty RJ, Beeghly-Fadiel A, Xiang YB, Long J, Cai Q, Wen W, Xu WH, Cai H, He J, Gao YT, Zheng W, Shu XO. Association of obesity-related genetic variants with endometrial cancer risk: a report from the Shanghai Endometrial Cancer Genetics Study. Am J Epidemiol. 2011;174:1115–1126. doi: 10.1093/aje/kwr233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chia VM, Newcomb PA, Trentham-Dietz A, Hampton JM. Obesity, diabetes, and other factors in relation to survival after endometrial cancer diagnosis. Int J Gynecol Cancer. 2007;17:441–446. doi: 10.1111/j.1525-1438.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- 5.Lukanova A, Zeleniuch-Jacquotte A, Lundin E, Micheli A, Arslan AA, Rinaldi S, Muti P, Lenner P, Koenig KL, Biessy C, Krogh V, Riboli E, Shore RE, Stattin P, Berrino F, Hallmans G, Toniolo P, Kaaks R. Prediagnostic levels of C-peptide, IGF-I, IGFBP -1, -2 and -3 and risk of endometrial cancer. Int J Cancer. 2004;108:262–268. doi: 10.1002/ijc.11544. [DOI] [PubMed] [Google Scholar]
- 6.Allen NE, Key TJ, Dossus L, Rinaldi S, Cust A, Lukanova A, Peeters PH, Onland-Moret NC, Lahmann PH, Berrino F, Panico S, Larranaga N, Pera G, Tormo MJ, Sanchez MJ, Ramon Quiros J, Ardanaz E, Tjonneland A, Olsen A, Chang-Claude J, Linseisen J, Schulz M, Boeing H, Lundin E, Palli D, Overvad K, Clavel-Chapelon F, Boutron-Ruault MC, Bingham S, Khaw KT, Bueno-de-Mesquita HB, Trichopoulou A, Trichopoulos D, Naska A, Tumino R, Riboli E, Kaaks R. Endogenous sex hormones and endometrial cancer risk in women in the European Prospective Investigation into Cancer and Nutrition (EPIC) Endocr Relat Cancer. 2008;15:485–497. doi: 10.1677/ERC-07-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu WH, Matthews CE, Xiang YB, Zheng W, Ruan ZX, Cheng JR, Gao YT, Shu XO. Effect of adiposity and fat distribution on endometrial cancer risk in Shanghai women. Am J Epidemiol. 2005;161:939–947. doi: 10.1093/aje/kwi127. [DOI] [PubMed] [Google Scholar]
- 8.Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol. 2004;159:1160–1167. doi: 10.1093/aje/kwh161. [DOI] [PubMed] [Google Scholar]
- 9.Saltzman BS, Doherty JA, Hill DA, Beresford SA, Voigt LF, Chen C, Weiss NS. Diabetes and endometrial cancer: an evaluation of the modifying effects of other known risk factors. Am J Epidemiol. 2008;167:607–614. doi: 10.1093/aje/kwm333. [DOI] [PubMed] [Google Scholar]
- 10.Friberg E, Orsini N, Mantzoros CS, Wolk A. Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia. 2007;50:1365–1374. doi: 10.1007/s00125-007-0681-5. [DOI] [PubMed] [Google Scholar]
- 11.Akhmedkhanov A, Zeleniuch-Jacquotte A, Toniolo P. Role of exogenous and endogenous hormones in endometrial cancer: review of the evidence and research perspectives. Ann N Y Acad Sci. 2001;943:296–315. doi: 10.1111/j.1749-6632.2001.tb03811.x. [DOI] [PubMed] [Google Scholar]
- 12.Crosbie EJ, Zwahlen M, Kitchener HC, Egger M, Renehan AG. Body mass index, hormone replacement therapy, and endometrial cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2010;19:3119–3130. doi: 10.1158/1055-9965.EPI-10-0832. [DOI] [PubMed] [Google Scholar]
- 13.Wild S, Pierpoint T, Jacobs H, McKeigue P. Long-term consequences of polycystic ovary syndrome: results of a 31 year follow-up study. Hum Fertil (Camb) 2000;3:101–105. doi: 10.1080/1464727002000198781. [DOI] [PubMed] [Google Scholar]
- 14.Daniilidis A, Dinas K. Long term health consequences of polycystic ovarian syndrome: a review analysis. Hippokratia. 2009;13:90–92. [PMC free article] [PubMed] [Google Scholar]
- 15.Fearnley EJ, Marquart L, Spurdle AB, Weinstein P, Webb PM Australian Ovarian Cancer Study Group and Australian National Endometrial Cancer Study Group. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: an Australian case-control study. Cancer Causes Control. 2010;21:2303–2308. doi: 10.1007/s10552-010-9658-7. [DOI] [PubMed] [Google Scholar]
- 16.Ashizawa N, Yahata T, Quan J, Adachi S, Yoshihara K, Tanaka K. Serum leptin-adiponectin ratio and endometrial cancer risk in postmenopausal female subjects. Gynecol Oncol. 2010;119:65–69. doi: 10.1016/j.ygyno.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Dal Maso L, Augustin LS, Karalis A, Talamini R, Franceschi S, Trichopoulos D, Mantzoros CS, La Vecchia C. Circulating adiponectin and endometrial cancer risk. J Clin Endocrinol Metab. 2004;89:1160–1163. doi: 10.1210/jc.2003-031716. [DOI] [PubMed] [Google Scholar]
- 18.Friedenreich CM, Langley AR, Speidel TP, Lau DC, Courneya KS, Csizmadi I, Magliocco AM, Yasui Y, Cook LS. Case-control study of markers of insulin resistance and endometrial cancer risk. Endocr Relat Cancer. 2012;19:785–792. doi: 10.1530/ERC-12-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burzawa JK, Schmeler KM, Soliman PT, Meyer LA, Bevers MW, Pustilnik TL, Anderson ML, Ramondetta LM, Tortolero-Luna G, Urbauer DL, Chang S, Gershenson DM, Brown J, Lu KH. Prospective evaluation of insulin resistance among endometrial cancer patients. Am J Obstet Gynecol. 2011;204:355, e351–357. doi: 10.1016/j.ajog.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER, Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudd ML, Price JC, Fogoros S, Godwin AK, Sgroi DC, Merino MJ, Bell DW. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011;17:1331–1340. doi: 10.1158/1078-0432.CCR-10-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, Liang H, Lu KH, Broaddus RR, Mills GB. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, Guo W, Scherer SE, Carter H, Westin SN, Dyer MD, Verhaak RG, Zhang F, Karchin R, Liu CG, Lu KH, Broaddus RR, Scott KL, Hennessy BT, Mills GB. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012;22:2120–2129. doi: 10.1101/gr.137596.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oh H, Irvine KD. Yorkie: the final destination of Hippo signaling. Trends Cell Biol. 2010;20:410–417. doi: 10.1016/j.tcb.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papazisis KT, Geromichalos GD, Dimitriadis KA, Kortsaris AH. Optimization of the sulforhodamine B colorimetric assay. J Immunol Methods. 1997;208:151–158. doi: 10.1016/s0022-1759(97)00137-3. [DOI] [PubMed] [Google Scholar]
- 28.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 29.Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst. 1990;82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 30.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev. 2012;33:981–1030. doi: 10.1210/er.2011-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lippman M, Bolan G, Huff K. The effects of estrogens and antiestrogens on hormone-responsive human breast cancer in long-term tissue culture. Cancer Res. 1976;36:4595–4601. [PubMed] [Google Scholar]
- 33.Osborne CK, Bolan G, Monaco ME, Lippman ME. Hormone responsive human breast cancer in long-term tissue culture: effect of insulin. Proc Natl Acad Sci U S A. 1976;73:4536–4540. doi: 10.1073/pnas.73.12.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gunter MJ, Hoover DR, Yu H, Wassertheil-Smoller S, Manson JE, Li J, Harris TG, Rohan TE, Xue X, Ho GY, Einstein MH, Kaplan RC, Burk RD, Wylie-Rosett J, Pollak MN, Anderson G, Howard BV, Strickler HD. A prospective evaluation of insulin and insulin-like growth factor-I as risk factors for endometrial cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:921–929. doi: 10.1158/1055-9965.EPI-07-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Folsom AR, Anderson KE, Sweeney C, Jacobs DR Jr. Diabetes as a risk factor for death following endometrial cancer. Gynecol Oncol. 2004;94:740–745. doi: 10.1016/j.ygyno.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 36.Berstein LM, Kvatchevskaya JO, Poroshina TE, Kovalenko IG, Tsyrlina EV, Zimarina TS, Ourmantcheeva AF, Ashrafian L, Thijssen JH. Insulin resistance, its consequences for the clinical course of the disease, and possibilities of correction in endometrial cancer. J Cancer Res Clin Oncol. 2004;130:687–693. doi: 10.1007/s00432-004-0587-2. [DOI] [PubMed] [Google Scholar]
- 37.Tsugane S, Inoue M. Insulin resistance and cancer: epidemiological evidence. Cancer Sci. 2010;101:1073–1079. doi: 10.1111/j.1349-7006.2010.01521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose DP, Vona-Davis L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr Relat Cancer. 2012;19:R225–241. doi: 10.1530/ERC-12-0203. [DOI] [PubMed] [Google Scholar]
- 39.Mu N, Zhu Y, Wang Y, Zhang H, Xue F. Insulin resistance: a significant risk factor of endometrial cancer. Gynecol Oncol. 2012;125:751–757. doi: 10.1016/j.ygyno.2012.03.032. [DOI] [PubMed] [Google Scholar]
- 40.Beauchamp MC, Yasmeen A, Knafo A, Gotlieb WH. Targeting insulin and insulin-like growth factor pathways in epithelial ovarian cancer. J Oncol. 2010;2010:257058. doi: 10.1155/2010/257058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergstrom A, Pisani P, Tenet V, Wolk A, Adami HO. Overweight as an avoidable cause of cancer in Europe. Int J Cancer. 2001;91:421–430. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1053>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 42.Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, Cahill DA, Goldstein BJ, White MF. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352:73–77. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- 43.Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MG Jr, Glasheen E, Lane WS, Pierce JH, White MF. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377:173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 44.Lavan BE, Fantin VR, Chang ET, Lane WS, Keller SR, Lienhard GE. A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J Biol Chem. 1997;272:21403–21407. doi: 10.1074/jbc.272.34.21403. [DOI] [PubMed] [Google Scholar]
- 45.White MF. Insulin signaling in health and disease. Science. 2003;302:1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H, Fagan DH, Zeng X, Freeman KT, Sachdev D, Yee D. Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene. 2010;29:2517–2527. doi: 10.1038/onc.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heuson JC, Legros N. Influence of insulin deprivation on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in rats subjected to alloxan diabetes and food restriction. Cancer Res. 1972;32:226–232. [PubMed] [Google Scholar]
- 48.De Pergola G, Silvestris F. Obesity as a major risk factor for cancer. J Obes. 2013;2013:291546. doi: 10.1155/2013/291546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 50.Murphy LJ, Ghahary A. Uterine insulin-like growth factor-1: regulation of expression and its role in estrogen-induced uterine proliferation. Endocr Rev. 1990;11:443–453. doi: 10.1210/edrv-11-3-443. [DOI] [PubMed] [Google Scholar]
- 51.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17:351–360. doi: 10.1677/ERC-09-0252. [DOI] [PubMed] [Google Scholar]
- 53.Larsson O, Morita M, Topisirovic I, Alain T, Blouin MJ, Pollak M, Sonenberg N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc Natl Acad Sci U S A. 2012;109:8977–8982. doi: 10.1073/pnas.1201689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]