


Abstract
Understanding telomere length maintenance mechanisms is central in cancer biology as their dysregulation is one of the hallmarks for immortalization of cancer cells. Important for this well-balanced control is the transcriptional regulation of the telomerase genes. We integrated Mixed Integer Linear Programming models into a comparative machine learning based approach to identify regulatory interactions that best explain the discrepancy of telomerase transcript levels in yeast mutants with deleted regulators showing aberrant telomere length, when compared to mutants with normal telomere length. We uncover novel regulators of telomerase expression, several of which affect histone levels or modifications. In particular, our results point to the transcription factors Sum1, Hst1 and Srb2 as being important for the regulation of EST1 transcription, and we validated the effect of Sum1 experimentally. We compiled our machine learning method leading to a user friendly package for R which can straightforwardly be applied to similar problems integrating gene regulator binding information and expression profiles of samples of e.g. different phenotypes, diseases or treatments.
INTRODUCTION
Telomeres protect the eukaryotic chromosomal ends against fusion, degradation and unwanted double-strand break repair mechanisms. The length and structure of telomeres is tightly controlled (1). Telomeric DNA is synthesized by telomerase, an enzyme not expressed in most somatic cells. In humans, the majority of cells lack telomerase activity and telomeres shorten gradually with each cell division. The accumulation of critically short telomeres leads to replicative senescence and eventual cell death. About 85–90% of primary tumors re-express telomerase activity, thereby enabling those cells to become immortal by maintaining their telomere length (2). Thus, understanding the mechanisms that maintain telomere length can have substantial medical implications, in particular for ageing and carcinogenesis. Saccharomyces cerevisiae is a well studied model organism with an active telomerase enzyme (3). Telomerase of S. cerevisiae is comprised of the RNA template, TLC1, and the ‘Ever shorter telomere’ proteins Est1, Est2 and Est3. Est2 is the catalytic subunit of telomerase, while Est1 and Est3 are TLC1-RNA-associated proteins (4). Cdc13 (Est4) is a sequence-specific telomere-DNA-binding protein, involved in telomere capping to protect the chromosomal ends from degradation and it interacts with Est1 to recruit the telomerase complex (2).
The yeast genome has close to 6000 recognized genes. By systematically deleting each individual non-essential gene, a collection of 4700 mutants (knockouts) was established [non-essential yeast mutant collection (5)]. This collection was later complemented by two additional libraries of mutants of all the essential genes (yeast has ∼1300 essential genes) whereby either hypomorphic (6) or temperature-sensitive alleles (7) of the genes were created. Systematic mutant screens can be carried out with these mutant collections even if the phenotype of interest is not selectable. Genome-wide screening efforts resulted in a comprehensive list of genes that, when mutated, affect telomere length in S. cerevisiae (8–12). These systematic screens revealed that ∼8% of the genes within the yeast genome affected (either directly or indirectly) telomeric length homeostasis. Indeed, a total of ∼500 of such telomere-length maintenance (TLM) genes were identified. About 60% of the identified TLM genes lead to short telomeres when mutated compared to the wild-type and the other 40% to elongated telomeres. TLM proteins have many different biochemical functions and localize to several compartments in the cell. Most of these were not known to play a role in homeostasis of telomere length, and their mechanism of action is only now starting to be studied.
In this study, we followed a computational approach and used this phenotypic information (aberrant telomere length) as a starting point to elucidate the transcriptional regulation of the telomerase holoenzyme. TLC1 was excluded from the analysis because no expression data was available. To predict the effect of putative regulators (transcription factors, chromatin modifiers) of the telomerase genes, we followed a Mixed Integer Linear Programming (MILP) approach we developed recently (13). MILP can be used as a powerful linear regression method. Compared to a lasso regression approach, the most prominent advantages of an MILP-based regression are that the error penalties are linear avoiding over-emphasizing outliers (L1 regression) and MILP allows integrating binary switches or discrete constraints [for details, see (13)]. We constructed regulatory models using the MILP approach and a comprehensive set of gene expression profiles of deletion strains of S. cerevisiae (14,15). To restrict the TLM list to those that are expected to impact on telomere length through a direct regulation of the EST genes, we focused on deletion strains of transcriptional regulators that lead to telomere shortening (short tlms). Putative binding interactions were inferred from ChIP experiments of regulators binding to their targets [taken from the database YEASTRACT (www.yeastract.com) and (16)]. To select the regulators being relevant specifically for telomere maintenance (and the corresponding telomere phenotype), we set up a discriminative machine learning algorithm and studied the regulation of the EST genes in regulator deletion strains with aberrant telomere length (short tlm mutants) compared to regulator deletion strains with normal telomere length (non-TLM genes or controls). We identified genes affecting histone levels and modifications as the main regulators of telomerase transcription in yeast, and we identified the transcription factors Sum1, Hst1 and Srb2 as most promising hits regulating EST1.
MATERIALS AND METHODS
Gene expression data
We used published microarray gene expression data of 269 yeast regulator deletion strains (strains BY4741, S288C and BYTET). This dataset was originally generated by Hu et al. and consisted of 588 two-color cDNA microarray hybridizations of 269 regulator mutants against a reference sample (14). The dataset of Hu et al. was re-analyzed by Reimand et al. (15), and we used the data from the latter. Briefly, all probes on the arrays which were not annotated as open reading frames were removed. For duplicated and triplicated probes the average was calculated. The re-analysis included a variance stabilizing normalization (15,17). Altogether, this pre-processed dataset of Reimand et al. consisted of expression values of 6253 protein-coding genes for 269 regulator deletion strains and was taken from Array Express (E-MTAB-109, www.ebi.ac.uk/arrayexpress/). For our model, we performed a z-score transformation for each gene across the whole dataset. To annotate each deletion strain as a deletion of a TLM gene, we used the results from (8–12) yielding knockout samples for 18 tlm mutants showing shortened telomeres (short tlm mutants), 11 showing elongated telomeres (long tlm mutants) and 240 non-TLM controls (normal telomere length) (Supplementary Table S1).
Constructing the regulatory network
To identify regulators of the EST genes, we first constructed a regulatory network consisting of 6728 nodes and 203 234 edges between 382 regulators and 6346 target genes. The network based on the binding information taken from the YEAst Search for Transcriptional Regulators And Consensus Tracking (YEASTRACT) database (www.yeastract.com) and a study of Yu and Gerstein (16). To date (August 2015), YEASTRACT bases on more than 1300 publications. We used only YEASTRACT entries annotated as ‘documented’ (DNA binding plus expression evidence) from high-throughput chromatin immunoprecipitation assays (ChIP-on-ChIP) and in silico refinements of this data (15,18,19). In addition, we used binding information from the study of Yu and Gerstein, who studied hierarchical structures in gene regulatory networks of yeast (16). In the following, these putative regulatory interactions of our regulatory network are denoted as ‘known binding’, the respective regulators ‘putative regulators’ and the targets ‘putative targets’.
Modeling EST regulation
Typically several regulators bind to a gene's promoter, each contributing to the expression of the target gene (13,20–25). We used a MILP approach to predict gene expression of EST genes to (i) address additive cooperativity; and (ii) to select the most relevant regulators of the EST genes.
As depicted in Supplementary Figure S1, the model contained the three EST genes regulated by n regulators R1-Rn. The predicted gene expression value  was calculated as:
was calculated as:
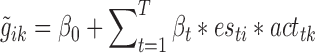 |
(1) |
where, β0 was an additive offset, T the number of all investigated regulators, βt was the optimization parameter for regulator t, esti was the edge strength between regulator t and its putative target gene i and acttk the activity of regulator t in sample k. To model strain specific effects of a regulator, the activity was calculated as:
 |
(2) |
acttk was the estimated effect of regulator t in strain k, esti the edge strength between regulator t and gene i, gik the gene expression of gene i in strain k. Equation (2) defines the activity based on the cumulative effect of a regulator on all its target genes, normalized by the sum of all target genes to balance regulators with high and low numbers of targets. As regulators may be post-transcriptionally regulated, we didn't use the expression values of a regulator to estimate their sample specific effect. Instead, similar to Balwierz et al. (26), we estimated the activity of a regulator in a deletion strain by the differential expression of their putative target genes. The basic idea is that a regulator is more likely to be active in the specific sample if the putative targets are differentially expressed. The edge strength esti was the edge weight between the regulators and the target genes. It was equal to 1 if gene i was reported to be a target of regulator t (known-binding, selected from YEASTRACT), and was zero otherwise.
The objective for the optimization problem was to minimize the difference of the measured transcript level (from the microarrays) and the predicted gene expression 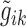 value, i.e. minimizing the error terms eik (L1 regression)
value, i.e. minimizing the error terms eik (L1 regression)
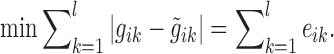 |
(3) |
Because the linear optimizer could not handle absolute values, the absolute values were transformed into two inequalities for each gene i and knockout sample k,
 |
(4) |
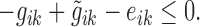 |
(5) |
To solve this optimization problem we used the optimizer Gurobi (www.gurobi.com, version 6.0–6.04). To gain a representative variety of models with different sizes, we constructed models constraining the number of regulators. For each EST gene, models were constructed starting by one regulator up to a maximum of n-2 putative regulators (with known binding), where n was the number of samples. After constraining the number of regulators, the prediction error was minimized by the optimizer [Equation (3)]. To gain an objective estimate for the performance and to circumvent overfitting, we performed cross-validation and resampling (see next section). The prediction performance of our model was estimated by the correlation between the measured (from the validation sets) and the predicted gene expression value (gained from the training sets).
The machine learning approach
A schematic overview of the workflow is given in Figure 1. To predict the transcript levels of each EST gene in the knockout strains which affect telomere length, we divided the dataset into data from knockout strains that showed short telomeres (short tlm mutants), long telomeres (long tlm mutants) and a control dataset showing normal telomere length. We mainly focused on the short telomere phenotype, because telomere elongation is an important hallmark of cancerogenesis. This resulted in a dataset of 18 short tlm mutants and 240 control knockout samples. For each target gene (EST1, EST2, EST3), we performed a ten-times sixfold cross-validation. Explicitly, the algorithm proceeded as follows:
Figure 1.

Schematic overview of the computational workflow. Using the expression profiles of short tlm knockout mutants as well as randomly selected control samples, a cross-validation was performed employing our linear modeling approach (inner loop for parameter optimization not shown). A significance test was performed to identify significant regulators being highly relevant for explaining expression of short tlm knockouts, while being not relevant for the controls.
We randomly selected 120 datasets of all control samples (drawing with replacement).
Datasets of the 18 short tlm mutants as well as of the 120 control samples were randomly divided into six equally sized partitions. Five sixths (15 short tlm samples, 100 controls) were used to train the regulatory model and the remaining sixth was used to validate the predictions.
- The modeling was done separately for the short tlm mutants and the control datasets. To obtain solutions of a large range of model sizes, we generated models of different sizes:
- we started with constraining the models to use only one regulator, i.e. the number of β-parameters was limited to a maximum of two (β0 and one β for the optimal regulator).
- This was repeated increasing the limit by one, now allowing one more β-parameter to be used by the model.
-
Step (b) was repeated until the allowed number of β-parameters reached eleven (for 10 regulators with known binding information to the target gene, plus β0).For each number of regulators we further did a fivefold inner cross-validation, which means the training dataset was divided into fifths and four fifths were used to determine the combination of regulators (training phase of the model) and the remaining one fifth to test the performance of the combination. The combination with the best performance was then used for the remaining steps.Running (a) to (c) yielded two lists of selected regulators, one list for the short tlm mutants, and one list for the controls.
Steps (ii) and (iii) were repeated six times covering all possible partitions to be training sets.
To estimate the performance of the models, we used the validation datasets and calculated the Pearson correlation of the predicted gene expression values (basing on the β-parameters yielded from the training sets) and the real expression values of the validation datasets from all six runs.
Steps (ii) to (v) were one cross-validation, and we repeated these steps ten-times.
For the control samples, steps (i) to (vi) were repeated ten-times to cover their large variety.
For each EST gene, altogether 60 different models were constructed for the short tlm mutants and 600 for the control samples. For each model, we counted how often each regulator was selected by the optimizer. With these distributions we did a one-sided Wilcoxon Test for each regulator in the list to identify significantly different regulators between the short tlm mutants and the control samples. Significant levels (P-values) were corrected for multiple testing using the Benjamini–Hochberg method (27).
Analyzing regulator activity correlation
To identify potential false negatives, we calculated the Pearson correlation between the activities of all pairs of regulators (t, t’) of the EST genes across the investigated samples. This was done separately for the dataset of short tlm mutants and the controls and led to two correlation matrices, one for each dataset, which we called TxT matrices. To get regulators that were active specifically in the short tlm mutants, we subtracted the TxT matrix of the controls from the TxT matrix of the short tlm mutants leading to a differential TxT matrix. All matrices can be found in the Supplementary Material (Supplementary Table S4). Additionally, we also calculated the correlation between the activity of all putative regulators (with known binding information) of each EST gene and the gene expression values of the corresponding EST gene, again separately for the short tlm mutants and the controls to get further hints for regulators which may have been disregarded in the modeling. The regulators were ranked by their correlations for both datasets and also for the correlation differences between the datasets. The results were then compared to the significant regulators obtained by our modeling and to the top entries of the differential TxT matrix to identify additional (potentially false negative) regulators.
Co-regulation
To identify regulators with synergistic effects (complex partners, similar activity values), we simulated regulator knockouts by disregarding the edge between the regulator and the target gene. We then calculated models for the short tlm samples and the control dataset as described above ('The machine learning approach'). This was done exemplarily for Hst1 and Sum1. We further constructed models mimicking cooperative activity as described elsewhere (28) calculating the geometric mean. The activity of the combination Sum1_Hst1 was calculated by
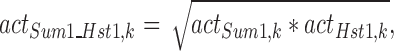 |
(6) |
of knockout sample k.
Experimental validation: EST1 expression analysis
Yeast RNA was extracted as described previously (29). The RNA was DNase I treated for 1 h at 37°C using the RNase-Free DNase Set (QIAGEN). A total of 3 μg RNA of wild-type cells (BY4741 Mat a, BY4742 Mat α) and sum1 mutants of both strains have been reverse transcribed with oligodT12–18 (12.5 ng/μl) following the protocol of the Superscript III RT Kit (Invitrogen). Respective minus-RT controls were used to assess the DNA backgrounds. The cDNA was diluted 2.5 times in H2O and analyzed by qPCR with the DyNAmo Flash SYBR Green qPCR Kit (Thermo Scientific) in technical triplicates: 10 min at 95°C, 35 cycles of 15 s at 95°C and 1 min at 60°C. qPCR-primers have been used at a final concentration of 500 nM (EST1-FWD: GCT GCC ACA ATG GGA AGT TTC G; EST1-REV: TGC CAG GAG GGT TTG ATG ACG; ACTIN-FWD: CCC AGG TAT TGC CGA AAG AAT GC; ACTIN-REV: TTT GTT GGA AGG TAG TCA AAG AAG CC). The EST1 values were normalized to ACTIN expression (ΔCt method). The expression (2−delta Ct) of EST1 in sum1 mutants relative to the respective wild-type strain (Mat a/α) is displayed for three biological replicates (Figure 2). Two-tailed unpaired t-tests with Welch's correction were performed to test for significant differences.
Figure 2.

Expression of EST1 in sum1 deletion strains of both mating-types (left: mating-type a; right: mating-type α) compared to the wild-type (measured by RT-qPCR). The expression values are given relative to the control (actin). The error bars indicate the standard error (SEM) over the three replicates. Two-tailed unpaired t-tests with Welch's correction were performed to test for significant differences.
RESULTS
Rationale
The TLM network represents a potentially useful target for anticancer therapy, as telomere length maintenance is a pre-requisite for the constant growth of cancer cells. In particular, we were interested in transcriptional regulators that lead to short telomeres when deleted (abbreviated as ‘short tlm mutants’ in the following). These mutants are defective for positive regulators of telomere elongation and have a high chance to modulate the expression of the EST genes, which encode the telomerase complex. Thus, we investigated regulator knockout strains which were short tlm mutants and analyzed their effects on the expression of the EST genes. As samples for the modeling we used yeast deletion strains of transcription factors or chromatin modifiers. These deletion strains were then divided into a short tlm dataset and a control dataset (and later also a long tlm dataset was used, see below). The sample sizes and their overlaps between the TLM genes, the regulator deletion strains and the putative regulators are shown in a Venn diagram (Supplementary Figure S2). The workflow for our study is depicted in Figure 1. We assumed that TLM genes that, when mutated, result in short telomeres, may participate in a transcriptional network culminating in the expression changes of the EST telomerase components. To identify specific regulators best explaining Est transcript levels in the short tlm knockout strains, we constructed linear models for each EST gene. The task for the linear models was to predict the gene expression of each EST gene in each sample using activity information (for each specific sample) of putative regulators. Putative regulators were selected from a database according to experimental evidence of the regulator's binding to the promoter of the EST gene (listed in Supplementary Table S2). We note that the calculated sample specific activity of a regulator is based on the cumulative differential expression of the regulator in the according sample. The models were optimized to select regulators which led to the best prediction of EST gene expression in each sample. This procedure was done twice, i.e. for the expression data of the short tlm knockout mutants, and for the expression data of the controls. We calculated models of different complexity for each dataset (short tlm knockout strains and the controls) employing cross-validation. From all these different runs, we counted how often each regulator was selected by the optimized models. This yielded two lists of regulator counts, one list for the short tlm mutants and one for the controls. To find regulators specifically explaining the regulation of EST genes in the short tlm knockout strains, the frequency of each regulator (Supplementary Figure S3) was compared between the short tlm mutants and the models for the controls. Performing a test of significance for each putative regulator led to the identification of regulators that specifically affect EST expression in the short tlm knockout mutants.
Investigating the predictions of the model
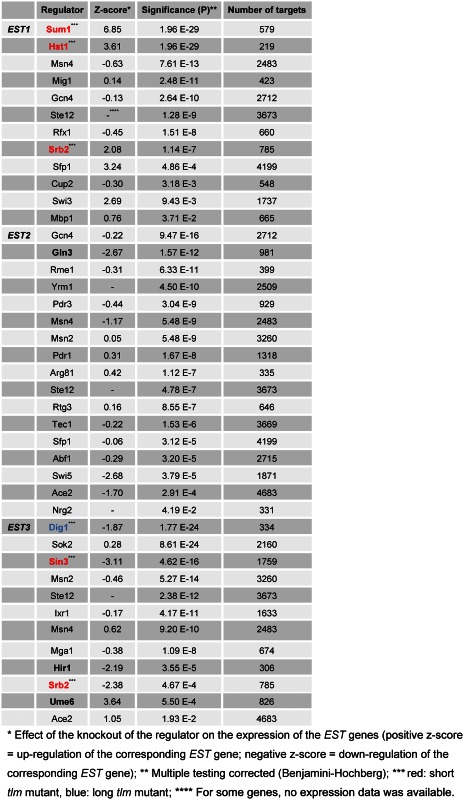
Running our models as described above yielded 32 significant regulators for Est1, Est2 and Est3, respectively (Table 1). The models were learned on training data. To estimate their performances, we used independent validation data for which we calculated the correlation of the modeled predicted expression values and the experimental expression values. For the models predicting Est1 regulators, we got a good overall Pearson correlation coefficient (PCC) of r = 0.51, for Est2 and Est3 the performance was lower (PCC r = 0.30 and r = 0.12, respectively). All correlations were highly significant (for all: P < 2.2 E-16; more details on performance estimates and model statistics, see Text S1 in the Supplementary Material). In the following, we focus on predicted regulators with a high impact on the expression of the predicted EST targets, i.e. we selected predicted regulators with less than 1000 putative targets and a strong knockout effect (absolute z-score > 1) or regulators which are TLM genes. For each EST gene, the main regulators detected are marked in bold in Table 1 (expression values are given by z-scores, also in the following). A high value of expression indicates an upregulation of the EST gene when the regulator was knocked out. This suggests an inhibitory effect of the regulator. Downregulation, in contrast, suggests an activating effect of the regulator. Interestingly, several of the regulators identified in our study are by themselves TLM genes, and when mutated affect telomere length. Regulators that cause short telomeres when mutated (8,10) were Sum1, Hst1, Srb2 and Sin3 and are marked in red in Table 1. One of the regulators of EST3 (Dig1, blue in Table 1), causes telomere elongation when deleted (10). To investigate if aberrant telomere length in general is putatively due to transcriptional regulation, we also investigated our models feeding them with expression data of short and long tlm mutants. Again, we compared the results to the control samples. Consistently, we yielded quite similar results as using the data of only the short tlm mutants (see Supplementary Table S3).
Table 1. Significant regulators of EST genes.
|
Regarding the results of the short tlm mutants, for EST1, we found Sum1 (P = 1.96 E-29), Hst1 (P = 1.96 E-29) and Srb2 (P = 1.14 E-7) to be highly significantly enriched in our predictions. For the predicted regulators, we investigated the literature (Pubmed, www.ncbi.org) in the context of ‘telomere’, ‘telomerase’ and each of the EST gene symbols. Sum1 is a general chromatin silencing factor, as well as an initiation factor of replication. Furthermore, it is involved in the regulation of middle-sporulation genes. Sum1 builds a complex with the sirtuin protein Hst1 and the protein Rfm1, repressing genes through histone deacetylation (30–33). The sirtuin family members Sir2 and Hst1 have been reported to show similarity with Sum1 in telomere maintenance because of their specific co-enriched binding sites and their interaction with Rap1, a protein binding at the telomeric repeat regions (30–33). Srb2 is a subunit of the RNA polymerase II mediator complex. It is either directly involved in TLC1 transcription or indirectly in TLC1 accumulation (34). In the data we analyzed, Sum1 influenced the regulation of EST1 most strikingly: for the sum1 knockout strain, EST1 showed the highest expression level among all knockout mutants investigated (z-score = 6.85, log-fold change = 0.93, see also Supplementary Figure S4a). EST1 expression was also distinctively upregulated in the hst1 (z-score = 3.61) and srb2 knockout mutants (z-score = 2.08). In summary, Sum1, Hst1 and Srb2 are very likely to regulate EST1 by inhibition. This result is unexpected, as strains deleted for SUM1, HST1 or SRB2 exhibit short telomeres (see ‘Discussion’ section). We experimentally validated this new finding exemplarily with Sum1 (see next section). Gln3 is the only significant regulator of EST2 fulfilling the z-score and the target number criteria. Gln3 is involved in Tor Complex 1 regulated telomere shortening upon starvation by controlling the level of the Ku heterodimer (35). For EST3, the regulators Ume6, Sin3, Srb2, Hir1 and Dig1 were highly significant. Sin3 interacts with Rpd3 and Rpd2 to form histone deacetylase complexes. It is involved in the transcriptional repression and activation of diverse processes (36). Sin3 plays a role in silencing, repair of DNA double-strand breaks, and telomere length maintenance. In the complex with Rpd3, Sin3 affects silencing at the telomeres (36). Ume6 is another interactor of Rpd3 and a key regulator of early meiotic genes. It is involved in chromatin remodeling and in the recruitment of Sin3 and Rpd3 subunits of the histone deacetylase complex (37). Thus, our approach identified two different proteins of the Rpd3-based histone deacetylase complexes. In summary, our machine learning based linear modeling predictions identified mainly regulators involved in the regulation of chromatin and histone modifications.
Experimental validation of the predicted effect of Sum1 on EST1 expression
Because the sum1 deletion strain showed the highest effect on EST1 expression (Supplementary Figure S4a) and was a distinctively significant hit of our predictions, we investigated gene expression of EST1 in the wild-type and the sum1 mutant. Because Sum1 is involved in mating-type regulation (38), the expression of EST1 was measured for sum1 mutants of both mating types, Mat a and Mat α, by RT-qPCR. The results are shown in Figure 2. For both sum1 mutants, EST1 was highly upregulated (4.37-fold ± 0.67 SEM for Mat a and 6.00-fold ± 0.48 SEM for Mat α). This high upregulation of EST1 in the sum1 mutants observed by individual PCR is comparable to our observation of the investigated microarray gene expression data (15), which we used for our modeling analysis. In summary, we could show that the sum1 knockout has a strong repressive effect on the expression of EST1.
Correlation analysis between regulator activities and EST expression
If the activity of a regulator was very similar to the activity of another regulator in each of the investigated samples, the model may have difficulties distinguishing between them and may neglect one of these regulators causing false negatives. To identify such potential false negatives, we calculated the correlation between all regulator activities (Supplementary Table S4) as well as between the regulator activities and the expression of the EST genes (Supplementary Tables S5–S7). This was done separately for the short tlm mutants and the controls, and the regulators with the largest differences were selected to obtain short tlm mutant specific regulators (Supplementary Table S8 shows the selected candidates, Supplementary Table S4 contains the correlations of activities of all potential pairs of regulators of short tlm mutants, controls and the differences, for details of this analysis, see Text S2 in the Supplementary Material). For EST1 and EST2 no further regulators were found. Interestingly, we found good correlation of Sum1 and Hst1 suggesting a cooperativity which was supported by a model for this (details, see Supplementary Text S3). For EST3, we found Gln3 as a potential further regulator with similar activity and expression correlation as Dig1 (Supplementary Tables S7 and S8).
Software implementation and availability
The method is implemented within the software package MIPRIP for R (www.r-project.org). It is freely available at http://www.leibniz-hki.de/en/miprip.html. MIPRIP is platform independent and runs on R version 3.1.2 together with RStudio version 0.98.1103 and Gurobi version 6.0.4. Instead of solving the Mixed Integer Linear Models directly, all analyses were implemented in R using the Gurobi R API. MIPRIP uses the standard CRAN R package slam.
DISCUSSION
Telomere length needs to be precisely controlled during embryogenesis and for cancer cell immortalization. Central for this well balanced control is the transcriptional regulation of the telomerase, a protein complex encoded in S. cerevisiae by the three EST genes and TLC1. S. cerevisiae is a well suited model organism to study telomere biology of cancer cells as more than a quarter of all yeast genes have human homologs and the telomerase is constitutively expressed and functional in yeast (3). The candidate regulators we selected mainly based on ChIP-binding information. Hence, the regulators of the EST genes we used for the modeling are able to bind to the promoters of the target genes. To identify regulators best explaining the expression of the EST genes we used our previously developed linear modeling approach based on MILP (13). This method was embedded into a machine learning procedure to identify regulators that specifically regulate telomerase. Models of knockout strains showing short telomeres were compared to controls, hypothesizing that the absence of these regulators may directly influence telomerase expression and hence reduce telomerase activity. Although telomerase is regulated at various post-transcriptional levels and by many signaling processes, our aim was to find novel transcriptional regulators of telomerase. Hence, a hidden assumption of our approach was that transcription levels of the EST genes could be a limiting factor in telomere maintenance and we investigated EST expression of strains with knocked out regulators which by themselves are TLM genes that show abnormal telomere length when deleted. For EST1, our most prominent hits were Sum1 and Hst1. Sum1 is a general chromatin silencing factor. In a complex with Hst1 and Rfm1, it represses gene expression through histone deacetylation at their promoters. In the context of telomeres, Sum1 is known to be involved in telomere maintenance through an interaction with Rap1, similarly to sirtuins Sir2 and Hst1 (30). However, a role for Sum1 in directly regulating the expression of telomerase genes has not been reported so far. Furthermore, we identified Srb2 to be highly significant for EST1 regulation. In addition, Gln3 is a significant regulator of EST2 and Sin3, Dig1, Srb2, Hir1 and Ume6 are regulators of EST3. Similarly, no previous reports linked these regulators to the regulation of telomerase expression. EST1 expression was highly upregulated in the sum1, hst1 and srb2 knockout strains (Supplementary Figure S4a); EST2 was highly downregulated in the gln3 mutant (Supplementary Figure S4b) and EST3 was highly upregulated in the ume6 mutant and downregulated in sin3, srb2, dig1 and hir1 (Supplementary Figure S4c). The different effects of deleting UME6 and SIN3, two genes that in meiosis usually act together to repress expression of meiotic genes (39) is surprising, although not unseen: previous work has shown that the repressive effect of Ume6 can be switched, so it can act as a positive regulator (40,41). Our results suggest that Sum1 and Hst1 may act in combination in EST1 regulation, consistent with the idea that they do so as part of the histone deacetylase complex they form together with Rfm1 (30–33). It was shown elsewhere that Rfm1 tethers Hst1 and Sum1 (32), hence it is likely that Rfm1 may be indirectly involved in EST regulation. Our quantitative RT-PCR results confirmed the upregulation of EST1 in a sum1 mutant, in line with the investigated microarray data from Reimand et al. (15). This implies a role for Sum1 in negatively regulating EST1. However, contrary to the expectations, deletion of SUM1 leads to a short telomere phenotype. We therefore speculated that high EST1 expression may have a negative effect on telomere length. We investigated EST1 expression in all 18 short tlm regulator knockouts and compared it with the non-TLM controls. Indeed, we found a significant difference (P = 0.047, Student's t-test): for the short tlm mutants the average expression of EST1 was z-score = 0.78, while it was z-score = -0.056 for the control group. Thus, EST1 seems to be upregulated in the short tlms. We speculate that excessive EST1 may cause an imbalance between the subunits of the telomerase holoenzyme, limiting telomerase activity, for example by titration of factors important for telomerase activity, such as the RNA template Tlc1 or the recruiting factor Cdc13. Alternatively, it may bind to telomeres (42) and compete with functional complexes. Such a mechanism suggests a further role of Sum1 and Hst1 in telomere maintenance besides their role in Rap1 dependent telomeric recruitment (30).
Although to date the identified regulators had not been shown to regulate the telomerase complex directly on a transcriptional level, some of the regulators we found were reported to be involved in telomere maintenance (by e.g. chromatin remodeling). Thus, we hypothesize the existence of feed-forward loops strengthening the regulatory signal. Such feed-forward loops have been intensively investigated for cellular networks, as a way to improve signal to noise ratios as they respond to rather persistent signals (43–46). We suggest that Sum1 together with Hst1 and EST1 form an incoherent feed-forward loop regulating telomere length, where Sum1/Hst1 and EST1 positively regulate telomeres and Sum1/Hst1 negatively regulates EST1 (Supplementary Figure S5) whose over expression may compromise normal telomere elongation activity.
In summary, we embedded our novel concept of linear regulation models based on MILP into a useful machine learning strategy which enabled us to identify novel regulators of the telomerase holoenzyme, where Sum1 is the most promising regulator of EST1.
Supplementary Material
Acknowledgments
We thank Karsten Rippe for fruitful discussion about telomere maintenance mechanisms in cancer.
Disclaimer: The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
FUNDING
German ministry for education and research (Bundesministerium für Bildung und Forschung, BMBF); CSCC/IFB [01EO1002, 01EO1502]; eBio/SYSMETBC [0316168D]; CancerSys/MYCNET [0316076C]; eMed/CancerTelSys [01ZX1302B]; Cooperation Program in Cancer Research of the German Cancer Research Center (Deutsches Krebsforschungszentrum (DKFZ)) and Israel's Ministry of Science, Technology and Space (MOST); Israel Cancer Association and the Israel Cancer Research Fund (to M.K.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Hug N., Lingner J. Telomere length homeostasis. Chromosoma. 2006;115:413–425. doi: 10.1007/s00412-006-0067-3. [DOI] [PubMed] [Google Scholar]
- 2.Kupiec M. Biology of telomeres: lessons from budding yeast. FEMS Microbiol. Rev. 2014;38:144–171. doi: 10.1111/1574-6976.12054. [DOI] [PubMed] [Google Scholar]
- 3.Teixeira M.T. Saccharomyces cerevisiae as a model to study replicative senescence triggered by telomere shortening. Front. Oncol. 2013;3:101. doi: 10.3389/fonc.2013.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taggart A.K., Zakian V.A. Telomerase: what are the Est proteins doing. Curr. Opin. Cell Biol. 2003;15:275–280. doi: 10.1016/s0955-0674(03)00040-1. [DOI] [PubMed] [Google Scholar]
- 5.Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 6.Breslow D.K., Cameron D.M., Collins S.R., Schuldiner M., Stewart-Ornstein J., Newman H.W., Braun S., Madhani H.D., Krogan N.J., Weissman J.S. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Aroya S., Coombes C., Kwok T., O'Donnell K.A., Boeke J.D., Hieter P. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol. Cell. 2008;30:248–258. doi: 10.1016/j.molcel.2008.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Askree S.H., Yehuda T., Smolikov S., Gurevich R., Hawk J., Coker C., Krauskopf A., Kupiec M., McEachern M.J. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8658–8663. doi: 10.1073/pnas.0401263101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-Shitrit T., Yosef N., Shemesh K., Sharan R., Ruppin E., Kupiec M. Systematic identification of gene annotation errors in the widely used yeast mutation collections. Nat. Methods. 2012;9:373–378. doi: 10.1038/nmeth.1890. [DOI] [PubMed] [Google Scholar]
- 10.Gatbonton T., Imbesi M., Nelson M., Akey J.M., Ruderfer D.M., Kruglyak L., Simon J.A., Bedalov A. Telomere length as a quantitative trait: genome-wide survey and genetic mapping of telomere length-control genes in yeast. PLoS Genet. 2006;2:e35. doi: 10.1371/journal.pgen.0020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shachar R., Ungar L., Kupiec M., Ruppin E., Sharan R. A systems-level approach to mapping the telomere length maintenance gene circuitry. Mol. Syst. Biol. 2008;4:172. doi: 10.1038/msb.2008.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ungar L., Yosef N., Sela Y., Sharan R., Ruppin E., Kupiec M. A genome-wide screen for essential yeast genes that affect telomere length maintenance. Nucleic Acids Res. 2009;37:3840–3849. doi: 10.1093/nar/gkp259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schacht T., Oswald M., Eils R., Eichmuller S.B., Konig R. Estimating the activity of transcription factors by the effect on their target genes. Bioinformatics. 2014;30:i401–i407. doi: 10.1093/bioinformatics/btu446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Z., Killion P.J., Iyer V.R. Genetic reconstruction of a functional transcriptional regulatory network. Nat. Genet. 2007;39:683–687. doi: 10.1038/ng2012. [DOI] [PubMed] [Google Scholar]
- 15.Reimand J., Vaquerizas J.M., Todd A.E., Vilo J., Luscombe N.M. Comprehensive reanalysis of transcription factor knockout expression data in Saccharomyces cerevisiae reveals many new targets. Nucleic Acids Res. 2010;38:4768–4777. doi: 10.1093/nar/gkq232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu H., Gerstein M. Genomic analysis of the hierarchical structure of regulatory networks. Proc. Natl. Acad. Sci. U.S.A. 2006;103:14724–14731. doi: 10.1073/pnas.0508637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huber W., von Heydebreck A., Sultmann H., Poustka A., Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18(Suppl. 1):S96–S104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- 18.Harbison C.T., Gordon D.B., Lee T.I., Rinaldi N.J., Macisaac K.D., Danford T.W., Hannett N.M., Tagne J.B., Reynolds D.B., Yoo J., et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., Hannett N.M., Harbison C.T., Thompson C.M., Simon I., et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 20.Bauer T., Eils R., Konig R. RIP: the regulatory interaction predictor–a machine learning-based approach for predicting target genes of transcription factors. Bioinformatics. 2011;27:2239–2247. doi: 10.1093/bioinformatics/btr366. [DOI] [PubMed] [Google Scholar]
- 21.Cheng C., Alexander R., Min R., Leng J., Yip K.Y., Rozowsky J., Yan K.K., Dong X., Djebali S., Ruan Y., et al. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res. 2012;22:1658–1667. doi: 10.1101/gr.136838.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Consortium F., Suzuki H., Forrest A.R., van Nimwegen E., Daub C.O., Balwierz P.J., Irvine K.M., Lassmann T., Ravasi T., Hasegawa Y., et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet. 2009;41:553–562. doi: 10.1038/ng.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong X., Greven M.C., Kundaje A., Djebali S., Brown J.B., Cheng C., Gingeras T.R., Gerstein M., Guigo R., Birney E., et al. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol. 2012;13:R53. doi: 10.1186/gb-2012-13-9-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliveira A.P., Patil K.R., Nielsen J. Architecture of transcriptional regulatory circuits is knitted over the topology of bio-molecular interaction networks. BMC Syst. Biol. 2008;2:17. doi: 10.1186/1752-0509-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Setty M., Helmy K., Khan A.A., Silber J., Arvey A., Neezen F., Agius P., Huse J.T., Holland E.C., Leslie C.S. Inferring transcriptional and microRNA-mediated regulatory programs in glioblastoma. Mol. Syst. Biol. 2012;8:605. doi: 10.1038/msb.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balwierz P.J., Pachkov M., Arnold P., Gruber A.J., Zavolan M., van Nimwegen E. ISMARA: automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res. 2014;24:869–884. doi: 10.1101/gr.169508.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 28.Lai X., Schmitz U., Gupta S.K., Bhattacharya A., Kunz M., Wolkenhauer O., Vera J. Computational analysis of target hub gene repression regulated by multiple and cooperative miRNAs. Nucleic Acids Res. 2012;40:8818–8834. doi: 10.1093/nar/gks657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luke B., Panza A., Redon S., Iglesias N., Li Z., Lingner J. The Rat1p 5′ to 3′ exonuclease degrades telomeric repeat-containing RNA and promotes telomere elongation in Saccharomyces cerevisiae. Mol. Cell. 2008;32:465–477. doi: 10.1016/j.molcel.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 30.Li M., Valsakumar V., Poorey K., Bekiranov S., Smith J.S. Genome-wide analysis of functional sirtuin chromatin targets in yeast. Genome Biol. 2013;14:R48. doi: 10.1186/gb-2013-14-5-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bedalov A., Hirao M., Posakony J., Nelson M., Simon J.A. NAD+-dependent deacetylase Hst1p controls biosynthesis and cellular NAD+ levels in Saccharomyces cerevisiae. Mol. Cell. Biol. 2003;23:7044–7054. doi: 10.1128/MCB.23.19.7044-7054.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCord R., Pierce M., Xie J., Wonkatal S., Mickel C., Vershon A.K. Rfm1, a novel tethering factor required to recruit the Hst1 histone deacetylase for repression of middle sporulation genes. Mol. Cell. Biol. 2003;23:2009–2016. doi: 10.1128/MCB.23.6.2009-2016.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zill O.A., Rine J. Interspecies variation reveals a conserved repressor of alpha-specific genes in Saccharomyces yeasts. Genes Dev. 2008;22:1704–1716. doi: 10.1101/gad.1640008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mozdy A.D., Podell E.R., Cech T.R. Multiple yeast genes, including Paf1 complex genes, affect telomere length via telomerase RNA abundance. Mol. Cell. Biol. 2008;28:4152–4161. doi: 10.1128/MCB.00512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ungar L., Harari Y., Toren A., Kupiec M. Tor complex 1 controls telomere length by affecting the level of Ku. Curr. Biol. 2011;21:2115–2120. doi: 10.1016/j.cub.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 36.Sun Z.W., Hampsey M. A general requirement for the Sin3-Rpd3 histone deacetylase complex in regulating silencing in Saccharomyces cerevisiae. Genetics. 1999;152:921–932. doi: 10.1093/genetics/152.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kadosh D., Struhl K. Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell. 1997;89:365–371. doi: 10.1016/s0092-8674(00)80217-2. [DOI] [PubMed] [Google Scholar]
- 38.Chi M.H., Shore D. SUM1–1, a dominant suppressor of SIR mutations in Saccharomyces cerevisiae, increases transcriptional silencing at telomeres and HM mating-type loci and decreases chromosome stability. Mol. Cell. Biol. 1996;16:4281–4294. doi: 10.1128/mcb.16.8.4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lardenois A., Stuparevic I., Liu Y., Law M.J., Becker E., Smagulova F., Waern K., Guilleux M.H., Horecka J., Chu A., et al. The conserved histone deacetylase Rpd3 and its DNA binding subunit Ume6 control dynamic transcript architecture during mitotic growth and meiotic development. Nucleic Acids Res. 2015;43:115–128. doi: 10.1093/nar/gku1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubin-Bejerano I., Mandel S., Robzyk K., Kassir Y. Induction of meiosis in Saccharomyces cerevisiae depends on conversion of the transcriptional represssor Ume6 to a positive regulator by its regulated association with the transcriptional activator Ime1. Mol. Cell. Biol. 1996;16:2518–2526. doi: 10.1128/mcb.16.5.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Washburn B.K., Esposito R.E. Identification of the Sin3-binding site in Ume6 defines a two-step process for conversion of Ume6 from a transcriptional repressor to an activator in yeast. Mol. Cell. Biol. 2001;21:2057–2069. doi: 10.1128/MCB.21.6.2057-2069.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Virta-Pearlman V., Morris D.K., Lundblad V. Est1 has the properties of a single-stranded telomere end-binding protein. Genes Dev. 1996;10:3094–3104. doi: 10.1101/gad.10.24.3094. [DOI] [PubMed] [Google Scholar]
- 43.Fu W., Ergun A., Lu T., Hill J.A., Haxhinasto S., Fassett M.S., Gazit R., Adoro S., Glimcher L., Chan S., et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat. Immunol. 2012;13:972–980. doi: 10.1038/ni.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mangan S., Alon U. Structure and function of the feed-forward loop network motif. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mangan S., Itzkovitz S., Zaslaver A., Alon U. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J. Mol. Biol. 2006;356:1073–1081. doi: 10.1016/j.jmb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Mangan S., Zaslaver A., Alon U. The coherent feedforward loop serves as a sign-sensitive delay element in transcription networks. J. Mol. Biol. 2003;334:197–204. doi: 10.1016/j.jmb.2003.09.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.