

Abstract
PrimPol is a DNA damage tolerant polymerase displaying both translesion synthesis (TLS) and (re)-priming properties. This led us to study the consequences of a PrimPol deficiency in tolerating mutagenic lesions induced by members of the APOBEC/AID family of cytosine deaminases. Interestingly, during somatic hypermutation, PrimPol counteracts the generation of C>G transversions on the leading strand. Independently, mutation analyses in human invasive breast cancer confirmed a pro-mutagenic activity of APOBEC3B and revealed a genome-wide anti-mutagenic activity of PRIMPOL as well as most Y-family TLS polymerases. PRIMPOL especially prevents APOBEC3B targeted cytosine mutations within TpC dinucleotides. As C transversions induced by APOBEC/AID family members depend on the formation of AP-sites, we propose that PrimPol reprimes preferentially downstream of AP-sites on the leading strand, to prohibit error-prone TLS and simultaneously stimulate error-free homology directed repair. These in vivo studies are the first demonstrating a critical anti-mutagenic activity of PrimPol in genome maintenance.
INTRODUCTION
DNA damage tolerance (DDT) is an integral part of the DNA damage response network that maintains the integrity of the genome (1–4). DDT enables replication to continue in the presence of a fork-stalling lesion. Principally, four distinct DDT pathways could be distinguished, (i) direct translesion synthesis (TLS) across the damaged template (‘on the flight’), (ii) repriming behind the damaged template followed by gap filling TLS (post-replicative TLS), (iii) direct template switching (TS), taking advantage of the intact template of the sister chromatid or (iv) repriming behind the damaged template, where the remaining gap is restored by homology directed repair. TLS enables direct replicative bypass of lesions that otherwise stall the replicative DNA polymerases. It involves a set of specialized proofread inactive TLS polymerases that can accommodate non-Watson/Crick base pairs in their catalytic centre. This capacity can lead to misinsertion errors and renders TLS potentially mutagenic. In contrast, TS avoids the damage by taking advantage of the intact template on the sister chromatid and therefore is relatively error-free. Repriming downstream of the lesion is another elegant way to relief acute replication stress. Common to all DDT pathways, they enable replication progression in the presence of otherwise replication blocking lesions and in this way contribute to genome stability by prevention replication fork collapses (1–4).
The capacity to tolerate lesions in the DNA template is essential for programmed mutation pathways that operate in higher eukaryotes. These intentional mutation processes are initiated by cytosine (C) deaminases of the APOBEC/AID family. Members of this family deaminate C to uracil (U) in RNA and single-stranded DNA. Replication over U generates C>T transitions. Alternatively, uracil glycosylases can process Us into non-instructive AP-sites, which upon TLS can give rise to both C transitions (C>T) and transversions (C>G, C>A). In the innate immune system cytosine deamination by APOBEC3A and APOBEC3B can mutagenize and inactivate retro-elements. In the adaptive antibody-dependent immune system, cytosine deamination by the Activation-Induced cytidine Deaminase (AID) induces somatic hypermutation (SHM) and class switch recombination (CSR) of Ig genes, enabling the generation of high affinity antibody variants (SHM) with new effector functions and tissue distribution (CSR).
SHM is triggered in antigen activated, centroblastic B cells of the germinal center, where the expression of AID becomes transiently induced (5–9). SHM is confined to the hypermutation domain, the transcribed regions encompassing the VDJ and VJ exon of rearranged Ig heavy and light chain genes, respectively (7–9). Mutations that increase the affinity between the clonotypic surface Ig and cognate antigen provide a selective advantage to the B cell. As a consequence high affinity antibodies dominate immune recall responses, a phenomenon known as antibody affinity maturation.
CSR is a programmed recombination process between two active switch regions of the Igh locus that enables antigen activated B cells to switch their IgH isotype and hence change the antibody effector function and tissue distribution (10,11).
Other members of the APOBEC/AID family have been implicated in establishing innate immunity, specifically by controlling retro-elements. For example, APOBEC3A and APOBEC3B can inactivate retroviruses by inducing predominantly G/C>A/T transitions in vitro as well in vivo (12). Aberrant targeting of APOBEC3B has been linked to active genome wide mutagenesis as well as kataegis in various cancers (13,14). Especially, breast cancer, cervical cancer, bladder cancer, lung squamous cell carcinoma, lung adenocarcinoma, and head and neck cancer are characterized by a high mutation load of cytosines at TpC dinucleotides, the preferred target sequence of APOBEC3A/B. In breast cancer specifically APOBEC3B has been identified as highly mutagenic (14–22).
PrimPol has recently been identified as a novel and unique DNA polymerase, which in vitro displays both primase and TLS activity (23–26). In vitro, it has been demonstrated that PrimPol cannot incorporate nucleotides across an AP site but potentially can reprime downstream of a replication-blocking lesion under nuclear like conditions. Alternatively, in the presence of high manganese ion concentration, a condition more closely resembling mitochondria, PrimPol can generate single nucleotide deletion in the presence of a template AP-site (25). In addition, like most DNA polymerases, PrimPol incorporates an A opposite a template U (23,26). Given the contribution of TLS polymerases in establishing somatic mutations at non-instructive AP-sites, we wondered whether PrimPol modulates APOBEC/AID-induced mutagenesis. This study focuses in particular on the in vivo role of PrimPol in establishing the characteristic somatic mutation spectra downstream of APOBEC/AID family member induced AP-sites in murine SHM and human invasive breast cancer. Our data reveal a strand-biased anti-mutagenic activity of PrimPol, where PrimPol reprimes efficiently downstream of AP-sites on the leading strand, thereby contributing to replication fork progression and simultaneously preventing error-prone TLS.
MATERIALS AND METHODS
Generation of PrimPol mouse
Zygotes isolated from C57BL/6N mice were co-injected with in vitro transcribed CRISPR/Cas9 mRNA and sgRNA (sequence TTAACAAATTGGCCAACCC-AGG, designed using the crispr.mit.edu tool) from pX330 plasmid (27). This sgRNA targets CRISPR/Cas9 to exon 5 of the PrimPol locus (ENSMUSE00001229433). Offspring was tested by PCR for PrimPol inactivating mutations using the primers (FW: GTC GGA CAA CCT AGG TTC AGT GC, Rev: CCC TGA ATT TCA TCT CAT TGT CTA C) and ABI® sequencing (3730 DNA analyzer, Applied Biosystems). A PrimPol mutant, carrying a 4-bp CCAA deletion in exon 5 was selected, backcrossed once onto C57BL/6J and maintained heterozygous. For genotyping the wild-type and mutant alleles are distinguished by MscI digestion, where the non-digested product is indicative for a mutant allele. Experiments were approved by an independent animal ethics committee of the Netherlands Cancer Institute (Amsterdam, Netherlands) (DEC number 13007) and executed according national and European guidelines.
cDNA synthesis and qPCR
Total RNA was isolated from wild-type and PrimPolΔ/Δ mouse embryonic fibroblasts (MEFs) using RNeasy mini (Qiagen). cDNA library was synthesized by Invitrogen Superscript III kit and oligo dT primers. A nested PCR was performed on the cDNA spanning exon 1–15 (fwd GCT CTG GTT CCC GCC ATC TCT, rev CTT TCT CTC CAG GCT CTG GGA CA) followed by a nested primer set spanning exon 3–14 (fwd TGG CCA AGC CAG AAG AAC CAT CCT, rev CGT CAT CCC AGG CAG CGG CA). Subsequently, the cDNA was cut using MscI enzyme (Thermo Scientific), verifing the CCAA deletion. The deletion removed the MscI site in the wild-type allele.
For qPCR total RNA was isolated from wild-type and PrimPolΔ/Δ MEFs using RNeasy mini (Qiagen). cDNA library was synthesized using Invitrogen Superscript III kit and random hexamer oligos. qPCR was performed with Fast SYBR® Green Master Mix (Thermo Scientific) and the LightCycler 480II (Roche). Normalization was performed to GAPDH (fwd: CAA TGA CCC CTT CAT TGA CC, rev: GAT CTC GCT CCT GGA AGA TG). PrimPol mRNA was amplified with primers specific for the exon-junction 4–5 (fwd: GAG TGC AAA AGG GGA AAT GG, rev: ATA ACT TCA TAG CAG TGC AAG AG) and the exon-junction 8–9 (fwd: CTA TCT TCC CTG GTG AGC AAT, rev: CTG AAG TGC CAG TAC TGT TAA A).
Derivation of primary pre-B cell cultures and mouse embryonic fibroblast cell lines
E14.5 embryos were isolated from intercrosses of heterozygous PrimPol mice. Single cell suspensions were generated from fetal livers and subsequently cultured on ST2 feeder cells in IL7-containing complete medium (Iscoves, 8% FCS and penicillin/streptomycin) for the generation of pre-B cell cultures. MEFs were isolated using Trypsin and cell strainers, according to (28,29). Primary MEF cultures were established under low oxygen (3%) conditions. MEFs were immortalized (2 per genotype) using lentiviral expression of a p53-specific shRNA (30). Immortalized MEFs were grown in complete IMDM medium and incubated at 37°C in 5% CO2 and normal oxygen levels.
Flow cytometry
Lymphoid cells from bone marrow, thymus and spleen were isolated from 8-week old mice. To reveal T cell progenitor subsets in the thymus, single cell suspensions were stained with conjugated antibodies, specific for CD3-FITC (Biolegend), CD4-APC, CD8a-PerCp-Cy5.5, CD25-PE, CD44-APCCy7 (Biolegend), TCRβ-Pacific Blue (Biolegend). To analyse lymphoid subsets in the spleen, single cell suspensions were stained with CD3-FITC, CD4-APC, CD8a-PerCp-Cy5.5, CD19-APCH7, CD45R(B220)-PacificBlue, IgD-PE (eBioscience), IgM-PECy7 (eBioscience). To define the B cell progenitor subsets, suspensions from the bone marrow were stained with IgD-FITC, CD25-PE, IgM-PECy7 (eBioscience), CD45R (B220)-PacificBlue, CD117 (cKit)-APC (eBioscience), CD19-APCH7. Dead cells were excluded from the analysis by propidium iodide staining. Antibodies were purchased from BD Pharmingen unless mentioned otherwise. Samples were measured on a FACS Fortessa® and analysed using FlowJo® software (Version: 10.0.8r1).
Class switch recombination
Single cells suspensions were prepared from the spleens of 8-week old PrimPolΔ/Δ and their wild-type littermates. After the lysis of red blood cells, naïve splenic B cells were enriched by CD43 depletion using biotinylated anti-CD43 (Clone S7, BD Biosciences), and the IMag® system (BD Biosciences), as described by the manufacturer. Purified B cells were labelled for 10 min at 37°C with 5 μM carboxy fluorescein diacetate succinimidyl ester (CFSE, Molecular Probes) in IMDM medium containing 2% FCS. After washing, cells were cultured in IMDM, supplemented with 8% FCS, 50 μM 2-mercapthoethanol, penicillin/streptomycin at a density of 105 cells/well in 24 well plates. CSR to IgG3 and IgG1 was induced by exposure to LPS (50 μg/ml Escherichia Coli LPS, 055:B5, Sigma) or LPS/rIL-4 (rIL4 20 ng/ml). Four days later, the proliferative capacity was determined by CFSE dilution; CSR frequencies were determined by staining with CD19-PercpCy5.5 (BD), IgM-APC, and IgG3-PE (LPS cultures) or IgG1-PE (LPS/rIL4 cultures). IgM-APC, IgG1-PE and IgG3-PE were purchased from Southern Biotech. Data were acquired by flow cytometry (Fortessa®, BD) and analysed using FlowJo software (Version: 10.0.8r1).
Somatic hypermutation and statistics
Viable (DAPI−) germinal centre B cells (CD19-APC+, PNAhigh-FITC, CD95-PE+) were sorted from Peyer's patches of 8-week old PrimPolΔ/Δ and their wild-type littermates on an Aria® sorter (Becton Dickinson). DNA was extracted using proteinase K treatment and ethanol precipitation. The JH4 intronic sequence of rearranged VHJ558 family members were amplified by PCR using Pfu Ultra® polymerase (Stratagene) (31). PCR products were purified using the QIAquick® Gel Extraction kit (Qiagen), cloned into the TOPO II blunt vector (Invitrogen Life Technologies). Plasmid DNA was isolated using High Pure Plasmid kit (Roche) and sequenced on a 3730 DNA analyzer (Applied Biosystems). Sequence alignments were performed using Seqman software (DNAStar version 12,2,0).
Calculations exclude non-mutated sequences, insertions and deletions. Clonally related sequences with identical mutations and complementary determining region 3 and duplicated sequences were counted only once. Statistic analysis of the mutation spectra was performed using the χ2-test, with the number of mutations of base x and number sequenced base x. For the corrected percentage, the number of mutations were adjusted such that each base contributes 25% of the sequence. Mutation frequencies are expressed as the percentage of a defined nucleotide substitution at base (X) relative to all sequenced bases (X).
DNA fibre analysis
Per well of a 6-well plate, 7.5 × 104 MEFs were seeded and cultured overnight in IMDM medium. Prior to UVC exposure (20 or 40 J/m2), MEFs were incubated in medium containing 25 μM 5-Chloro-2′-deoxyuridine (CldU) for 20 min at 37°C. After UVC exposure, medium containing 500 μM 5-Iodo-2′-deoxyuridine (IdU) was added, resulting in a final concentration of 250 μM IdU and 12.5 μM CldU. After 20 min at 37°C, cells were trypsinized, 2 μl of a suspension of 3 × 105 MEFs/ml was spotted onto a microscope slide, incubated for 5 min and lysed with 7 μl lysis buffer (200 mM Tris-HCl pH7.4, 50 mM EDTA, 0.5% SDS) for 3 min. Slides were tilted to 15°C to allow the DNA to run down the slide. Next, slides were air dried and subsequently fixed in methanol-acetic acid (3:1). After rehydration, fixed DNA fibres were denatured in 2.5M HCl for 75 min. Incorporation of CldU was detected using rat-α-BrdU antibodies (1:500; BU1/75, AbD Serotec) and Alexafluor-555-labelled goat-α-rat antibodies (1:500; Molecular Probes), whereas incorporated IdU was detected using mouse-α-BrdU antibodies (1:750; Clone B44, BD) and Alexafluor-488-labelled goat-α-mouse antibodies (1:500; Molecular Probes). Finally, slides were mounted in Fluoro-Gel (Electron Microscopy Sciences). Microscopy was performed using a fluorescent microscope (Zeiss Axiovert S 100).
Pre-B cell survival assay
2 × 104 pre-B cells were seeded on ST2 feeder cells in 24 well plates in 0.5 ml complete medium and IL7 prior to UV-C irradiation (254 nm, UVC irradiation chamber, Dr Gröbel UV-Elektronik, GmbH, Germany). After 15 min, cells were irradiated and cultured in 1 ml complete medium and IL7. For determining the survival, pre-B cells were harvested after 3 days of culture and live (propidium iodine negative) pre-B cells were counted by on a FACSArray (Becton Dickinson). Data were analysed using FlowJo software.
Mutation and gene expression analysis of TCGA invasive breast cancer (BRCA)
To analyse the impact of PrimPol expression on the mutation load, we mined the mutation and gene expression data from The Cancer Genome Atlas (TCGA). Normalized RNAseqV2 containing RNA expression and level-2 exome mutation data from invasive breast cancers (BRCA) were retrieved and analysed in R using R Studio (Version 0.98.983). Duplicates in the data sets were taken out. For statistical analyses of the mutational frequencies between all tumours and PRIMPOL homozygous deleted tumours we applied the Mann-Whitney test. To correlate the mutation load of each tumour to the level of gene expression we applied the Spearman's correlation.
For mutational analysis of BRCA, the sense (C>G) and antisense (G>C) mutations were taken together, since there was no difference detected in total numbers.
RESULTS
CRISPR/Cas9 mediated inactivation of PrimPol in the mouse germline
In mice, the PrimPol locus locates on chromosome 8 and the spliced transcript comprises 15 exons encoding a single mRNA of 3812 nt (Figure 1A). To define the consequences of a PrimPol deficiency in mice, we took advantage of the CRISPR/Cas9 genome editing technology to inactivate PrimPol (27,32). To minimize potential confounding issues associated with long-term embryonic stem cell (ES) culturing, backcrossing and conventional gene targeting (33,34), we choose for zygote injection (35). Cas9 mRNA and a single guide RNA (sgRNA) for exon 5 of PrimPol (ENSMUSE00001229433 at position 362–365 in the ORF of PrimPol) was injected in 143 zygotes. Of these, 126 developed into two-cell embryos and were implanted in eight surrogate mothers. In total, 34 pups were born and screened for insertion/deletion mutations. A founder was identified with a 4-nt deletion (CCAA) in exon 5 causing a frame-shift and the deletion of the highly conserved N122 in the catalytic domain (Supplementary Figure S1A). PCR amplification of cDNA from MEFs of PrimPol mutants with external primers (spanning exon 1–15) in combination with a nested primer set (spanning exon 3–14) confirmed the presence of a single transcript with the expected 4 nt deletion, which destroys a MscI site in the wild-type allele (Figure 1B). This mutant PrimPol allele (PrimPolΔ) results in a premature stop, giving rise to a truncated polypeptide of 127 amino acids that lacks a critical portion of the archaeo-eukaryotic primase domain and the entire C-terminal Zn-finger regions essential for the priming activity of PrimPol (Supplementary Figure S1A) (25,26). As determined by qPCR the premature stop codon resulted in a 50% reduction of PrimPol mRNA compared to wild-type MEFs (Supplementary Figure S1B). PrimPol heterozygous and homozygous mice were viable and born at the expected Mendelian frequency (data not shown).
Figure 1.

CRISPR/Cas9-mediated inactivation of PrimPol. (A) Schematic presentation of the CRISPR/Cas9-based inactivation of PrimPol by CRISPR/Cas9 and sgRNA (cursive, PAM in blue). A four-base pair CCAA (red) deletion in exon 5 renders the single mRNA of PrimPol out of frame and destroys the indicated MscI restriction site. (B) The PrimPol gene has a single mRNA transcript. A nested PCR was performed on the cDNA spanning exon 1–15, followed by a nested primer set spanning exon 3–14. Subsequently, the cDNA was cut using MscI enzyme (Thermo Scientific), to verify the CCAA deletion that destroyed the MscI site in the PrimPol Δ allele. (C) UV-C sensitivity of PrimPol deficient primary pre-B cells. The UV-C sensitivity of wild-type (WT) and PrimPol deficient pre-B cells was determined by measuring survival in response to increasing UV-C doses. PCNAK164R/K164R mutant cells were included as positive controls (28). (D and E) Replication speed and fork progression after UV-C treatment. For DNA fibre assays, MEFs were first labelled with CldU for 20 min and then 20 min with IdU. After CldU, cells were exposed to 20 J/m2 or 40 J/m2 of UV-C light. The lengths of CldU tracks were measured and the average replication speed (kb/min, error bar indicates standard deviation) on a non-damaged template was measured. (D) For analysis of replication speed of WT and PrimPolΔ/Δ in absence of DNA damage, we measured the length of the CldU track and calculated the fork speed in kb/min. (E) For analysis of replication progression upon UV-C induced DNA damage, IdU:CldU ratios of mock and UV-C treated WT and PrimPolΔ/Δ MEFs were calculated. At least 50 DNA fibres were analysed per experiment. A representative experiment is shown (of two). The Mann-Whitney test was applied to determine statistical significance. NS means not significant, **** indicates P < 0.0001.
UV-survival and replication fork progression in the absence of PrimPol
Primary pre-B cell lines were established and assessed for their sensitivity to UV (Figure 1C). Consistent with a previous knockdown study in human fibroblasts (23), PrimPol deficient and proficient cells displayed equal sensitivity to UV-C induced DNA damage.
To directly determine the impact of UV-C induced lesions on DNA replication speed and progression in a PrimPol deficient condition, we performed DNA fibre assays. The DNA fibre assay allows the visualization of individual replicons on a single molecule level (36,29). Consistent with the concept that PrimPol is a genuine DDT polymerase (24–26), PrimPol was found dispensable in determining the replication speed during unperturbed replication (Figure 1D). This observation is consistent with an independent report by Wan et al. (37), however, contrasts a report by Mourón et al. (24), suggesting that the lack of PrimPol in unperturbed Hela cells significantly reduces fork rate. Next, we analysed the relevance of PrimPol in maintaining replication progression in the presence of increased UV-C damage. While PrimPol proficient and deficient cells were equally sensitive to UV-C induced DNA damage inflicted by 20 J/m2, at 40 J/m2 the IdU:CldU ratio was further reduced indicating impaired replication fork progression in the PrimPol deficient setting (Figure 1E). A limited redundancy in the remaining DDT system may explain this threshold in revealing the PrimPol deficiency.
T and B cell development proceeds normally in the absence of PrimPol
To investigate potential effects of a PrimPol deficiency on early B and T cell development in bone marrow and thymus respectively, the cellularity and composition of the B and T cell precursor subsets of PrimPolΔ/Δ and wild-type litters were compared.
To determine the size of specific B cell precursor subsets in the bone marrow, we distinguished pro-B cells (CD19+, CD45R-low, CD117+, IgM-), pre-B cells (CD19+, CD25+, CD45R-low, CD117-, IgM-), immature B cells (CD19+, CD25-, CD45R-low, CD117-, IgM+) and mature B cells (CD19+, CD45R-high, IgM+), by staining with a cocktail of IgM, CD19, CD25, CD45R and CD117 specific antibodies. Dead cells were excluded from the analysis by propidium iodide staining.
To reveal T cell progenitor subsets in the thymus, single cell suspensions were stained with conjugated antibodies, specific for CD3, CD4, CD8a, CD25, CD44 and TCRβ, to measure the size of the double negative (DN, CD4-/CD8-), double positive (DP, CD4+/CD8+) and single positive compartments (CD4+/CD8- and CD4-/CD8+). Furthermore, the developmental subsets within the CD4-, CD8- (double negative compartment were further distinguished by CD44- and CD25-specific staining to reveal DN 1 (CD44+25-), DN 2 (CD44+, CD25+), DN 3 (CD44-, CD25+) and DN 4 (CD44-, CD25-) subsets.
These analyses revealed no alterations in bone marrow and thymus (Supplementary Figure S2A and B). Likewise, the mature B and T cell compartments in the spleen, as determined by CD4 for helper T cells, CD8 for cytotoxic T cells and CD19 for B cells, and CD19-, CD3- for non B/T cells, were indistinguishable between these genotypes (Supplementary Figure S2C). We conclude that the inactivation of PrimPol has no critical impact on the development of B or T cell in the bone marrow, thymus and spleen.
PrimPol is dispensable during class switch recombination
CSR takes place in G1 of the cell cycle and strongly depends on the formation of AID-induced staggered DSBs in active Igh switch regions (10,11). The resulting DSBs enable CSR between the two switch regions, where the intervening DNA is circularized and the recombined DNA ends are re-joined by non-homologous or alternative end joining (38). To investigate whether PrimPol controls CSR, we determined the CSR frequencies of CFSE loaded, naïve B cells (CD43-) isolated from the spleen of PrimPolΔ/Δ and wild-type littermates. CSR to IgG3 and IgG1 of antigen-inexperienced B cells exposed to LPS alone or LPS and IL4, respectively, were found unaffected (Supplementary Figure S2D). Likewise, proliferation, as measured by CSFE dilution, was indistinguishable between both genotypes (Supplementary Figure S2E). Apparently, PrimPol does not impair the proliferative capacity of B cells and is dispensable for CSR.
PrimPol exerts a strand-biased anti-mutagenic activity during SHM
To determine if PrimPol modulates the mutational profile of hypermutated Ig genes, Peyer's patches were collected from the ileum of PrimPol deficient and proficient littermates. After isolation of the genomic DNA from sorted wild-type and PrimPolΔ/Δ germinal centre B cells, the JH4 intronic region was amplified by PCR and non-selected mutations were determined as described previously (31). The two SHM data sets were built from mutated sequences of germinal centre B cells from 10 PrimPolΔ/Δ and 10 wild-type littermate controls and provided a total number of 773 and 757 mutations in 68 770 and 61 254 base pairs, respectively (Figure 2D). The average frequency of point mutations in mutated JH4 intron of germinal centre B cells from these mice was comparable: 1.12% in the wild type and 1.24% in the PrimPolΔ/Δ setting (Figure 2D) and followed a highly similar distribution over the sequenced region and mutation load per sequence (Figure 2A, Supplementary Figure S3A). Likewise, no effects on the frequency of single and tandem mutations were observed (Figure 2D). Due to low numbers, insertions and deletions were not analysed (data not shown).
Figure 2.

SHM in rearranged JH4 intronic sequences from wild-type (WT) and PrimPolΔ/Δ B cells. (A) The distribution of somatic mutations along the JH4 intron is displayed as a percentage of mutations per total number of mutated sequences. (B) Mutation spectra in hypermutated JH4 introns of GC B cells from WT and PrimPolΔ/Δ mice. The numbers, percentages and percentages corrected for the base pair compositions are depicted. Only mutated sequences were considered. C>G transversions increased compared to WT to a level comparable to G>C transversions in PrimPolΔ/Δ B cells (χ2-test, P = 0.0019 significant changes are marked in dark grey (C>G), and G>C is marked in light grey). (C) The C>G and G>C transversion bias found in WT is absent in germinal centre B cells of PrimPolΔ/Δ mice. The asterisk indicates significant changes (χ2-test, in WT: P = 0.00089, PrimPolΔ/Δ: P = 0.78). (D) Data set generation and SHM frequencies in PrimPolΔ/Δ GC B cells. The number of mice, mutated sequences, base pairs sequenced, number of mutations, tandem mutations and average mutation frequencies are indicated per genotype.
Most remarkably, when comparing the spectra of nucleotide substitutions a prominent inter-genotypic difference was found (Figure 2B and C, Supplementary Figure S3B). The absence of PrimPol resulted in a selective increase of C>G transversions, leaving other G/C as well as A/T mutations unaffected. Apparently, PrimPol is capable of prohibiting selectively the generation of C>G transversions at AP-sites in the (+) strand, which according to previous studies provides the template for the leading strand synthesis in mature B cells (39–41). A close examination of the G/C mutations in the SHM spectra of wild-type mice revealed a yet uncharacterized 3.4-fold G>C over C>G transversion bias (Figure 2B and C). This finding was consistent with the mutations spectra in the JH4 regions described previously (8,28) (Supplementary Figure S3C and D). In contrast, we did not observe a G>C over C>G bias in the PrimPolΔ/Δ mice. The loss of the bias was due to a selective increase of C>G transversions in the PrimPolΔ/Δ setting, implicating a selective strand-biased PrimPol activity in tolerating AP-sites on the leading strand (Figure 2B and C).
PRIMPOL has an anti-mutagenic role in human invasive breast cancer
The molecular characteristics of PRIMPOL and its anti-mutagenic activity during SHM imply a critical genome-wide role for PRIMPOL in genome maintenance. To further explore on the anti-mutagenic activity of PRIMPOL in an independent system, we first determined a human cancer type that frequently lost the PRIMPOL genomic loci. The highest number of PRIMPOL deficient tumours was observed in breast cancer patients diagnosed with invasive breast cancer (1.7%; 14 out of 817), consisting of invasive lobular carcinoma or invasive ductal carcinoma, or a mixed group (42). If PRIMPOL exerts a critical anti-mutagenic activity, the overall frequency of point mutations, as determined by exome sequencing, is expected to increase in PRIMPOL deficient tumours as compared to the majority of PRIMPOL proficient tumours. Indeed, as expected and observed the overall mutation load nearly doubled in PRIMPOL deficient tumours (P-value<10−4, Mann-Whitney test) (Figure 3A). Furthermore, point mutations were significantly increased at G/C and A/T base pairs and deletions in PRIMPOL deficient tumours (Figure 3B). The high load of C/G mutations in invasive breast cancer likely reflects damages caused by APOBEC3B-induced U/G mismatches and AP-sites at C/G base pairs as well as other G/C base damages. In summary, these data indicate a critical genome-wide anti-mutagenic activity of PRIMPOL in vivo.
Figure 3.

PRIMPOL expression and mutation load in human invasive breast cancers. (A) The point mutation load in the exomes of invasive breast cancers compared to the PRIMPOL deficient subset. The number of mutations is depicted as log10 scale, boxes contain 50% of all values and bars indicate the highest and lowest values. PrimPol deficient tumours display significantly more point mutations. Asterisks indicate significant differences (P > 0.0001). (B) Specific base substitutions, insertions and deletions in the exomes of invasive breast cancers and the PRIMPOL deficient subset. The number of mutations is depicted as log10 value, boxes contain 50% of all values and bars indicate the highest and lowest values. Asterisks indicate significant differences. P-values as determined by the Mann-Whitney test were as follows: C>G: P > 0.0001, C>T: P > 0.0001, C>A: P > 0.0001, A>T: P = 0.0013, A>G: P = 0.0003, A>C: P > 0.0001, insertions: P = 0.057, deletions: P = 0.0029. (C) Spearman's rank correlation of the PRIMPOL expression or APOBEC3B expression with defined mutations in 817 human invasive breast cancers. The Spearman's rank correlation was determined for all point mutations as well as specific base substitutions, small insertions and small deletions. The Rho values of the specific correlations are indicated. These analyses suggest a pro-mutagenic activity of APOBEC 3B and an anti-mutagenic activity of PRIMPOL in invasive breast cancer. Stars indicate P-value < 0.01, exact P-values are depicted in Supplementary Figures S5 and 6. (D) APOBEC3B target sequence TpC mutation is increased in PRIMPOL deficient tumours. All TpC mutations increase in PRIMPOL deficient tumours, though C>G and C>A increase by ±2.2 times, while C>T transitions increases by 1.6 times. P-values as determined by Mann-Whitney test were as follows: C>G: P > 0.0001, C>T: P > 0.0008, C>A: P > 0.0001. (E) Spearman's rank correlation of APOBEC3B and PRIMPOL expression and TpC mutations per tumour. APOBEC3B has a positive correlation with TpC mutations. PRIMPOL has a negative correlation on all mutation types. Stars indicate P-value <0.01, exact P-values can be found in Supplementary Figure S11.
As reported before, APOBEC3B mRNA expression has a positive correlation with total load of mutations and is associated with an overall poor prognosis (Figure 3C) (13,16,43). We found APOBEC3B expression directly associated with a strong increase in C>G, C>T, C>A as well as mutations at A/T base pairs and deletions (Figure 3C). This suggests that increased APOBEC3B can contribute to the entire spectrum of point mutations. A finding consistent with the notion that APOBEC3B-induced U/G mismatches lead to mutations around the initial U/G mismatch. This observation supports the role of mismatch repair in generating mutations in nucleotides flanking cytosine targeted by APOBEC/AID family (8,44–45). These data further support a pro-mutagenic activity of APOBEC3B in invasive breast cancers.
In contrast, extending the same analyses implicated a clear anti-mutagenic activity for PRIMPOL. PRIMPOL expression was found to correlate inversely with the mutation load. This observation affected all substitutions and deletions, and was strongest for C>G transversions, and increased 3-fold in PRIMPOL deficient tumours. To exclude that gains and losses of the genomic locus of PRIMPOL underlies the correlation with mutation load, we extended the analysis to neighbouring genes of PRIMPOL. Expression of the neighbouring genes CASP3 and CENPU genes revealed a positive correlation, indicating that the correlation of PRIMPOL expression does not depend on gains and losses of the genomic locus (Supplementary Figure S4).
PRIMPOL prevents APOBEC3B-mediated mutagenesis
APOBEC3B contributes to invasive breast cancer mutagenesis and cytosines within TpC dinucleotides have been identified as preferred APOBEC3B deamination targets (13,16,43,46). Consistent with these notions, we observed that the majority of mutations at C occurred in TpC dinucleotides (54%, 30 out of 56 C mutations per tumour). Most relevant, in PRIMPOL deficient tumors these mutations at TpC further increased 2.2-fold for TpC>G and TpC>A transversions and 1.6-fold for C>T transitions (Figure 3D, Supplementary Figure S14).
As an independent approach in assessing the anti-mutagenic activity of PRIMPOL and the pro-mutagenic activity of APOBEC3B, we determined the relation between PRIMPOL and APOBEC3B expression with the mutation load, specifically TpC>G, TpC>T and TpC>A mutations. Consistent with the pro-mutagenic activity of APOBEC3B, Spearman's correlations revealed that high levels of APOBEC3B expression increased TpC>G mutations, but also TpC>T and TpC>A mutations (Figure 3E). As suggested previously, C deamination by APOBEC3B predominantly generates C>T transitions, and in invasive breast cancer this is the most abundant point mutation.
Consistent with the anti-mutagenic activity of PrimPol during SHM, the level of PRIMPOL expression correlated negatively with the mutation load in invasive breast cancers. High levels of PRIMPOL expression counteracted TpC>G, TpC>T and TpC>A mutations (Figure 3E).
In summary, our analyses reveal an anti-mutagenic activity of PRIMPOL in genome maintenance. Consistent with the anti-mutagenic activity of PrimPol in prohibiting transversions at AID-induced AP-sites in germinal centre B cells, mutation spectra in human invasive breast cancers strongly suggest that this activity also applies to APOBEC3B-induced AP-sites and likely all AP-sites generated during Base Excision Repair.
Y-family TLS polymerases display anti- and pro-mutagenic activity
While TLS polymerases enable direct replicative bypass of DNA lesions that otherwise stall the replicative DNA polymerases, the activity of TLS can come at the risk of increased mutagenesis (1–4). The question whether in general TLS as component of the DNA damage response network leads to an increased or decreased mutagenesis remains to be addressed. To assess this important issue in an unbiased manner, we also correlated the mutation load (point mutations, deletions and insertions) with the expression level of the Y-family TLS polymerases, POLH, POLI, POLK and REV1. These analyses revealed that especially POLK and POLI displayed a strong anti-mutagenic pattern on all types of base substitutions and deletions, indicating a prominent anti-mutagenic role for POLK and POLI in genome maintenance (Figure 4A). In contrast, REV1 expression level had only a slight anti-mutagenic correlation with C>T mutations, which however was not significant at TpC>T dinucleotides mutations and other base substitutions (Figure 4B). This observation likely relates to a dual pro- and anti-mutagenic role of REV1, which apart from its selective deoxycytidyl transferase activity is capable of recruiting other TLS polymerases (47,48). Interestingly, REV1 expression does correlate negatively with insertions and deletions, indicating that REV1 likely counteracts the generation of these mutations. Remarkably, a positive correlation was found between POLH expression and C transversions but not C transitions. This also accounted for TpC>G and TpC>A mutations, indicating that POLH contributes to the increased TpC>G and TpC>A transversions in APOBEC3B expressing invasive breast cancers.
Figure 4.

Y-family TLS polymerases correlate both positively and negatively with mutations in invasive breast cancer. (A) Spearman's rank correlation of Y-family TLS polymerases with specified mutations. Stars indicate P-value <0.01, exact P-values can be found in Supplementary Figures S7–10. (B) Spearman's rank correlation of Y-family TLS polymerases with TpC mutations per tumour. Stars indicate P-value <0.01, exact P-values can be found in Supplementary Figures S12 and 13.
In summary, these observations reveal TLS as an important arm of the DDR network that primarily prevent mutagenesis and therefore contribute to genome maintenance.
DISCUSSION
PrimPol displays (re-) priming and TLS capabilities, and is thought to have a function during DDT (23–25). These unique properties led us to study the consequences of PrimPol loss of function in vivo. Active mutagenesis processes, which are based on cytosine deamination by APOBEC/AID family members, were used as independent readouts to determine DDT activity in vivo (8,28,49–53).
SHM is a programmed mutation process initiated by AID. The mutation spectra of Ung2;Msh2 and K164R;Ung2 double mutants revealed an AID footprint, where G/C transitions appear to arise at similar frequencies on both strands (8,45), suggesting there is no strong strand bias at the level of C deamination by AID. Accordingly, AP-sites are expected to arise at similar frequencies on both strands and block replicative polymerases that cannot tolerate AP-sites. In contrast to these notions, the SHM spectra of the JH4 intron of rearranged Igh revealed a clear biased G>C over C>G mutation. This was lost in PrimPol deficient mice, due to increased C>G mutation frequency. PrimPol deletion does not affect C>A/G>T transversions. This observation likely relates to the existence of two alternative pathways that generate C transversions. All transversions depend on the U glycosylase Ung2 and a large proportion (∼50%) also on Msh2 (8). Rev1 acts downstream of Ung2, but not downstream the Ung2/Msh2 hybrid pathway (54). Rev1 generates C>G transversions during SHM (51,54) and is likely counteracted by PrimPol to prevent C>G mutations on the leading strand. The TLS polymerase responsible for C>A transversions is not known. However, as there is no G>T over C>A transversion bias, the generation of these transversions may follow a different route.
As C transversions depend on uracil glycosylase Ung2, these data indicate a strand-biased anti-mutagenic activity of PrimPol at AP-sites, prohibiting C>G transversions downstream of AID and Ung2 in the leading strand. A strand-biased repriming activity of PrimPol is likely responsible in establishing the G>C over C>G strand bias of somatically mutated Igh loci. As G/C transversions are Ung2 dependent, PrimPol likely prohibits mutagenic bypass of AP-site. The strand bias likely relates to an anti-mutagenic activity of PrimPol on the leading strand. This specific C>G transversion effect is consistent with a strong replication origin, which in mature B cells resides around the 3′ regulatory region (39–41). The localization of the replication origin in the 3′ regulatory region places PrimPol anti-mutagenic repriming activity on the leading strand, thereby suppressing C>G transversions (Figure 5). This model where PrimPol acts primarily on the leading strand independently supports a recent report (55). Furthermore, our observations highlight a delicate balance of pro- and anti-mutagenic processing of AID-induced lesions during SHM. In conclusion, the G>C over C>G transversion bias characteristic for hypermutated JH4 region is established at the level of strand-biased PrimPol-dependent inhibition of mutagenic TLS. The TLS polymerase responsible for C/G>G/C transversions is likely Rev1, a member of the Y-family of DNA polymerases (51,54).
Figure 5.
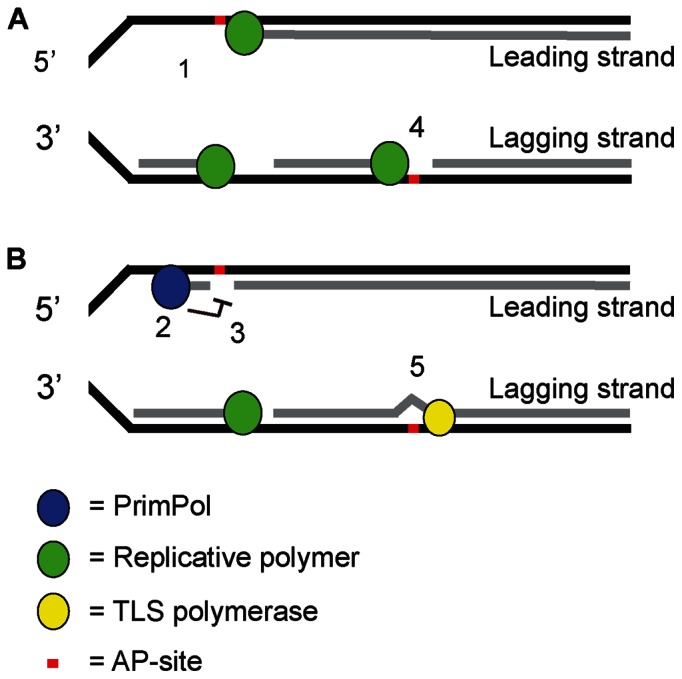
Model: PrimPol prevents mutagenic DDT of AP-sites induced by APOBEC/AID family members. Members of the APOBEC/AID family of cytosine deaminases are associated with programmed mutagenesis. Deamination of C generates a highly mutagenic U, as it instructs a template T. Further processing of U by a uracil glycosylase generates a non-instructive AP-sites that besides C>T transitions can induce C>G and C>A transversions. During SHM, the strand bias regarding G>C over C>G transversions primarily relates to strand-biased PrimPol-dependent error-free DDT of AP-sites on the leading strand. While these findings are based on local SHM spectra in Ig genes, our studies in invasive human breast cancers suggest that PrimPol exerts genome-wide anti-mutagenic DDT activity that likely relates to its repriming capacity on the leading strand. In our model, these AP-sites stall high fidelity replicative DNA polymerases on the leading and lagging strand (1 and 4). Our data imply that PrimPol reprimes downstream of AP-sites in the leading strand to enable fork progression (2) and simultaneously prevents error-prone TLS (3). The remaining gap with the AP-site likely stimulates homology directed error-free repair using the intact template of the sister chromatid. Finally, an AP endonuclease and Polβ restore the remaining AP-site, completing canonical BER initiated by a uracil glycosylase. In contrast, on the lagging strand, repriming of PrimPol at AP-sites does not occur, and AP-sites are tolerated by an AP tolerant TLS polymerase, e.g. Rev1 (5).
Apart from AID-induced SHM in rearranged Ig genes, the pro-mutagenic nature of APOBEC3B appears critical in controlling innate immunity of retro-elements. Invasive breast cancers have a high load of APOBEC3B-dependent mutations with a typical TpC dinucleotide signature, suggesting a derailed APOBEC activity during tumourigenesis (16). Here we took advantage of this signature as readout for DDT at APOBEC3B-induced AP-sites in the genomes of invasive breast cancers. Consistent with the anti-mutagenic activity of PrimPol during SHM, the mutation load, including TpC mutations, was significantly increased in the PRIMPOL deficient tumour subset. In addition, an inverse correlation between PRIMPOL expression and specific point mutations was observed, supporting a critical anti-mutagenic activity and function of PRIMPOL in genome maintenance. The effect of PRIMPOL on mutations at TpC dinucleotides was strongest for C transversions (TpC>G, TpC>A), which is in line with anti-mutagenic activity of PRIMPOL in response to AP-sites generated by APOBEC3B in these tumours.
However, the difference between murine SHM and APOBEC3B mutagenesis in human invasive breast cancer is that PRIMPOL affects C/G transversions and transition in invasive breast cancer, whereas only C>G transversion are affected by PrimPol in our SHM readout. This might be explained by differential processing of AP-sites specific between tissues or species.
In contrast, we observed a strong pro-mutagenic effect of APOBEC3B for all point mutations and deletions, most strongly for C mutations. Similar to the processing of AID-induced DNA lesions during SHM, APOBEC3B expression was also found to correlate with mutations at template A/T in invasive breast cancer. These A/T mutations are likely caused by error-prone repair of U–G mismatches, which can involve error-prone TLS polymerase activity even of an undamaged template around the initial U/G mismatch (8,44–45).
As mentioned TLS polymerases enable direct replicative bypass of lesions that otherwise stall the replicative DNA polymerases. In general, TLS is often associated with increased mutagenesis (1–4,56). However, the net effect of TLS polymerase expression on the mutation load has not been determined thus far. Therefore, the question whether in general TLS as a critical component of the DNA damage response network leads to an increased or decreased mutation load remained to be addressed. To approach this important issue in an unbiased manner, we extended our analysis to the entire Y-family of TLS polymerases by comparing the mutation load (point mutations, deletions and insertions) with the expression level of individual Y-family TLS polymerases, POLH, POLI, POLK and REV1. Our studies revealed an overall anti-mutagenic role for POLI, POLK for all point mutations and deletions, but not insertions. On the contrary, REV1 expression did except for C>T mutations not significantly correlate with other point mutations. This lack of correlations on point mutations might be due to balanced anti- and pro-mutagenic role of REV1, such as the recruitment of other TLS polymerases and its CMP transferase activity. Interestingly, like POLK and POLI, REV1 appears to prevent deletions, but remarkably was identified as the only Y-family polymerase that prevents insertions. In contrast to all other family members, POLH expression showed a positive pro-mutagenic correlation with both C and TpC transversions, suggesting an involvement of POLH in bypassing APOBEC3B-induced AP-sites in invasive breast cancer. This notion is in line with a previous SHM study where in a catalytic-inactive Rev1 setting C>G transversions depended on Polη (57).
Based on these findings we propose a model in which PrimPol exerts a genome-wide anti-mutagenic activity at AP-sites on the leading strand, and likely at other replication blocking lesions. Our model is supported by an independent recent finding, which indicates that APOBEC induced mutations are enriched on the lagging strand (58). We here provide insights into the mechanism underlying these and our observations. While our readouts take advantage of APOBEC/AID family member induced AP-sites, it likely applies to all other AP-sites. On the lagging strand, Polα is the main primase, possibly making repriming by PrimPol obsolete on this strand. Similar to TLS, repriming by PrimPol allows replication to continue effectively. To explain the anti-mutagenic activity of PrimPol, we propose that the remaining gap is either restored by homology directed repair and subsequently canonical repair of the fork-stalling lesion, or a PrimPol-dependent error-free TLS process. However, since AP-sites are non-instructive, it is unlikely that TLS would suddenly become error-free after repriming of PrimPol. Therefore, an error-free repair of the remaining gap by homology directed repair is likely responsible for the anti-mutagenic activity of PrimPol. After homology directed repair, base excision repair can restore the AP-site.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge R. Agami and H. te Riele for comments on the manuscript, E. van Dijk for providing the nucleotide context of BRCA mutations, R. Bin Ali from the Mouse Clinic for Cancer and Aging research (MCCA) Transgenic Facility for his help in generating PrimPol mutant mice, A. Pfauth, F. van Diepen, and M. van Baalen for cell sortings, Roelof Pruntel for sequencing, and the animal caretaker team of the Netherlands Cancer Institute-Antoni van Leeuwenhoek Hospital for biotechnical assistance.
FUNDING
Dutch cancer foundation; Netherlands Organization for Scientific Research [KWF NKI-2012–5243, ZonMW Top 91213018 to H.J.]; National Roadmap grant for Large-Scale Research Facilities by NWO [to I.J.H.]. Funding for open access charge: Dutch cancer foundation; Netherlands Organization for Scientific Research [KWF NKI-2012–5243, ZonMW Top 91213018 to H.J.]; National Roadmap grant for Large-Scale Research Facilities by NWO [to I.J.H.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Sale J.E., Lehmann A.R., Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohmori H., Friedberg E.C., Fuchs R.P., Goodman M.F., Hanaoka F., Hinkle D., Kunkel T.A., Lawrence C.W., Livneh Z., Nohmi T., et al. The Y-family of DNA polymerases. Mol. Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 3.Prakash S., Johnson R.E., Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg E.C. Suffering in silence: the tolerance of DNA damage. Nat. Rev. Mol. Cell Biol. 2005;6:943–953. doi: 10.1038/nrm1781. [DOI] [PubMed] [Google Scholar]
- 5.Victora G.D., Nussenzweig M.C. Germinal centers. Annu. Rev. Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 6.McHeyzer-Williams L.J., McHeyzer-Williams M.G. Antigen-specific memory B cell development. Annu. Rev. Immunol. 2005;23:487–513. doi: 10.1146/annurev.immunol.23.021704.115732. [DOI] [PubMed] [Google Scholar]
- 7.Di Noia J.M., Neuberger M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 8.Krijger P.H., Langerak P., van den Berk P.C., Jacobs H. Dependence of nucleotide substitutions on Ung2, Msh2, and PCNA-Ub during somatic hypermutation. J. Exp. Med. 2009;206:2603–2611. doi: 10.1084/jem.20091707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme [see comments] Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 10.Stavnezer J., Guikema J.E., Schrader C.E. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaudhuri J., Alt F.W. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 12.Harris R.S., Liddament M.T. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 13.Burns M.B., Temiz N.A., Harris R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L., et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakofsky C.J., Roberts S.A., Malc E., Mieczkowski P.A., Resnick M.A., Gordenin D.A., Malkova A. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 2014;7:1640–1648. doi: 10.1016/j.celrep.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burns M.B., Lackey L., Carpenter M.A., Rathore A., Land A.M., Leonard B., Refsland E.W., Kotandeniya D., Tretyakova N., Nikas J.B., et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor B.J., Nik-Zainal S., Wu Y.L., Stebbings L.A., Raine K., Campbell P.J., Rada C., Stratton M.R., Neuberger M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife. 2013;2:e00534. doi: 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A., et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts S.A., Gordenin D.A. Clustered and genome-wide transient mutagenesis in human cancers: Hypermutation without permanent mutators or loss of fitness. Bioessays. 2014;36:382–393. doi: 10.1002/bies.201300140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts S.A., Lawrence M.S., Klimczak L.J., Grimm S.A., Fargo D., Stojanov P., Kiezun A., Kryukov G.V., Carter S.L., Saksena G., et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burch L.H., Yang Y., Sterling J.F., Roberts S.A., Chao F.G., Xu H., Zhang L., Walsh J., Resnick M.A., Mieczkowski P.A., et al. Damage-induced localized hypermutability. Cell Cycle. 2011;10:1073–1085. doi: 10.4161/cc.10.7.15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan K., Roberts S.A., Klimczak L.J., Sterling J.F., Saini N., Malc E.P., Kim J., Kwiatkowski D.J., Fargo D.C., Mieczkowski P.A., et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 2015;47:1067–1072. doi: 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bianchi J., Rudd S.G., Jozwiakowski S.K., Bailey L.J., Soura V., Taylor E., Stevanovic I., Green A.J., Stracker T.H., Lindsay H.D., et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell. 2013;52:566–573. doi: 10.1016/j.molcel.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mouron S., Rodriguez-Acebes S., Martinez-Jimenez M.I., Garcia-Gomez S., Chocron S., Blanco L., Mendez J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013;20:1383–1389. doi: 10.1038/nsmb.2719. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Gomez S., Reyes A., Martinez-Jimenez M.I., Chocron E.S., Mouron S., Terrados G., Powell C., Salido E., Mendez J., Holt I.J., et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell. 2013;52:541–553. doi: 10.1016/j.molcel.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keen B.A., Jozwiakowski S.K., Bailey L.J., Bianchi J., Doherty A.J. Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Res. 2014;42:5830–5845. doi: 10.1093/nar/gku214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krijger P.H., van den Berk P.C., Wit N., Langerak P., Jansen J.G., Reynaud C.A., de Wind N., Jacobs H. PCNA ubiquitination-independent activation of polymerase eta during somatic hypermutation and DNA damage tolerance. DNA Repair (Amst) 2011;10:1051–1059. doi: 10.1016/j.dnarep.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Wit N., Buoninfante O.A., van den Berk P.C., Jansen J.G., Hogenbirk M.A., de Wind N., Jacobs H. Roles of PCNA ubiquitination and TLS polymerases kappa and eta in the bypass of methyl methanesulfonate-induced DNA damage. Nucleic Acids Res. 2015;43:282–294. doi: 10.1093/nar/gku1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dirac A.M., Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J. Biol. Chem. 2003;278:11731–11734. doi: 10.1074/jbc.C300023200. [DOI] [PubMed] [Google Scholar]
- 31.Jolly C.J., Klix N., Neuberger M.S. Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res. 1997;25:1913–1919. doi: 10.1093/nar/25.10.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H., Wang H., Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc. 2014;9:1956–1968. doi: 10.1038/nprot.2014.134. [DOI] [PubMed] [Google Scholar]
- 33.Hogenbirk M.A., Heideman M.R., Velds A., van den Berk P.C., Kerkhoven R.M., van Steensel B., Jacobs H. Differential programming of B cells in AID deficient mice. PLoS One. 2013;8:e69815. doi: 10.1371/journal.pone.0069815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanden Berghe T., Hulpiau P., Martens L., Vandenbroucke R.E., Van Wonterghem E., Perry S.W., Bruggeman I., Divert T., Choi S.M., Vuylsteke M., et al. Passenger mutations confound interpretation of all genetically modified congenic mice. Immunity. 2015;43:200–209. doi: 10.1016/j.immuni.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H., Wang H., Shivalila C.S., Cheng A.W., Shi L., Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merrick C.J., Jackson D., Diffley J.F. Visualization of altered replication dynamics after DNA damage in human cells. J. Biol. Chem. 2004;279:20067–20075. doi: 10.1074/jbc.M400022200. [DOI] [PubMed] [Google Scholar]
- 37.Wan L., Lou J., Xia Y., Su B., Liu T., Cui J., Sun Y., Lou H., Huang J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013;14:1104–1112. doi: 10.1038/embor.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boboila C., Alt F.W., Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 39.Norio P., Kosiyatrakul S., Yang Q., Guan Z., Brown N.M., Thomas S., Riblet R., Schildkraut C.L. Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during B cell development. Mol. Cell. 2005;20:575–587. doi: 10.1016/j.molcel.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 40.Guan Z., Hughes C.M., Kosiyatrakul S., Norio P., Sen R., Fiering S., Allis C.D., Bouhassira E.E., Schildkraut C.L. Decreased replication origin activity in temporal transition regions. J. Cell Biol. 2009;187:623–635. doi: 10.1083/jcb.200905144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Demczuk A., Gauthier M.G., Veras I., Kosiyatrakul S., Schildkraut C.L., Busslinger M., Bechhoefer J., Norio P. Regulation of DNA replication within the immunoglobulin heavy-chain locus during B cell commitment. PLoS Biol. 2012;10:e1001360. doi: 10.1371/journal.pbio.1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciriello G., Gatza M.L., Beck A.H., Wilkerson M.D., Rhie S.K., Pastore A., Zhang H., McLellan M., Yau C., Kandoth C., et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sieuwerts A.M., Willis S., Burns M.B., Look M.P., Meijer-Van Gelder M.E., Schlicker A., Heideman M.R., Jacobs H., Wessels L., Leyland-Jones B., et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm. Cancer. 2014;5:405–413. doi: 10.1007/s12672-014-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J., Miller B.F., Furano A.V. Repair of naturally occurring mismatches can induce mutations in flanking DNA. Elife. 2014;3:e02001. doi: 10.7554/eLife.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rada C., Di Noia J.M., Neuberger M.S. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 46.Burns M.B., Leonard B., Harris R.S. APOBEC3B: pathological consequences of an innate immune DNA mutator. Biomed. J. 2015;38:102–110. doi: 10.4103/2319-4170.148904. [DOI] [PubMed] [Google Scholar]
- 47.Guo C., Fischhaber P.L., Luk-Paszyc M.J., Masuda Y., Zhou J., Kamiya K., Kisker C., Friedberg E.C. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin W., Xin H., Zhang Y., Wu X., Yuan F., Wang Z. The human REV1 gene codes for a DNA template-dependent dCMP transferase. Nucleic Acids Res. 1999;27:4468–4475. doi: 10.1093/nar/27.22.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng X., Winter D.B., Kasmer C., Kraemer K.H., Lehmann A.R., Gearhart P.J. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 50.Delbos F., De Smet A., Faili A., Aoufouchi S., Weill J.C., Reynaud C.A. Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2005;201:1191–1196. doi: 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jansen J.G., Langerak P., Tsaalbi-Shtylik A., van den Berk P., Jacobs H., de Wind N. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Exp. Med. 2006;203:319–323. doi: 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saribasak H., Maul R.W., Cao Z., Yang W.W., Schenten D., Kracker S., Gearhart P.J. DNA polymerase zeta generates tandem mutations in immunoglobulin variable regions. J. Exp. Med. 2012;209:1075–1081. doi: 10.1084/jem.20112234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langerak P., Nygren A.O., Krijger P.H., van den Berk P.C., Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007;204:1989–1998. doi: 10.1084/jem.20070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krijger P.H., Tsaalbi-Shtylik A., Wit N., van den Berk P.C., de Wind N., Jacobs H. Rev1 is essential in generating G to C transversions downstream of the Ung2 pathway but not the Msh2+Ung2 hybrid pathway. Eur. J. Immunol. 2013;43:2765–2770. doi: 10.1002/eji.201243191. [DOI] [PubMed] [Google Scholar]
- 55.Schiavone D., Jozwiakowski S.K., Romanello M., Guilbaud G., Guilliam T.A., Bailey L.J., Sale J.E., Doherty A.J. PrimPol is required for replicative tolerance of g quadruplexes in vertebrate cells. Mol. Cell. 2016;61:161–169. doi: 10.1016/j.molcel.2015.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jansen J.G., de Wind N. Biological functions of translesion synthesis proteins in vertebrates. DNA Repair (Amst) 2003;2:1075–1085. doi: 10.1016/s1568-7864(03)00119-8. [DOI] [PubMed] [Google Scholar]
- 57.Kano C., Hanaoka F., Wang J.Y. Analysis of mice deficient in both REV1 catalytic activity and POLH reveals an unexpected role for POLH in the generation of C to G and G to C transversions during Ig gene hypermutation. Int. Immunol. 2012;24:169–174. doi: 10.1093/intimm/dxr109. [DOI] [PubMed] [Google Scholar]
- 58.Seplyarskiy V.B., Soldatov R.A., Popadin K.Y., Antonarakis S.E., Bazykin G.A., Nikolaev S.I. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016;26:174–182. doi: 10.1101/gr.197046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.