

Abstract
Endogenous double-stranded RNA (dsRNA) must be intricately regulated in mammals to prevent aberrant activation of host inflammatory pathways by cytosolic dsRNA binding proteins. Here, we define the long, endogenous dsRNA repertoire in mammalian macrophages and monocytes during the inflammatory response to bacterial lipopolysaccharide. Hyperediting by adenosine deaminases that act on RNA (ADAR) enzymes was quantified over time using RNA-seq data from activated mouse macrophages to identify 342 Editing Enriched Regions (EERs), indicative of highly structured dsRNA. Analysis of publicly available data sets for samples of human peripheral blood monocytes resulted in discovery of 3438 EERs in the human transcriptome. Human EERs had predicted secondary structures that were significantly more stable than those of mouse EERs and were located primarily in introns, whereas nearly all mouse EERs were in 3′ UTRs. Seventy-four mouse EER-associated genes contained an EER in the orthologous human gene, although nucleotide sequence and position were only rarely conserved. Among these conserved EER-associated genes were several TNF alpha-signaling genes, including Sppl2a and Tnfrsf1b, important for processing and recognition of TNF alpha, respectively. Using publicly available data and experimental validation, we found that a significant proportion of EERs accumulated in the nucleus, a strategy that may prevent aberrant activation of proinflammatory cascades in the cytoplasm. The observation of many ADAR-edited dsRNAs in mammalian immune cells, a subset of which are in orthologous genes of mouse and human, suggests a conserved role for these structured regions.
Double-stranded RNA (dsRNA) binding proteins (dsRBPs) bind highly base-paired, rod-like dsRNA molecules in a sequence-indiscriminate manner (Tian et al. 2004). In mammals, multiple dsRBPs activate the inflammatory response upon interaction with dsRNA, including toll-like receptor 3 (TLR3), melanoma differentiation-associated protein 5 (MDA5), EIF2AK2 (also known as PKR), and DEXD/H-box helicase 58 (DDX58, also known as RIG-I) (de Faria et al. 2013). These proteins were classically defined by their roles in initiation of an antiviral response, but recent data suggest that they are also activated by aberrantly regulated endogenous RNA (Hartner et al. 2009; Pan et al. 2011; Mannion et al. 2014; Liddicoat et al. 2015; Youssef et al. 2015). Although the endogenous dsRNA pool is not well defined, host systems that regulate levels of dsRNA in the cell are known. The endoribonuclease DICER1 recognizes dsRNA and cleaves it into smaller fragments for the host miRNA and siRNA pathways (Hannon 2002; Otsuka et al. 2007). Double-stranded RNA-specific adenosine deaminase (ADAR) enzymes deaminate adenosines to inosines (A-I editing) in dsRNA, converting AU base pairs to IU mismatches (Savva et al. 2012). ADAR editing targets a wide range of transcripts, and A-I modifications appear as adenosine to guanosine changes in cDNA, allowing for the identification of editing events in transcriptome-wide bioinformatics approaches from RNA-seq data sets (Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004).
Mammalian ADARs have an anti-inflammatory role in the innate immune response. Activation of mouse and rat alveolar macrophages, mouse C2C12 muscle precursor cells, or primary human muscle precursor cells with bacterial lipopolysaccharide (LPS), a major component of the gram-negative bacterial outer membrane, results in an acute inflammatory response and a rapid increase in the interferon-inducible ADAR1 p150 isoform (Rabinovici et al. 2001; Wu et al. 2009; Meltzer et al. 2010). Further evidence of the anti-inflammatory action of ADAR1 derives from studies of ADAR1 knockout stem cells, where interferon-inducible genes are globally up-regulated, reminiscent of a viral inflammatory response (Hartner et al. 2009). In mice, deletion of ADAR1 is lethal at ∼E11.5 due to rapid disintegration of the liver and severe defects in hematopoiesis (Wang et al. 2000; Hartner et al. 2004). Deletion of the adaptor protein MAVS partially rescues lethality, with signaling occurring through MDA5, and not DDX58 (Mannion et al. 2014; Pestal et al. 2015). Interestingly, addition of exogenous inosine-containing dsRNA in cell culture experiments inactivates DDX58 and prevents activation of inflammation (Scadden 2007), although whether this occurs with endogenous dsRNA is unknown. In ADAR1 knockout mice, it is hypothesized that a dysregulated pool of long, endogenous dsRNA leads to aberrant activation of the MAVS signaling cascade through MDA5, but the specific dsRNA ligands are unknown (Hartner et al. 2009; Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015). A recent paper suggests that ribosomal-derived dsRNA contributes to the activation of DDX58 and MDA5 in the absence of ATP hydrolysis by the helicase domain, but this finding only explains dysregulation of the dsRNA pool in certain disease states (Lassig et al. 2015). Several other host receptors also bind specific endogenous dsRNA ligands under sterile conditions, including TLR3 and EIF2AK2, again emphasizing the importance of dsRNA in aberrant inflammation (Cavassani et al. 2008; Green et al. 2012; Youssef et al. 2015). In these cases, a few endogenous ligands are known, including U2 RNA, snoRNAs, and tRNAs (Bernard et al. 2012; Youssef et al. 2015), but the complete pool of long dsRNA remains elusive.
In addition to acting as a substrate for dsRBP sensors, dsRNA structures also mediate gene regulation in some cases. During normal growth of mouse macrophages, an alternatively polyadenylated form of the Slc7a2 mRNA, termed CTN-RNA, is sequestered in nuclear paraspeckles by a structured, ADAR-edited 3′ UTR (Prasanth et al. 2005). Slc7a2 encodes a solute transporter essential for the nitric oxide response, and its rapid expression is required upon recognition of pathogen-associated molecular patterns like LPS. Addition of LPS to macrophages results in the endonucleolytic cleavage of the edited Slc7a2 3′ UTR and release of the mRNA into the cytosol for rapid translation. The Slc7a2 unprocessed transcript is quite long (∼60 kb), and the authors hypothesize that nuclear retention of CTN-RNA allows for rapid cleavage and transport, decreasing the time from stress recognition to response by skipping a lengthy transcription step (Prasanth et al. 2005). Giving some credence to this theory, additional cleaved, structured 3′ UTRs have been predicted, in silico, for both mouse and human, suggesting that the phenotype of nuclear retention is more widespread than appreciated (Osenberg et al. 2009).
Here, we characterize the long, edited dsRNAomes of mouse bone marrow-derived macrophages (BMDMs) and human peripheral blood monocytes (PBMs). We find interesting differences and similarities, including dsRNA structures in orthologs of both mouse and human genes. Analysis of the nuclear-cytoplasmic distribution of EER-containing transcripts suggests that a subset, like Slc7a2, is sequestered in the nucleus.
Results
Identification of long dsRNA by RNA-seq of LPS-stimulated macrophages and monocytes
As a first step toward understanding how dsRNA is regulated during inflammation, we defined the repertoire of long dsRNA in mammalian immune cells during inflammation. BMDMs were stimulated with bacterial LPS for 0, 6, and 12 h, followed by RNA extraction. Total RNA was depleted of rRNA, and a fraction was enriched for dsRNA via dsRNA-immunoprecipitation (dsRIP) with the J2 dsRNA-specific antibody. Libraries were prepared from total RNA and dsRIP samples and sequenced using Illumina 101-bp paired-end sequencing (Fig. 1A). As a comparison, we also aligned raw sequencing reads from a recent study that sequenced RNA isolated from LPS-stimulated human peripheral blood monocytes derived from patients of different ages using Illumina 50-bp single-end sequencing (Fig. 1A; Lissner et al. 2015). The inclusion of the human data set allowed for comparison of the long dsRNA repertoire between mouse and human, in a cell type that responds similarly to LPS-stimulation (Hambleton et al. 1996). While the preparation of the libraries was not completely identical (see Methods), the additional data set allowed for comparison of key features between the mouse and human long dsRNAomes of activated immune cells.
Figure 1.

Identification of EERs in murine macrophages and human monocytes. Experimental setup for RNA-seq analysis of LPS-stimulated (A) mouse BMDMs and human PBMs. Dashed line indicates analysis of existing data sets (Lissner et al. 2015). (B) Brief description of identification of EERs, detailed in Supplemental Figure S1. Folding free energy (ΔG) versus length (nt) plot of (C) mEERs (P = 0.1581) and (D) hEERs (P < 0.0001) compared to length-matched controls. P-values determined by Wilcoxon matched-pairs signed rank test. Weblogos for A-I editing sites in (E) mEERs and (F) hEERs using sites in ≥1% of reads of EERs for all samples combined (Seq2Logo Sequence Logo Generator) (Thomsen and Nielsen 2012). Known SNPs were removed from analyses.
Sequencing reads were aligned using an RNA editing-aware version of GNUMAP-bs that tolerates A-to-G mismatches during alignment (Supplemental Table S1; Hong et al. 2013). ADARs will only deaminate rod-like, unbranched dsRNA, and the longer the dsRNA, the more adenosines that will be edited (Bass 1997). Typically, dsRNA must be at least 30 bp to activate an immune response (Weber et al. 2006; Schlee 2013), but when viral dsRNA structures are investigated, they are usually at least 100 bp long (Pfaller et al. 2015). To specifically identify the long cellular dsRNA that might intersect with an immune response, we searched for clusters of editing, observed as A-to-G mismatches in RNA-seq data (Editing Enriched Regions [EERs]), using protocols similar to those previously described (Fig. 1B; Supplemental Fig. S1; Whipple et al. 2015). EERs were first defined as 50-nt windows containing ≥3 editing sites. Editing sites were only counted when ≥1% of the reads were edited. Each editing site also required ≥5 reads to be included in the analysis. Overlapping 50-nt windows were combined, and EERs within 2500 nt were classified as one EER to allow distant binding partners to form native structures. Visual inspection of EERs indicated that a 2500-nt gap combined EERs within a transcript (intramolecular interactions) but did not create EERs spanning multiple transcripts. As a final filtering step, we required that each EER be comprised of at least two combined windows within 2500 nt, to increase the likelihood of finding long structured regions. This editing pipeline identified 342 mouse EERs (mEERs) and 3438 human EERs (hEERs) (Table 1; Supplemental Tables S2, S3).
Table 1.
Identification of EERs
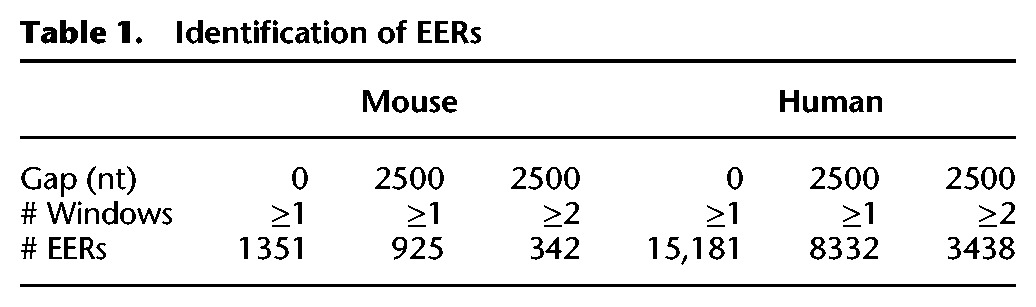
The mouse dsRIP experiment identified additional mEERs relative to the total RNA samples, but no significant enrichment of expression was observed for specific EERs in the dsRIP samples compared to the total RNA samples (Supplemental Fig. S2). Possibly, mEERs lack structures typically bound by the J2 dsRNA antibody (≥40 bp of dsRNA). Alternatively, the J2 antibody may not bind highly edited RNA efficiently. As an additional control, we trimmed the mouse reads using the cutadapt application (http://cutadapt.readthedocs.org) to create a 50-bp single-end library more directly comparable to the human library. By this method, we identified fewer mEERs (156 mEERs vs. 342 from 101-bp paired-end reads) that still exhibited the same trends (Supplemental Fig. S2D) as the larger data set summarized in Figure 3 below. The trimmed data set agrees with our analysis determined from the full-length 101-bp paired-end reads, and as such, we included only the EERs determined from full-length mouse reads in the remainder of these analyses.
hEERs are longer and more structured than mEERs
hEERs were significantly longer than mEERs, with a median length of 845 nt relative to a median length of 546 nt for mEERs (Supplemental Fig. S3A). mEERs did not exhibit significantly more stable structures than length-matched controls that were selected with the same read-depth criteria as the EERs (Fig. 1C). In contrast, hEERs exhibited structures with significantly increased stability (decreased folding free energy) relative to length-matched controls (Fig. 1D). In addition, hEERs exhibited more stable folding free energy per nucleotide (ΔG/nt) than mEERs (Supplemental Fig. S3B). However, visual inspection of structures predicted with folding algorithms (RNAfold, Mfold) (Zuker 2003) revealed regions of double-stranded structure that correlated with regions of editing for both human and mouse EERs. Structures often showed regions of editing separated by large, unstructured loops (e.g., see Fig. 4E,F below). Examination of individual EERs, created without using a 2500-nt gap parameter to combine EERs, indicated that hEERs were composed of significantly longer, and a greater number of, individual regions of editing, in part explaining the increased stability observed in hEERs relative to mEERs (Supplemental Fig. S3C,D). Repetitive elements in the mouse genome are more divergent in sequence than those of the human genome, resulting in fewer pairing partners and decreased levels of dsRNA compared to the human transcriptome, which is composed primarily of Alu repeats (Neeman et al. 2006).
Figure 4.

A subset of mEAGs also have EERs in the human ortholog. (A) Intersection of mEAGs with hEAGs (P < 0.0001 by χ2 test). (B) Primary intersecting feature of conserved EAGs defined as 3′ UTR, Intron, or Other. Gene structure, EER location, base-fraction editing (red, BFE 0%–100%), and relative expression (blue, FPKM, 0–10 reads) are shown for mouse (C) and human (D) Sppl2a/SPPL2A orthologs. Data are for 6 h post-LPS treatment of mouse BMDMs and human PBMs. Mfold-predicted EER structures are compared to the right (E,F).
mEERs and hEERs exhibit features expected of double-stranded regions
For both mouse and human (Fig. 1E,F), editing sites in EERs showed nearest neighbor preferences strongly resembling those from the literature (ADAR1-5′ U > A > C > G; 3′ G > C = A > U; ADAR2-5′ U > A > C > G; 3′ G > C > U = A) (Eggington et al. 2011). These preferences remained stable throughout LPS-stimulation in both organisms (data not shown). In addition, we confirmed editing of EERs by Sanger sequencing of cDNA derived from mouse BMDMs, RAW264.7 cultured macrophages, and phorbol myristate acetate (PMA)-differentiated human THP-1 macrophages (Supplemental Fig. S4A,B). Finally, as additional support for the structure of hEERs, we intersected human EER-associated genes (hEAGs) with known DICER1 binding sites from the literature and found that many of the hEAGs are in fact bound by human DICER1 (observed 719/2792 hEAGs, expected 579/2792, P < 0.0001 by χ2 test) (Rybak-Wolf et al. 2014).
mEERs and hEERs are spread throughout the genome
We mapped the chromosomal locations of EERs using the Idiographica program (Kin and Ono 2007). EERs were spread across all mouse and human chromosomes, including the X and Y Chromosomes (Fig. 2A,B). Consistent with the fact that mice and humans do not have holocentric chromosomes, EERs did not cluster on chromosome arms as observed for Caenorhabditis elegans (Whipple et al. 2015), but we did observe an abundance of EERs on human Chromosome 19. Chromosome 19 is dense with genes and repeats (Grimwood et al. 2004), likely driving the higher abundance of EERs. We then intersected EERs with chromosomal locations of all genes generated by the UCSC Table Browser. The majority of EERs were in annotated genes, with a small subset in intergenic space, >1 kb from a gene (Fig. 3A–C). Interestingly, EERs were in genes that were longer than expected (Supplemental Fig. S4C). To determine the location of EERs within protein-coding genes, we annotated the location of each EER using gene features for that organism obtained from the UCSC Table Browser. Most mEERs were within 3′ UTRs, and hEERs were primarily in introns, consistent with previous studies (Fig. 3D,E; Lev-Maor et al. 2008; Hundley and Bass 2010; Gu et al. 2012; Liddicoat et al. 2015). A small fraction of mEERs were in introns in our analysis, but visual inspection indicated most of these were 3′ UTR isoforms of splice variants, again suggesting that mouse dsRNA is predominantly located in 3′ UTRs. As observed for gene length, mouse and human EERs were in longer than expected introns and 3′ UTRs (Fig. 3F,G); however, EER length did not correlate with the length of the 3′ UTR or intron, indicating that increased length did not simply derive from EER length (Supplemental Figs. S5, S6). Gene ontology analyses comparing mouse EER-associated genes (mEAGs) with all expressed genes using GOrilla indicated no enrichment in mEAGs for any particular category, with only five enrichment categories meeting low levels of significance (Supplemental Table S4; Eden et al. 2007, 2009). A similar analysis performed for hEERs versus expressed genes indicated a slight enrichment in immune processes, not surprising given that monocytes are effectors of the immune response, and further, that LPS treatment increases the expression of many immune-related genes (Supplemental Table S5).
Figure 2.

Chromosomal location of EERs. Chromosomal location of (A) mEERs and (B) hEERs using Idiographica web program (Kin and Ono 2007). EERs are denoted with a red bar corresponding to the location of the EER on each numbered chromosome, shown as an oval with top and bottom corresponding to the + and − strands, respectively.
Figure 3.
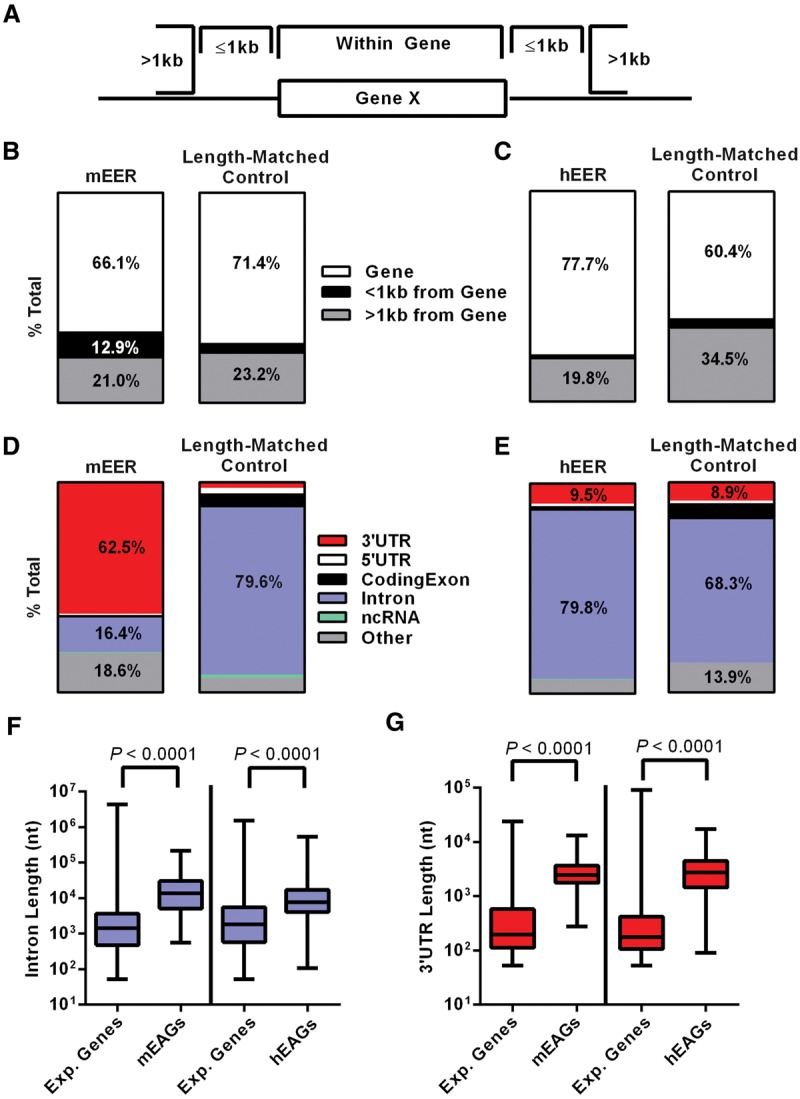
EERs are largely located in mouse 3′ UTRs and human introns. (A) Schematic of potential EER locations described in “parts of whole” bar plot for (B) mEERs and (C) hEERs, relative to length-matched controls. Location of EERs in a gene as 3′ UTR, 5′ UTR, coding exon, intron, ncRNA, or other feature (including unannotated space) for (D) mEERs and (E) hEERs, compared to length-matched controls. Overlap is reported as the percentage of EER bases overlapping an annotation, and plots indicate all categories >8% with text labels. (F) Length of introns containing EERs for mEAGs and hEAGs relative to introns in all expressed genes (Exp. Genes) in our analysis >53 nt (smallest EER) for each data set. (G) Length of 3′ UTRs containing EERs for mEAGs and hEAGs relative to 3′ UTRs in all expressed genes (Exp. Genes) in our analysis >53 nt (smallest EER) for each data set. P-values determined by Mann-Whitney U test.
A subset of mEAGs have EERs in the human ortholog
Two hundred eighty-five of 342 mEERs intersected with an annotated RefSeq gene (Supplemental Table S2). Of these mEAGs, 74 of 285 (observed 74; expected 24 based on mouse/human expression patterns; P < 0.0001 by χ2 test) also showed an EER in the orthologous human gene (Fig. 4A). The EERs were not necessarily in the same location for each ortholog (3′ UTR or intron) (Fig. 4B), and only two EAGs had partial nucleotide sequence conservation (Cds2, 92% identity over 30 nt, 85% identity over 37 nt; Abhd2 85% identity over 109 nt). We focused on two orthologous pairs of EAGs that lacked nucleotide sequence conservation, Sppl2a/SPPL2A and Tnfrsf1b/TNFRSF1B (mouse/human ortholog) (Fig. 4C,D; Supplemental Fig. S7). Sppl2a encodes an intra-membrane aspartic protease important for cleavage of type II membrane signal peptides such as TNF alpha, while Tnfrsf1b encodes a TNF alpha receptor that directs auxiliary functions of TNF alpha signaling, including cellular apoptosis (Holtmann et al. 2002; Friedmann et al. 2006). Sppl2a contained an EER in an annotated 3′ UTR for mouse and a region that showed contiguous read coverage with the 3′ UTR of human SPPL2A, likely an unannotated 3′ UTR (Fig. 4C,D). Each EER folded into a predicted structure with significant stretches of dsRNA; however, consistent with previous analyses (Fig. 1C,D), the hEER appeared considerably more structured than the mEER (cf. Fig. 4E,F). Both of the predicted structures showed base-pairing between edited regions that are distantly located in the 3′ UTR, with intervening, unedited regions characterized by numerous loops and mismatches. In the mouse, the intervening unstructured region was a larger fraction of the EER than that of the human transcript, consistent with the lower stability predicted for mEERs compared to hEERs (see Supplemental Fig. S3B). Folding the entire 3′ UTR of both Sppl2a transcripts resulted in similarly complex structures with editing sites mapping to the predicted dsRNA regions, indicating that the entire 3′ UTR is likely involved in folding (data not shown). Tnfrsf1b showed similar EERs to Sppl2a, except the human transcript contained three separate EERs, within the 3′ UTR and two introns (Supplemental Fig. S7). Again, the predicted 3′ UTR-containing EER of human Tnfrsf1b showed more dsRNA regions than the mEER (Supplemental Fig. S7).
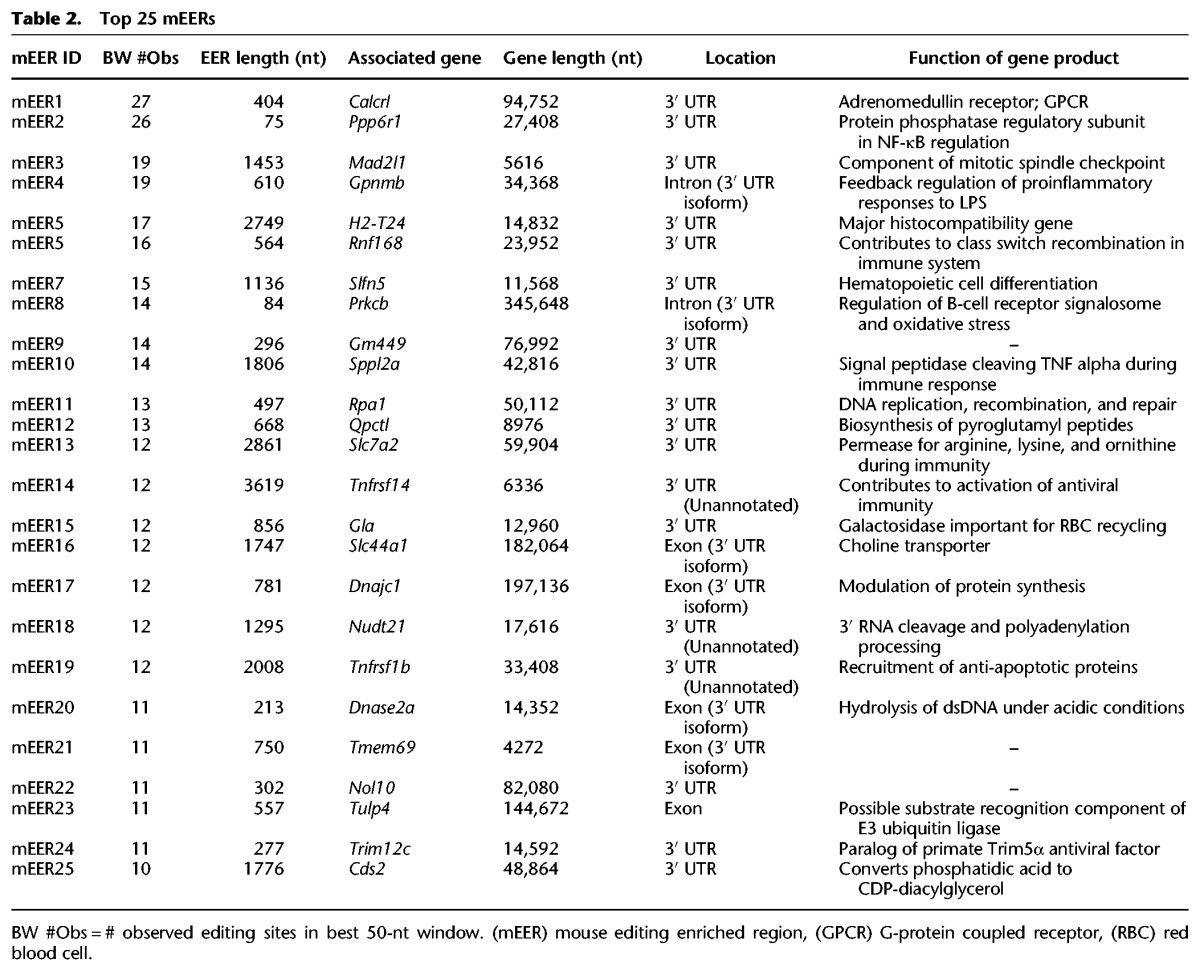
Several highly edited mEAGs are important for TNF alpha regulation
We focused on a list of the top 25 (“TOP25”) mEERs as ranked by best window of editing (highest number of editing sites in 50 nt) (Table 2). The highest ranking mEER by this metric was in the 3′ UTR of the Calcrl gene. The Calcrl EER showed 27 editing sites in a single 50-nt window. mEERs in the TOP25 list were all associated with protein-coding genes, and 24 of 25 were within an annotated 3′ UTR, unannotated 3′ UTR, or a 3′ UTR created by alternative splicing. Several TOP25 mEAGs encode proteins involved in TNF alpha signaling, including SPPL2A, TNFRSF1B, the TNF alpha receptor TNFRSF14, and the TNF alpha-regulated protein GPNMB (Friedmann et al. 2006; Tomihari et al. 2009; Yang et al. 2012; Shui and Kronenberg 2014). Interestingly, we also observed the Slc7a2 gene in the TOP25 list. As mentioned, an alternatively polyadenylated form of Slc7a2, known as CTN-RNA, contains a structured, edited 3′ UTR that contributes to its nuclear retention (Prasanth et al. 2005). During the LPS response, the structured 3′ UTR of CTN-RNA is cleaved by an unknown RNase, and the mRNA is released from the nucleus to mediate the nitric oxide response. Of possible relevance, the Tnfrsf1b transcript is cleaved in its 3′ UTR by ZC3H12A during the inflammatory response, again raising the possibility that 3′ UTR structure contributes to post-transcriptional regulation of inflammatory pathways (Uehata et al. 2013).
Table 2.
Top 25 mEERs
A subset of mEAGs is differentially edited and expressed
We considered the possibility that mEAGs might be regulated like Slc7a2 and performed several analyses to look for similarities. Slc7a2 regulation involves alternative polyadenylation, and we found that 73/342 mEERs contained an alternative polyA site (P < 0.0001 by χ2 test) (Supplemental Table S2; alternative polyA sites from the GenXPro APADBv2 database). Two hundred fifty-five of 316 mEAGs contained alternative polyA sites, not necessarily overlapping the mEER (P < 0.0001 by χ2 test) (Supplemental Table S2). Using RNA-seq data from our input samples, we confirmed that several TOP25 mEAGs were differentially expressed in response to LPS, similar to Slc7a2 (Supplemental Fig. S8A). Slc7a2 showed the highest fold induction at 6 h post-LPS treatment, and H2-T24, a mouse-specific histocompatibility locus, the highest up-regulation at 12 h post-LPS treatment. However, many TOP25 mEAGs were down-regulated at 6 and 12 h post-LPS induction, including Gm449 (2900026A02Rik), Rpa1, and Mad2l1. Editing was not correlated with altered expression, and an increase in expression led to either an increase or decrease in editing depending on the gene (Supplemental Fig. S8B).
A fraction of mEAGs is localized to the nucleus
Given the similarities to Slc7a2, we determined the localization of mEAGs within the cell. We utilized a published data set where BMDMs were activated, fractionated, and deep-sequenced (Bhatt et al. 2012). We aligned the raw reads (FASTQ files) using the GNUMAP-bs aligner and calculated FPKM scores for each transcript using the USeq application DefinedRegionDifferentialSeq (Nix et al. 2008). From this analysis, we determined a relative cytoplasmic/nuclear abundance for the TOP25 mEAGs in unstimulated cells. As a control, Actb and U2 RNA were shown to be enriched in the cytoplasm and nucleus, respectively (Fig. 5A). Fourteen of 24 (∼58%) TOP25 mEER-associated transcripts that met read coverage requirements were skewed toward the nucleus relative to the cytoplasm, whereas random protein-coding genes were almost exclusively in the cytoplasm (Fig. 5B). These findings were validated by monitoring levels of Actb, Snord50a, Nudt21, Mad2l1, H2-T24, and Sppl2a transcripts by qRT-PCR of fractionated RAW264.7 macrophages (Fig. 5C). Snord50a, a C/D box class snoRNA, served as a more consistent marker than U2 of nuclear retention in our experiments in RAW264.7 macrophages. An analysis of all mEAGs indicated that 73 of 241 (30%) genes with sufficient read coverage to be included in the analysis were skewed toward nuclear localization (Fig. 5D). Orthologous hEAGs were significantly more numerous in nuclear-localized mEAGs relative to cytoplasmic mEAGs (Fig. 5E), despite no significant difference in folding free energy for the two populations of mEAGs (Fig. 5F).
Figure 5.

A subset of mEAG transcripts is enriched in the nucleus. Data sets for unstimulated BMDMs (Bhatt et al. 2012) were used to determine the cytoplasmic to nuclear ratio of transcripts for (A) controls and TOP25 mEAGs and (B) 10 randomly selected protein-coding genes. (C) Validation of controls and mEAGs using qRT-PCR of RNA isolated from nuclear or cytoplasmic fractions of unstimulated RAW264.7 macrophages. (D) Predominant cellular localization for all mEAGs using Bhatt et al. data sets. (E) Predominant cellular localization of EAGs found in orthologs of both mouse and human. P = 0.01242 by Z test for two population proportions. (F) Folding free energy per nucleotide (ΔG/nt) for cytoplasmic and nuclear-localized mEERs. P = 0.9109 using Mann-Whitney U test.
hEERs display similar characteristics to mEERs and Slc7a2
We wanted to compare hEERs to mEERs for key features of Slc7a2 regulation. In our analysis, 473 of 3438 hEERs contained an alternative polyA site (P < 0.0001 by χ2 test) (Supplemental Table S3; GenXPro APADBv2 Database), a key feature of mouse Slc7a2 regulation. Of 2792 hEAGs, 2467 contained alternative polyA sites, not necessarily overlapping the EER sequence (P < 0.0001 by χ2 test) (Supplemental Table S3). We compared our hEERs to the in silico analysis of Osenberg et al. that identified putative human cleaved RNAs, similar to Slc7a2. We observed overlap with 28 of the 566 putatively cleaved segments (Osenberg et al. 2009). These findings suggest that a small subset of human mRNAs may undergo regulation similar to the mouse Slc7a2 transcript, as postulated (Osenberg et al. 2009). Recent work from the Mayr group shows that many human genes utilize multiple 3′ UTRs to control gene expression. Intersection of hEAGs with this data set indicated that 1025 of 2792 hEAGs are in transcripts that utilize alternative polyadenylation to produce multiple 3′ UTRs, again similar to the model for Slc7a2 (P < 0.0001 by χ2 test) (Supplemental Table S3; Lianoglou et al. 2013). In addition, many intriguing genes contained hEERs that are directly related to dsRNA biology, including MAVS, EIF2AK2, DDX58, IFNAR1, IFNAR2, IFNGR1, and IFNGR2 (Supplemental Table S3).
Discussion
The mechanisms by which ADARs contribute to the inflammatory response are an area of intense study (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015), yet the endogenous targets of ADAR during normal growth and subsequent inflammation are unknown. In this study, we set out to define the endogenous pool of long, edited dsRNAs that serve as substrates for dsRBPs, including ADAR. Using both experimentally generated data and publicly available data sets, we identified 342 regions of predicted structure in the mouse and 3438 regions from human samples, during the inflammatory response to LPS (Table 1). The overabundance of EERs in human transcripts is in agreement with the large number of edited Alu elements in the human genome (Kim et al. 2004; Price et al. 2004). We observed that many editing sites within hEERs (53%) intersected Alu elements. However, in contrast to a recent report (Bahn et al. 2015), we did not see a difference in the number of hEER-editing sites intersecting Alus in introns versus 3′ UTRs (52% versus 58%). This difference could relate to many factors, including differences in analysis, cell type, and the dsRNA structures in question.
Mice have a more divergent repertoire of repetitive sequences, which includes an Alu-like element (B1 repeat) and several other types of repeats. This divergence drives a decrease in editing levels due to fewer potential pairing partners (∼30- to 40-fold lower than in human) (Neeman et al. 2006). Thirty-six percent of mEER-associated editing sites intersected with repetitive elements as well, most commonly the B1 repeat. Although the difference in absolute numbers for EERs between mouse and human was somewhat expected, the difference in genomic location was surprising. The majority of hEERs were within introns, whereas mEERs were almost exclusively in 3′ UTRs, a feature also observed in a smaller mouse data set from the literature (Fig. 3D,E; Liddicoat et al. 2015). It is possible that deeper sequencing of the mouse transcriptome will reveal additional EERs. Previous work from our laboratory using the nematode C. elegans also revealed many EERs located within intronic sequences, suggesting that structured regions in the mouse may serve additional or different regulatory functions than those in the worm and human (Whipple et al. 2015). Although the human transcriptome contained a lower fraction of EERs within 3′ UTRs relative to introns, the absolute number of EERs within 3′ UTRs is higher in human than mouse (307 hEERs versus 109 mEERs).
Intriguingly, ∼26% of mEAGs (74 EERs) had an EER in the orthologous human gene (Fig. 4A), a startling finding given that only 59 conserved editing sites are observed between mouse and human (Pinto et al. 2014). To our knowledge, this is the first time long structures have been documented in orthologous genes. We observed 74 conserved EERs that were composed of 183 individual EERs (created without a gap parameter). Of these 183 individual EERs, only two showed sequence conservation between mouse and human, and this conservation was limited to very short regions of the associated gene (Cds2, 92% identity over 30 nt, 85% identity over 37 nt; Abhd2 85% identity over 109 nt). The conservation of EERs in orthologous genes suggests a key role for these structures in regulation of gene function, and the lack of sequence conservation suggests it is the dsRNA structure that is most important.
As a first step toward determining the functional importance of EERs in gene regulation, we considered the example of Slc7a2, where a structured 3′ UTR regulates nuclear localization of the transcript (Prasanth et al. 2005). We determined the localization of mEAGs using a published data set from the laboratories of Doug Black and Steve Smale (Bhatt et al. 2012). Realignment of these data using our pipeline indicated that 58% of the TOP25 mEERs, and a significant percentage (30%) of all mEAGs, are enriched in the nuclear fraction (Fig. 5D). Thirty-eight percent of these nuclear-retained RNAs have an EER in the orthologous human gene, again implying an evolutionarily conserved importance for nuclear retention of structured RNA (Fig. 5E). Since induction of interferon by viral dsRNA occurs in the cytoplasm, it is intriguing to consider that nuclear retention of endogenous dsRNA serves to prevent aberrant activation of host inflammatory cascades; however, further experiments will be necessary to definitively test this hypothesis. Many EERs also contained, or were in proximity to, alternative polyA sites in both mouse and human, supporting the idea that some EERs undergo regulation similar to Slc7a2. In addition, intersection of our data with that from the Mayr group indicated that many hEERs are in genes that utilize alternative 3′ UTRs (Lianoglou et al. 2013), another feature of the Slc7a2 mechanism of regulation. We were particularly interested in immune-relevant EAGs, such as Sppl2a and Tnfrsf1b. We determined that these transcripts, important for TNF alpha signaling, contained EERs in their human orthologs as well. It is also possible that EERs influence other processes within the cell. For example, circularization of exons to create circular RNAs (circRNAs) is promoted by complementary Alu repeats in flanking introns (Dubin et al. 1995; Liang and Wilusz 2014; Zhang et al. 2014; Ivanov et al. 2015). Not surprisingly, these Alu repeats are hyperedited, and knockdown of ADAR1 increases the levels of a subset of circRNAs (Ivanov et al. 2015). In fact, an intersection of hEERs with circRNAs derived from circBase (circBase.org) revealed that 1424 of 3438 hEERs intersect at least one circRNA (observed 1424; expected 1244; P < 0.0001 by χ2 test), whereas no mEERs intersected circRNAs. Since production of circRNAs often requires sequences in introns, the latter is consistent with our observation that very few mEERs are in introns.
As expected for targets of ADAR editing, hEERs were predicted to fold into structures that were significantly more stable than length-matched controls; surprisingly, this difference was not observed for mEERs, and mEERs and length-matched controls showed similar folding free energies (Fig. 1C,D). Slight differences in human and mouse ADAR protein sequences could explain this difference, but no overt differences were observed in the catalytic or dsRNA binding domains (data not shown), and ADAR nearest neighbor preferences were also similar (Fig. 1E,F). We did observe that hEERs were comprised of longer, more numerous individual EERs (Supplemental Fig. S3C,D). Additionally, more distant pairing partners for mEERs correlated with intervening regions comprised of long unstructured regions that might lower stability and increase predicted folding free energies, as noted for Sppl2a and Tnfrsf1b (Fig. 4E,F; Supplemental Fig. S7).
We identified 3438 hEERs that were primarily located in introns of protein-coding genes (Fig. 3E). GO analysis of these genes showed a plethora of immune-relevant genes, as expected given the importance of monocytes in the immune response. hEERs are in a variety of dsRBPs and signaling molecules, including EIF2AK2, DDX58, and MAVS. An intriguing possibility is that molecules important for sensing dsRNA are also regulated by dsRNA content in the cell and within their own message. In addition to dsRBPs, many important immune factors, including IFNAR1/2 receptors, IFNGR1/2, multiple STAT proteins, MAPK2K1/2, and multiple IRAK1/2/3/4 proteins have hEERs within their sequences (Supplemental Table S3). The editing observed in immune transcripts is interesting for several reasons, including the intimate relationship between dsRNA and innate immunity and the known intersection of hADAR1 with immunity and disease. With the identification of the mouse and human long dsRNAomes, the stage is now set to define the targets of ADAR, and other dsRBPs, that are dysregulated during disease to produce an inflammatory response.
Methods
Reagents
All chemical reagents were purchased from Sigma-Aldrich unless noted.
Cell culture
All mice utilized in this study were handled in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Utah (Protocol number 12-11008). These protocols follow US federal guidelines described by the Office of Laboratory Animal Welfare (OLAW) as described in the Guide for the Care and Use of Laboratory Animals, 8th Edition.
BMDMs were isolated and differentiated from femurs and tibias of 7- to 8-wk-old male C57BL/6 mice (Jackson Laboratory), as described (Lochhead et al. 2012). Macrophages were differentiated for 7 d in differentiation media (RPMI 1640, Life Technologies; 20% horse serum, Life Technologies; 2 mM L-glutamine, Life Technologies; 20 µg/mL gentamicin; β-mercaptoethanol, Invitrogen; 30% L929 fibroblast supernatant containing macrophage-colony stimulating factor) and stored in a 37°C + 5% CO2 humidified incubator. Differentiation media was replaced every 2–3 d for 1 wk. Macrophage activation was performed at ∼144 h after initiation of differentiation in fresh media using 500 ng/µL ultrapure LPS from Salmonella minnesota R595 (InvivoGen #tlrl-smlps) for 6 or 12 h in 10-cm non-tissue-culture-treated plates.
RAW264.7 macrophages were cultured in Dulbecco's Modified Eagle's Media (Life Technologies) with 10% fetal bovine serum (Atlas Biologicals) and a mix of 10 U/mL penicillin and 10 µg/mL streptomycin in 10-cm tissue-culture-treated plates. Experiments were performed when cells were 70%–80% confluent.
Human THP-1 monocytes were grown in RPMI-1640 media supplemented with 10% fetal bovine serum (Atlas Biologicals) and 0.05 mM β-mercaptoethanol. THP-1 monocytes were differentiated into macrophages using 5 ng/mL PMA treatment for 48 h.
RNA isolation and immunoprecipitation of dsRNA
RNA was extracted from 10-cm plates of stimulated mouse macrophages using TRIzol Reagent (Life Technologies). Samples were then treated with TURBO DNase (Life Technologies) and ethanol-precipitated. RNA was resuspended in water and stored at −80°C. Purified RNA was immunoprecipitated using the J2 dsRNA-specific antibody (English and Scientific Consulting; Lot #J2-1104). In brief, the product of 9 µg of RiboZero-treated RNA was incubated in binding buffer (150 mM NaCl, 50 mM TRIS pH 8.0, 1 mM EDTA, 1% NP-40) with 5 µg of J2 antibody and RNaseOUT (Life Technologies) rotating overnight at 4°C. J2-bound dsRNA was incubated in binding buffer with 25 µL of prewashed protein-A/G agarose beads for 4 h at 4°C, followed by 5× washes in cold binding buffer. RNA was then extracted with TRIzol Reagent as above.
Library preparation and high-throughput sequencing
After RNA fragmentation, cDNA libraries were generated from total and J2-precipitated RNA using the Illumina TruSeq Stranded Total RNA Library Prep Kit according to the manufacturer's specifications (Illumina). DMSO (2.5%) was added to the cDNA synthesis reaction to promote read-through of structured RNA. Libraries were evaluated for the appropriate size distribution using a Bioanalyzer DNA 1000, followed by 101-cycle paired-end sequencing using the Illumina HiSeq 2000 platform at the Huntsman Cancer Center High Throughput Genomics Core. hEERs were identified using published FASTQ files (SRP045352) (Lissner et al. 2015). According to the authors, strand-specific libraries were created using the deoxyuridine triphosphate method and sequenced on an Illumina HiSeq 2000 machine, with single-end sequencing reads of 50 bp in length (Lissner et al. 2015).
Sequence alignment with GNUMAP-bs
Alignments were performed as described (Whipple et al. 2015), with slight adaptations for mouse and human genomes. Paired-end sequencing reads were aligned using the RNA editing-aware version of GNUMAP-bs to the Mus musculus GRCm38/mm10 genome and human GRCh37/hg19 (GNUMAP-bs only) (Hong et al. 2013). GNUMAP-bs was run as previously described with arguments “--lib_type wt1 --read_type rna --num_threads 12 --mpi 0 --nt_conv a2i --top_k_hash 10 --map_quality sensitive.” Alignments were filtered using the SamTranscriptomeParser application of the USeq package (http://useq.sourceforge.net) with options to remove excessive mismatches (“-a 10000”), reverse strands of paired reads (“-r”), and merge paired reads (“-p”). Analyses were completed with (“-n 1000000”) and without (“-n 1”) repetitive sequences, with no major differences. As such, data for replicate mapping reads were included in all analyses to take into account the repetitive nature of mammalian genomes (Price et al. 2004; Neeman et al. 2006). AlignmentEndTrimmer, a USeq application (Whipple et al. 2015), was used to trim low quality bases at ends of reads and eliminate all reads with >1 non-A-to-G mismatch. The error rates after trimming were comparable to the expected Illumina HiSeq 2000 error rate of 0.001 (average rate for mouse, 0.0011; average rate for human, 0.0004; Illumina.com).
Detection of EERs through RNA editing
The RNA editing pipeline was performed as described (Whipple et al. 2015). In brief, BAM files produced by SamTranscriptomeParser were fed into samtools mpileup (http://www.htslib.org/) (Li et al. 2009) to create pileup files containing genome position information. In addition to disabling base alignment quality (BAQ) computation, anomalous read pairs were used to generate pileup files. Next, the USeq application RNAEditingPileupParser was utilized to parse pileup files for A-to-G transitions with ≥5 overlapping reads on the same strand. RNAEditingScanSeqs, an additional USeq application, was then used to scan the parsed pileup files for 50-nt windows containing ≥3 editing sites where ≥1% of the reads at that site were edited. Overlapping editing-enriched windows were combined. EERs within 2.5 kb were merged using the EnrichedRegionMaker USeq tool. The distance of 2.5 kb was chosen by manually curating output and identifying a distance that would combine EERs within a single transcript but not combine EERs from separate transcripts. The final output was adapted to create a six column BED file containing all identified EERs.
Bioinformatics analysis of EERs
For annotation of EERs, BED files of Ensembl genome annotations (GRCm38/mm10 or GRCh37/hg19) were obtained from the UCSC Genome Browser and intersected with EERs using the annotateBed tool of the BEDTools suite (bedtools.readthedocs.org/) (Quinlan and Hall 2010; Quinlan 2014). The percentage of EER bases overlapping an annotation is reported as overlap. Output was manually curated to prevent overlap of features; precedence was given to noncoding RNA > coding exons > 3′ UTR > 5′ UTR > introns. In all cases, features and EERs were required to be on the same strand to be considered intersected. If an EER did not intersect any known region, it was considered intergenic.
The intersection of EER lists with published data sets of interest was performed using the USeq applications FileMatchJoiner or IntersectRegions as appropriate. BEDTools intersectBed, annotateBed, and genomeCoverageBed tools were used where appropriate to gather additional information as noted in text.
Validation of EERs
Total RNA was isolated by TRIzol extraction of activated BMDMs, RAW264.7 macrophages, or PMA-differentiated THP-1 macrophages and treated with TURBO DNase (Life Technologies) as described above. cDNA was generated using SuperScript III (Life Technologies) and amplified by PCR. PCR products were purified using a QIAquick PCR Purification Kit (Qiagen) and submitted for Sanger sequencing at the University of Utah DNA Sequencing Core (cores.utah.edu) after confirming the presence of only one PCR product. All primers are listed in Supplemental Table S6.
Subcellular fractionation and nuclear localization
FASTQ files from Bhatt et al. (SRP008831) (Bhatt et al. 2012) were aligned using GNUMAP-bs, and FPKM values were determined for cytoplasmic and nuclear fractions of mEAGs using the USeq program DefinedRegionDifferentialSeq. A ratio of cytoplasmic to nuclear FPKM was then calculated to represent the relative localization of genes of interest. To verify findings, cytoplasmic and nuclear fractions from RAW264.7 macrophages were collected as described (Bhatt et al. 2012). Briefly, monolayers of cells in 10-cm plates were washed twice with cold PBS + 1mM EDTA, followed by gentle scraping of cells into 1 mL of PBS + 1 mM EDTA. Plasma membranes were incubated in 300 µL of lysis buffer (10 mM Tris-HCl [pH 7.5], 0.15% NP40, and 150 mM NaCl) for 5 min. Lysates were layered over a chilled 24% sucrose cushion made in lysis buffer without detergent and centrifuged for 10 min at 4°C at 14,000 rpm. Four hundred microliters of supernatant were collected and served as the cytoplasmic fraction. The nuclei pellet was rinsed with ice-cold PBS + 1 mM EDTA and resuspended in 200 µL prechilled glycerol buffer (20 mM Tris-HCl [pH 7.9], 75 mM NaCl, 0.5 mM EDTA, 0.85 mM DTT, 0.125 mM PMSF, and 50% glycerol). An equal volume (200 µL) of cold nuclei lysis buffer (10 mM HEPES [pH 7.6], 1 mM DTT, 7.5 mM MgCl2, 0.2 mM EDTA, 0.3 M NaCl, 1 M Urea, and 1% NP40) was added to the sample, briefly vortexed, and incubated on ice for 2 min; this sample was the nuclear fraction.
Lysates from subcellular fractions were treated with proteinase K before RNA extraction to facilitate separation of RNA from RNA-Protein complexes. Four hundred-microliter aliquots of cellular fractions were brought to 0.1% SDS and 4 µL of RNA-grade proteinase K was added, followed by 60 min at 37°C. After proteinase K treatment, RNA was extracted using an equal volume of TRIzol as described above. Equal volumes of RNA were then reverse-transcribed using SuperScript III according to the manufacturer's protocol with an extension time of 1 h at 55°C. cDNA was diluted 1:5 in RNA-grade water, and 8 µL were utilized in a 20-µL qPCR reaction (10 µL SYBR green and 2 µL of 5 µM diluted primer mix) on a LightCycler480 machine.
Statistical analysis
Statistical analyses were performed using Prism 6.04 unless indicated (GraphPad Software). Student's t-tests were performed with Welch's correction for unequal variances when appropriate. Data distribution normality (Gaussian) was not assumed, such that nonparametric tests were also used where appropriate. P values of less than 0.05 were deemed significant for all experiments. χ2 tests were used where appropriate to determine observed versus expected significance. A Z test for two population proportions was utilized to compare abundance of samples in two populations where appropriate.
Data access
High-throughput sequencing data and processed data files from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) (Edgar et al. 2002) under SuperSeries accession number GSE75155.
Acknowledgments
We thank David Nix, Tim Mosbruger, and Darren Ames for thoughtful conversations and bioinformatics consultation throughout this project. We thank Joseph Whipple, Daniel Reich, Sarah Altschuler, and Osama Youssef for thoughtful conversations on data and techniques. We thank the laboratories of Ryan O'Connell and Matthew Mulvey for providing cell culture lines and protocols. B.L.B. and M.G.B. were supported by grants from the National Institutes of Health (NIH): National Institute of Aging grant 5DP1AG044162 and National Institute of General Medical Sciences grant 5R01GM044073. In addition, M.G.B. was supported by an NIH microbial pathogenesis training grant T32AI055434. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.203992.116.
References
- Bahn JH, Ahn J, Lin X, Zhang Q, Lee JH, Civelek M, Xiao X. 2015. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat Commun 6: 6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL. 1997. RNA editing and hypermutation by adenosine deamination. Trends Biochem Sci 22: 157–162. [DOI] [PubMed] [Google Scholar]
- Bernard JJ, Cowing-Zitron C, Nakatsuji T, Muehleisen B, Muto J, Borkowski AW, Martinez L, Greidinger EL, Yu BD, Gallo RL. 2012. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat Med 18: 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DM, Pandya-Jones A, Tong AJ, Barozzi I, Lissner MM, Natoli G, Black DL, Smale ST. 2012. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow M, Futreal PA, Wooster R, Stratton MR. 2004. A survey of RNA editing in human brain. Genome Res 14: 2379–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. 2008. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med 205: 2609–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Faria IJ, Olmo RP, Silva EG, Marques JT. 2013. dsRNA sensing during viral infection: lessons from plants, worms, insects, and mammals. J Interferon Cytokine Res 33: 239–253. [DOI] [PubMed] [Google Scholar]
- Dubin RA, Kazmi MA, Ostrer H. 1995. Inverted repeats are necessary for circularization of the mouse testis Sry transcript. Gene 167: 245–248. [DOI] [PubMed] [Google Scholar]
- Eden E, Lipson D, Yogev S, Yakhini Z. 2007. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol 3: e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL. 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann E, Hauben E, Maylandt K, Schleeger S, Vreugde S, Lichtenthaler SF, Kuhn PH, Stauffer D, Rovelli G, Martoglio B. 2006. SPPL2a and SPPL2b promote intramembrane proteolysis of TNFα in activated dendritic cells to trigger IL-12 production. Nat Cell Biol 8: 843–848. [DOI] [PubMed] [Google Scholar]
- Green NM, Moody KS, Debatis M, Marshak-Rothstein A. 2012. Activation of autoreactive B cells by endogenous TLR7 and TLR3 RNA ligands. J Biol Chem 287: 39789–39799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwood J, Gordon LA, Olsen A, Terry A, Schmutz J, Lamerdin J, Hellsten U, Goodstein D, Couronne O, Tran-Gyamfi M, et al. 2004. The DNA sequence and biology of human chromosome 19. Nature 428: 529–535. [DOI] [PubMed] [Google Scholar]
- Gu T, Buaas FW, Simons AK, Ackert-Bicknell CL, Braun RE, Hibbs MA. 2012. Canonical A-to-I and C-to-U RNA editing is enriched at 3′UTRs and microRNA target sites in multiple mouse tissues. PLoS One 7: e33720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambleton J, Weinstein SL, Lem L, DeFranco AL. 1996. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci 93: 2774–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon GJ. 2002. RNA interference. Nature 418: 244–251. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279: 4894–4902. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. 2009. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10: 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmann MH, Schuchmann M, Zeller G, Galle PR, Neurath MF. 2002. The emerging distinct role of TNF-receptor 2 (p80) signaling in chronic inflammatory disorders. Arch Immunol Ther Exp (Warsz) 50: 279–288. [PubMed] [Google Scholar]
- Hong C, Clement NL, Clement S, Hammoud SS, Carrell DT, Cairns BR, Snell Q, Clement MJ, Johnson WE. 2013. Probabilistic alignment leads to improved accuracy and read coverage for bisulfite sequencing data. BMC Bioinformatics 14: 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundley HA, Bass BL. 2010. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci 35: 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, et al. 2015. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 10: 170–177. [DOI] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. 2004. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res 14: 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin T, Ono Y. 2007. Idiographica: a general-purpose web application to build idiograms on-demand for human, mouse and rat. Bioinformatics 23: 2945–2946. [DOI] [PubMed] [Google Scholar]
- Lassig C, Matheisl S, Sparrer KM, de Oliveira Mann CC, Moldt M, Patel JR, Goldeck M, Hartmann G, Garcia-Sastre A, Hornung V, et al. 2015. ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. eLife 4: e10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005. [DOI] [PubMed] [Google Scholar]
- Lev-Maor G, Ram O, Kim E, Sela N, Goren A, Levanon EY, Ast G. 2008. Intronic Alus influence alternative splicing. PLoS Genet 4: e1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Wilusz JE. 2014. Short intronic repeat sequences facilitate circular RNA production. Genes Dev 28: 2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lianoglou S, Garg V, Yang JL, Leslie CS, Mayr C. 2013. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev 27: 2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissner MM, Thomas BJ, Wee K, Tong AJ, Kollmann TR, Smale ST. 2015. Age-related gene expression differences in monocytes from human neonates, young adults, and older adults. PLoS One 10: e0132061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead RB, Sonderegger FL, Ma Y, Brewster JE, Cornwall D, Maylor-Hagen H, Miller JC, Zachary JF, Weis JH, Weis JJ. 2012. Endothelial cells and fibroblasts amplify the arthritogenic type I IFN response in murine Lyme disease and are major sources of chemokines in Borrelia burgdorferi-infected joint tissue. J Immunol 189: 2488–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer M, Long K, Nie Y, Gupta M, Yang J, Montano M. 2010. The RNA editor gene ADAR1 is induced in myoblasts by inflammatory ligands and buffers stress response. Clin Transl Sci 3: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeman Y, Levanon EY, Jantsch MF, Eisenberg E. 2006. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 12: 1802–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix DA, Courdy SJ, Boucher KM. 2008. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics 9: 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenberg S, Dominissini D, Rechavi G, Eisenberg E. 2009. Widespread cleavage of A-to-I hyperediting substrates. RNA 15: 1632–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka M, Jing Q, Georgel P, New L, Chen J, Mols J, Kang YJ, Jiang Z, Du X, Cook R, et al. 2007. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 27: 123–134. [DOI] [PubMed] [Google Scholar]
- Pan ZK, Fisher C, Li JD, Jiang Y, Huang S, Chen LY. 2011. Bacterial LPS up-regulated TLR3 expression is critical for antiviral response in human monocytes: evidence for negative regulation by CYLD. Int Immunol 23: 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller CK, Mastorakos GM, Matchett WE, Ma X, Samuel CE, Cattaneo R. 2015. Measles virus defective interfering RNAs are generated frequently and early in the absence of C protein and can be destabilized by adenosine deaminase acting on RNA-1-like hypermutations. J Virol 89: 7735–7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto Y, Cohen HY, Levanon EY. 2014. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol 15: R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL. 2005. Regulating gene expression through RNA nuclear retention. Cell 123: 249–263. [DOI] [PubMed] [Google Scholar]
- Price AL, Eskin E, Pevzner PA. 2004. Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res 14: 2245–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR. 2014. BEDTools: the Swiss-Army tool for genome feature analysis. Curr Protoc Bioinformatics 47: 11.12.1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici R, Kabir K, Chen M, Su Y, Zhang D, Luo X, Yang JH. 2001. ADAR1 is involved in the development of microvascular lung injury. Circ Res 88: 1066–1071. [DOI] [PubMed] [Google Scholar]
- Rybak-Wolf A, Jens M, Murakawa Y, Herzog M, Landthaler M, Rajewsky N. 2014. A variety of dicer substrates in human and C. elegans. Cell 159: 1153–1167. [DOI] [PubMed] [Google Scholar]
- Savva YA, Rieder LE, Reenan RA. 2012. The ADAR protein family. Genome Biol 13: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD. 2007. Inosine-containing dsRNA binds a stress-granule-like complex and downregulates gene expression in trans. Mol Cell 28: 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M. 2013. Master sensors of pathogenic RNA–RIG-I like receptors. Immunobiology 218: 1322–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui JW, Kronenberg M. 2014. HVEM is a TNF receptor with multiple regulatory roles in the mucosal immune system. Immune Netw 14: 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MC, Nielsen M. 2012. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res 40: W281–W287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Bevilacqua PC, Diegelman-Parente A, Mathews MB. 2004. The double-stranded-RNA-binding motif: interference and much more. Nat Rev Mol Cell Biol 5: 1013–1023. [DOI] [PubMed] [Google Scholar]
- Tomihari M, Hwang SH, Chung JS, Cruz PD Jr, Ariizumi K. 2009. Gpnmb is a melanosome-associated glycoprotein that contributes to melanocyte/keratinocyte adhesion in a RGD-dependent fashion. Exp Dermatol 18: 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, Satoh T, Mino T, Suzuki Y, Standley DM, et al. 2013. Malt1-induced cleavage of regnase-1 in CD4+ helper T cells regulates immune activation. Cell 153: 1036–1049. [DOI] [PubMed] [Google Scholar]
- Wang Q, Khillan J, Gadue P, Nishikura K. 2000. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290: 1765–1768. [DOI] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80: 5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipple JM, Youssef OA, Aruscavage PJ, Nix DA, Hong C, Johnson WE, Bass BL. 2015. Genome-wide profiling of the C. elegans dsRNAome. RNA 21: 786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wang H, Zhang J, Ma X, Meng J, Li Y, Hou Z, Luo X. 2009. Adenosine deaminase that acts on RNA 1 p150 in alveolar macrophage is involved in LPS-induced lung injury. Shock 31: 410–415. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang X, Moore DR, Lightfoot SA, Huycke MM. 2012. TNF-α mediates macrophage-induced bystander effects through Netrin-1. Cancer Res 72: 5219–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef OA, Safran SA, Nakamura T, Nix DA, Hotamisligil GS, Bass BL. 2015. Potential role for snoRNAs in PKR activation during metabolic stress. Proc Natl Acad Sci 112: 5023–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. 2014. Complementary sequence-mediated exon circularization. Cell 159: 134–147. [DOI] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]