

Abstract
Although human LINE-1 (L1) elements are actively mobilized in many cancers, a role for somatic L1 retrotransposition in tumor initiation has not been conclusively demonstrated. Here, we identify a novel somatic L1 insertion in the APC tumor suppressor gene that provided us with a unique opportunity to determine whether such insertions can actually initiate colorectal cancer (CRC), and if so, how this might occur. Our data support a model whereby a hot L1 source element on Chromosome 17 of the patient's genome evaded somatic repression in normal colon tissues and thereby initiated CRC by mutating the APC gene. This insertion worked together with a point mutation in the second APC allele to initiate tumorigenesis through the classic two-hit CRC pathway. We also show that L1 source profiles vary considerably depending on the ancestry of an individual, and that population-specific hot L1 elements represent a novel form of cancer risk.
Human LINE-1 (L1) elements are autonomous retrotransposons that continue to produce new “offspring” L1 insertions in human genomes (Beck et al. 2010, 2011; Ewing and Kazazian 2010; Huang et al. 2010; Iskow et al. 2010; Stewart et al. 2011; The 1000 Genomes Project Consortium 2015; Sudmant et al. 2015). Until recently, L1 elements were thought to be mobilized primarily in the germline and then silenced in somatic cells throughout adulthood. However, several recent reports have shown that L1 elements are active in at least some adult somatic tissues, including the brain (Muotri et al. 2005; Coufal et al. 2009; Baillie et al. 2011; Evrony et al. 2012; Upton et al. 2015) and epithelial somatic tumors (Miki et al. 1992; Iskow et al. 2010; Lee et al. 2012; Solyom et al. 2012; Shukla et al. 2013; Helman et al. 2014; Tubio et al. 2014; Doucet-O'Hare et al. 2015; Ewing et al. 2015; Rodic et al. 2015). These observations have led to the suggestion that L1 might play a role in initiating human cancers by mutating specific oncogenes or tumor suppressor genes in somatic cells.
However, several broad surveys of somatic L1 insertions in human cancers have detected only a few strong L1 driver candidates (Lee et al. 2012; Shukla et al. 2013; Helman et al. 2014; Tubio et al. 2014), and it is unclear whether any of these insertions could have initiated tumorigenesis in the cancers in which they were discovered (see Discussion). Thus, somatic L1 drivers that affect the earliest stages of tumorigenesis have been elusive in these studies, and it is presently unclear whether L1 has the capacity to initiate tumorigenesis in somatic cells. This could, in principle, reflect a lack of knowledge of the oncogenes and tumor suppressor genes that act at the earliest stages of tumorigenesis in human cancers. However, it might instead indicate that L1 elements are not generally capable of initiating tumorigenesis in somatic cells because they are effectively repressed in most cells. An alternative possibility is that somatic L1 mobilization generally occurs only after an L1-permissive environment is established in an emerging tumor. Under this scenario, L1 could not actually initiate tumorigenesis but might instead become active during the more advanced stages of tumor progression and metastasis (e.g., Rodic et al. 2015).
In perhaps the strongest study implicating L1 in tumor initiation thus far, a somatic L1 insertion was identified in a case of colorectal cancer (CRC) that disrupted the APC tumor suppressor gene (Miki et al. 1992). The APC gene is the earliest gatekeeper that is mutated in the majority (∼85%) of CRC cases and, in fact, both copies of APC must be mutated to initiate CRC through this classic two-hit pathway (Kinzler and Vogelstein 1996; Fearon 2011). In the case of familial adenomatous polyposis (FAP) and other CRC syndromes, one mutated copy of APC is inherited in the germline, and the other is acquired during the lifetime of the individual. In the sporadic form of the disease, both copies of APC must be mutated independently in a somatic cell in order to initiate tumorigenesis (Kinzler and Vogelstein 1996; Fearon 2011). In the remaining 15% of CRC cases, tumorigenesis is instead initiated by a hypermutation phenotype that is associated with microsatellite instability (MSI) and faulty DNA repair (Fearon 2011). APC also can be mutated in this less common form of MSI-CRC, but it does not promote the earliest stages of tumor initiation in such cases.
Although one of the two APC alleles clearly was disrupted by L1 in the sporadic case of CRC described by Miki et al. (1992), the status of the second APC allele was not investigated in that study. Furthermore, the MSI phenotype of the tumor was not investigated, leaving open the possibility that tumorigenesis was initiated by a hypermutation phenotype associated with faulty DNA repair, rather than by the more common pathway involving biallelic mutations in APC (Fearon 2011). It also is unclear how the L1 insertion in APC could have been generated sufficiently early to initiate tumorigenesis in a normal colon cell. Thus, many questions remain unanswered as to the genesis of this somatic L1 insertion in APC and its impact on tumorigenesis (Miki et al. 1992).
Results
A novel somatic L1 insertion disrupts the APC tumor suppressor gene
Inspired by Miki et al. (1992), we screened CRC samples obtained from our institution to identify additional somatic L1 insertions in APC and other genes that have been implicated in CRC (Methods; Iskow et al. 2010). Among the first 10 tumors that we screened with L1-seq assays, we discovered a somatic L1 insertion in patient 20444 that disrupted the open reading frame (ORF) of the APC tumor suppressor gene (Fig. 1). PCR assays and subsequent Sanger sequencing confirmed that this novel somatic L1 insertion disrupted the sixteenth exon of the APC gene at codon 1396 (p.F1396L1 in GenBank gene ID NM_000038; hg19 coordinate Chr 5:112,175,479) (Table 1; Fig. 1; Supplemental Tables S1, S5; Supplemental Methods). This insertion was identified in the same exon as the Miki et al. (1992) insertion, just 388 bp upstream of that insertion (Fig. 1). We determined that the new L1 insertion is 1387 bp in length, ends with a ∼27-bp poly(A) tail, and is flanked by a 14-bp target site duplication (TSD) with the sequence 5′-CACTTGATAGTTTT-3′ (Fig. 1A; Supplemental Table S1). The insertion is truncated at the 5′ end and also contains a 5′ inversion and a small internal deletion at the inversion junction that presumably were caused by twin priming (Ostertag and Kazazian 2001). PCR validation assays confirmed that the APC L1 insertion was found only in the tumor and was absent from normal adjacent tissues, thus confirming it as a true somatic L1 insertion (Supplemental Methods).
Figure 1.

Mutagenesis of APC by a somatic L1 insertion. (A) A schematic of the L1 insertion in APC. The top diagram shows the location of the APC gene (vertical red bar) in band q22.2 of Chromosome 5. The diagram below depicts the 1387-bp somatic L1 insertion (dark and light green) in exon 16 of APC with the associated hallmarks of retrotransposition, including a flanking 14-bp TSD (orange), poly(A) tail (T in reverse order; red) and evidence for twin priming (two green boxes separated by an 18-bp deletion at the inversion point). (B) The two diagrams show the inactivating mutations that were discovered in both alleles of APC in the tumor. The top allele is inactivated by the L1 insertion at codon 1396 (multicolored bar; p.F1396L1) and has the reference codon at position 1450 (gray circle). The bottom allele is inactivated by the p.R1450* stop codon (red circle) and does not have an L1 insertion (gray circle). (C) The diagram shows the sites that are affected by these mutations within the APC protein. Also depicted is the L1 insertion identified by Miki et al. (1992) (red square; p.P1526L1). All three of these mutations occur within or near the somatic mutation cluster region (black bar) (Miyoshi et al. 1992) and are similar to other inactivating APC mutations in CRC (see Discussion). This image is adapted from the output of the Protein Painter tool (http://explore.pediatriccancergenomeproject.org/proteinPainter). The APC protein is 2843 amino acids long and consists of the following domains: Suppressor APC (red; involved in nuclear export and other functions); Armadillo/beta-catenin-like repeats (orange; mediate protein–protein interactions); APC cysteine-rich regions (yellow; bind beta-catenin); SAMP (green; binds axin); APC basic (blue; interacts with microtubules); and MAPRE1-binding (aka EB1-binding; purple; binds the microtubule-associating protein MAPRE1).
Table 1.
Twenty-seven somatic L1 insertions identified in the patient's tumor
Whole-genome sequencing (WGS) confirms the L1 insertion in APC and reveals additional somatic mutations
We next performed Illumina whole-genome sequencing (WGS) on the normal/tumor pair from patient 20444 to gain a better understanding of how this L1 insertion might have worked together with other somatic mutations to promote tumorigenesis (Supplemental Data S1, S2; Supplemental Tables S1, S6; Supplemental Methods). We began by scanning the WGS data to identify additional somatic mobile element insertions (MEIs) that might have been generated in the tumor using the Mobile Element Locator Tool (MELT) that we developed for the 1000 Genomes Project (Methods; Sudmant et al. 2015). MELT confirmed the somatic L1 insertion in the APC gene with the features outlined above and detected 26 additional somatic L1 insertions in the tumor that were absent from adjacent normal tissues (for a total of 27 somatic L1 insertions) (Table 1; Supplemental Table S1). All 27 were validated as somatic insertions in the tumor with at least one junction PCR and/or sequencing assay (Supplemental Table S5; Supplemental Methods). In addition, we amplified and sequenced the entire insertion and flanking regions for 16 of these somatic L1 insertions, including the APC insertion (Supplemental Tables S1, S5; Supplemental Methods). Our data revealed that all 16 of these fully sequenced insertions terminated in poly(A) tails and were flanked by TSDs, indicating that they were generated by target primed reverse transcription (TPRT) as expected for genuine L1 retrotransposition events (Table 1; Supplemental Table S1; Luan et al. 1993; Jurka 1997).
Vogelstein and colleagues have described the landscape of genes that are frequently mutated in CRC and also have mapped the temporal order in which these genes are mutated (Kinzler and Vogelstein 1996; Fearon 2011; The Cancer Genome Atlas Network 2012). Therefore, we next identified somatic mutations in the tumor, including single nucleotide variants (SNVs) and short insertions/deletions (indels), to determine how L1 might have worked together with other types of mutations to initiate and drive tumorigenesis. We detected a C to T somatic SNV that created a premature stop codon in the sixteenth exon of APC just 160 bp downstream from the somatic L1 insertion (p.R1450* in GenBank gene ID NM_000038; hg19 coordinate Chr 5:112,175,639) (Fig. 1B; Supplemental Data S1). By manually inspecting the WGS Illumina mate pairs in the region using the Integrative Genomics Viewer (IGV) (Thorvaldsdottir et al. 2013), we determined that the L1 insertion and the stop codon were on mutually exclusive chromosomes and thus together would disrupt both APC alleles. We further confirmed that the L1 and stop codon affected two different APC alleles using a PCR-based strategy (Fig. 1B; Supplemental Table S5; Supplemental Methods). The p.R1450* somatic mutation has been reported 21 times previously in CRC by The Cancer Genome Atlas (TCGA) project (The Cancer Genome Atlas Network 2012) and has been independently documented in the COSMIC database (Forbes et al. 2014). In fact, codon 1450 is one of the most frequently mutated APC sites in patients with sporadic CRC (Fearon 2011). These data indicate that L1 was responsible for inactivating one APC allele, and the p.R1450* stop codon was responsible for inactivating the second APC allele.
A microsatellite stable CRC
TCGA and others have identified two subtypes of CRC: one subtype with microsatellite stability (MSS) and a second subtype with high levels of microsatellite instability (MSI) (Fearon 2011; The Cancer Genome Atlas Network 2012). These two CRC subtypes also progress along different pathways: The MSS subtype is initiated by mutations in APC, whereas the MSI subtype is initiated by a hypermutation phenotype that is associated with faulty DNA repair (Markowitz and Bertagnolli 2009; Fearon 2011). Thus, we next sought to assign our CRC case to one of these two subtypes. We used the MSIsensor tool (Niu et al. 2014), together with the Illumina WGS data described above, to measure microsatellites in our tumor and adjacent normal control tissues (Methods). The microsatellite mutation rate in our CRC sample (0.01%) was clearly within the MSS range (Niu et al. 2014) and was well below the rates observed in tumors that have MSI phenotypes (3.5%–41%). Thus, our CRC sample was unambiguously assigned to the MSS subtype. Likewise, no somatic mutations were detected in DNA repair genes that previously have been linked to hypermutation phenotypes in CRC (MLH1, MLH3, MSH2, MSH3, MSH6, PMS2, POLE) (The Cancer Genome Atlas Network 2012). Collectively, these data demonstrate that our CRC case progressed along the classic MSS route whereby APC serves as the earliest gatekeeper. Therefore, these data formally demonstrate that the L1 insertion, combined with the p.R1450* stop codon, were responsible for initiating tumorigenesis in this case.
Other somatic mutations supporting an L1-initiated pathway of tumorigenesis
As outlined above, Vogelstein and colleagues and TCGA previously have defined the landscape of genes that are frequently mutated in CRC (including genes mutated in MSS-CRC), and we identified somatic mutations in three additional MSS-CRC genes in our tumor (Supplemental Data S1; Supplemental Methods). First, we identified a somatic point mutation in the PIK3CA gene (CAT to CGT; p.H1047R) that causes an amino acid substitution of histidine to arginine at codon 1047 of the encoded protein. PIK3CA is mutated in 18% of MSS-CRCs, and this precise mutation has been found previously in five independent CRCs by TCGA (The Cancer Genome Atlas Network 2012). We also identified an 18-bp tandem-duplication in the KRAS gene that includes codons 59 and 61 and retains the ORF of the encoded protein (p.L56_E62dup). KRAS is mutated in 43% of MSS-CRCs, and this mutation likely contributed to tumorigenesis since activating point mutations affecting codons 59 and 61 have been reported previously for this gene (The Cancer Genome Atlas Network 2012; Forbes et al. 2014). This specific tandem duplication has not been identified previously by TCGA nor is it present in the COSMIC database. However, COSMIC has several entries of somatic indels spanning positions 59 and 61 that previously have been implicated in a variety of cancer types, suggesting that indels in this region can activate KRAS (Forbes et al. 2014). We also identified a 2-bp somatic deletion in the ACVR1B gene that causes a frameshift in the third exon of this gene (p.K177fs). ACVR1B is mutated in 4% of MSS-CRC cases (The Cancer Genome Atlas Network 2012).
The p.R1450* mutation in APC, along with the PIK3CA, KRAS, and ACVR1B mutations outlined above, were validated through PCR, cloning, and Sanger capillary sequencing (Supplemental Table S5; Supplemental Methods). RNA-seq analysis of the normal and tumor specimens also revealed that the mutant alleles of these genes were all expressed in the tumor but were not expressed in the adjacent normal control tissues, as would be expected of somatic mutations (including the L1 insertion allele of APC, the p.R1450* allele of APC, and the mutant alleles of PIK3CA, KRAS, and ACVR1B described above) (Supplemental Table S3; Supplemental Methods). As an independent validation of the somatic mutations, we also performed Illumina WGS on a second genomic DNA preparation from a separate region of the tumor and confirmed the presence of the L1 insertion in APC along with the other somatic mutations outlined above (p.R1450* in APC, and the somatic mutations in PIK3CA, KRAS, and ACVR1B) (Supplemental Data S1, S2; Supplemental Table S6). Our data suggest that these mutations were clonal driver mutations, because they were found in two separate tumor locations and thus, must have occurred early in tumorigenesis. Likewise, at least 25 of 27 (92.6%) of the somatic L1 insertions were found in two separate tumor locations (Supplemental Data S2), suggesting that most (if not all) of these insertions also occurred at early stages of tumor development.
L1 source elements generating somatic offspring insertions
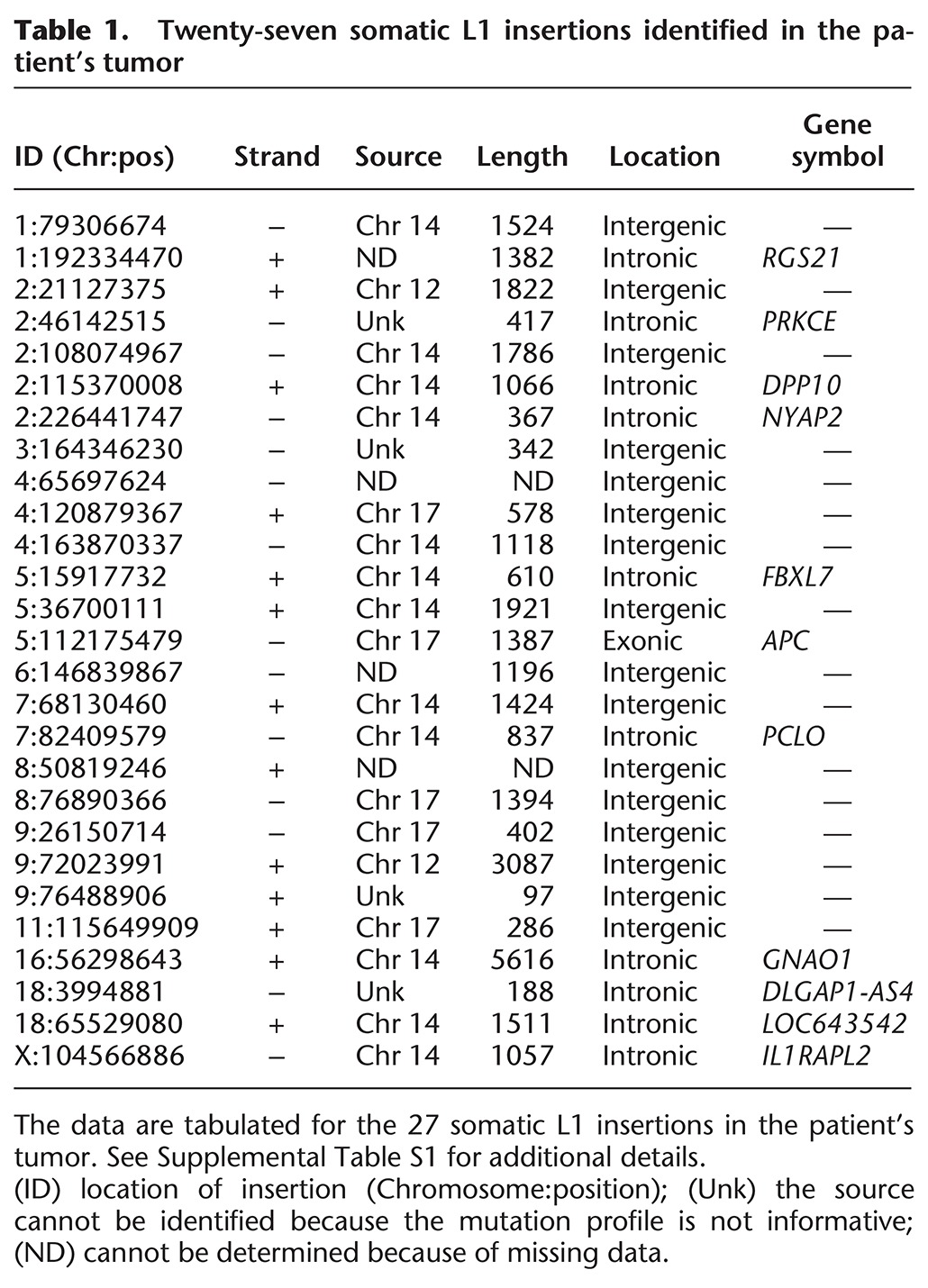
We next identified the full-length L1 (FL-L1) source elements that generated most of the somatic L1 insertions in the tumor, including the source element that produced the APC insertion. To accomplish this goal, we leveraged interior mutations within FL-L1s, which are abundant, as markers to identify source elements that produced specific offspring insertions (Fig. 2). Our analysis was focused on Human-specific (Hs) full-length elements (FL-L1Hs), since all active L1s in the human genome are found within this group. Using MELT and associated MELT tools, we identified a total of 308 FL-L1Hs elements in the patient's genome and analyzed the mutation profiles of these elements as follows (Methods): The full sequences of 264 reference FL-L1Hs copies were available in the hg19 human genome reference sequence (Smit et al. 1996–2010; Lander et al. 2001), and the sequences of 31 of 44 additional nonreference FL-L1Hs elements were determined using a combination of long-range PCR and Sanger or PacBio sequencing (Methods, Supplemental Tables S2, S5). By examining the internal mutation patterns of 295 of 308 (95.8%) of the FL-L1Hs elements compared to the L1.3 reference L1 element (GenBank ID L19088) (Dombroski et al. 1993), we learned that all but five of these elements had a unique singleton mutation that alone could be used to distinguish each individual element from all other FL-L1Hs elements in the patient's genome (Fig. 2B,C; Supplemental Table S2). Moreover, the complete mutational signatures of these elements, which ranged from 7 to 147 mutations, were unique for each FL-L1Hs element in this patient (Fig. 2A; Supplemental Table S2).
Figure 2.
Interior mutations in 295 FL-L1Hs source elements in the patient's genome. (A) The total number of mutations in each FL-L1Hs source element is depicted, grouped by pre-Ta and Ta subfamilies (Boissinot et al. 2000). (B) The Chr 17 FL-L1Hs source element profile is compared to the three closest FL-L1Hs elements in the patient's genome. Although the three most similar elements have 15, 14, and 14 mutations in common with the Chr 17 source element, respectively (middle of Venn diagram), they have 18 total differences (Δ) in all three examples. Similar results were obtained with the remaining elements in the patient's genome (Supplemental Table S2). (C) Mutation frequencies in FL-L1Hs source elements. Individual mutations are plotted by the total number of FL-L1Hs elements in which they are found. A total of 2788 mutations are confined to a single source element (leftmost bar), whereas only a few mutations are shared by the majority of the 295 FL-L1Hs elements in the patient's genome (right bars). This large collection of singleton mutations, and the profiles that are generated by combining these mutations, has allowed us to identify source elements that generated specific somatic offspring insertions in the tumor (Fig. 3) and also allowed us to evaluate the expression of these elements (Fig. 4). (D–F) Mutation profiles for the Chr 17 (D), Chr 14 (E), and Chr 12 (F) source elements. All three of these source elements are heterozygous in our patient's genome. Differences from the reference L1.3 element (GenBank ID L19088) (Dombroski et al. 1993) are marked in green, red, blue, and yellow and represent mutations to A, T, C, or G, respectively. We also determined the “allele frequencies” at which mutations appear in the FL-L1Hs source elements from the patient's genome and have depicted these in pie charts above each mutation. Mutations that uniquely tag a single element are marked with a star (*). Black bars above the 3′ ends depict the signatures of mutations that were used to identify somatic offspring insertions in the tumor.
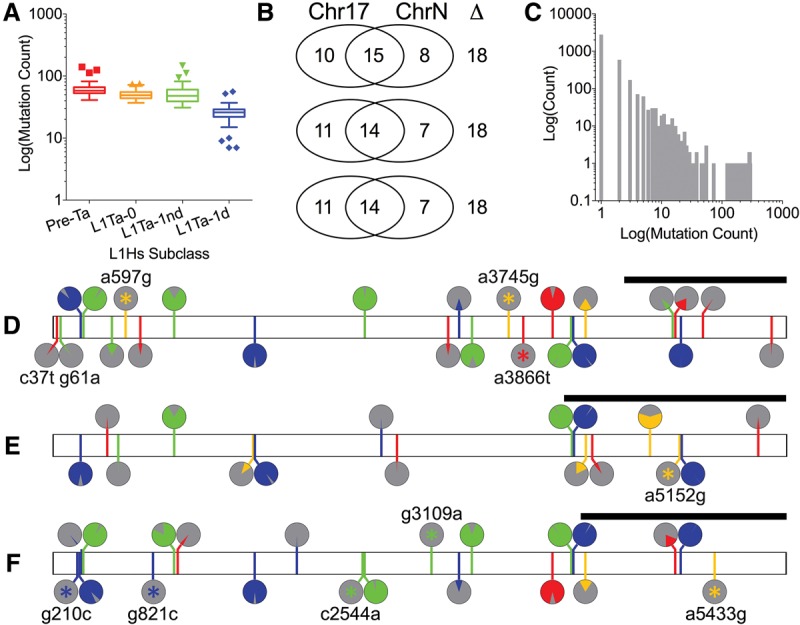
By comparing these FL-L1Hs profiles with the mutation profile of the L1 insertion in APC, we identified a candidate source element on Chromosome 17 (Chr 17:18776467) that shared an identical pattern of interior mutations with the L1 insertion in APC (Figs. 2D, 3A). This Chr 17 FL-L1Hs element had a profile of 25 mutations that was unique among FL-L1Hs elements in the patient's genome and also was consistent with this source giving rise to the APC insertion along with four additional somatic L1 insertions in the tumor (Figs. 2D, 3A; Supplemental Table S2). We identified two additional FL-L1Hs source elements on Chromosomes 14 (Chr 14:59160899) and 12 (Chr 12:117814460) that accounted for most of the remaining somatic L1 insertions in the tumor (Table 1; Figs. 2E,F, 3B,C). The unique signatures within these source elements allowed us to unambiguously assign 18 of 27 offspring insertions to one of these three source elements (Fig. 3; Table 1; Supplemental Table S1). One additional offspring was mapped to the Chr 12 source element using a 3′ transduction that was associated with the offspring insertion (Fig. 3C; Moran et al. 1999). The eight remaining insertions could not be mapped unambiguously to source elements, because we had limited sequence information for these elements or they did not span regions containing unique mutations.
Figure 3.
Source elements that gave rise to somatic L1 insertions in the tumor. As in Figure 2, the bar diagrams in A–C depict mutations in the L1 sequences of source elements and somatic offspring relative to the reference element L1.3. Colored vertical lines represent single nucleotide mutations as outlined in Figure 2. The Circos plots show the somatic offspring L1 insertions that were generated by each FL-L1Hs source element. (A) The Chr 17 source element gave rise to five somatic insertions, including the insertion in APC. The mutation profile of the APC insertion uniquely and perfectly matches that of the Chr 17 FL-L1Hs source element to the extent that the APC insertion spans the 3′ region of the Chr 17 source element. Two of the Chr 17 somatic offspring (denoted by *) had extreme 5′ truncations and thus only had one mutation (C5788T) compared to L1.3. Although the mutation profiles of these offspring do not exclusively match that of the Chr 17 source element, the one remaining possible source element for these somatic offspring (ID 1:86392759) was ruled out due to lack of intact ORFs. (B) The Chr 14 source element gave rise to 12 somatic insertions. The mutation profiles for 11 of 12 (91.7%) of these offspring uniquely and perfectly match the mutation profile of the Chr 14 source element. The remaining somatic offspring (denoted by #) had one additional mutation (T4250G) that was not present in the Chr 14 source element. This mutation does not match any other source element and most likely was introduced during retrotransposition (which is error prone) (Gilbert et al. 2005). The blurry end of somatic offspring 16:56298643 represents ambiguity of the 5′ end because we did not sequence that end. (C) The Chr 12 source element gave rise to two somatic insertions. One was assigned to the Chr 12 source element using mutation profiles as outlined for the Chr 17 and Chr 14 source elements above, whereas the other was assigned using a 3′ transduction (purple box flanked by poly(A) tails in green) (Moran et al. 1999). (D) Circos plot depicting all 27 somatic insertions discovered in this tumor, including the somatic offspring with known source elements depicted above (A–C) and eight additional somatic offspring from an unknown source element: (green) unknown chromosome (Un).
The interior sequences of the Chr 17, Chr 14, and Chr 12 FL-L1Hs source elements revealed two intact ORFs and also indicated that all three are L1Ta-1d elements, which include the most active “hot” L1s in humans (Supplemental Table S2; Boissinot et al. 2000; Brouha et al. 2003; Beck et al. 2010). All three also are nonreference insertions that are absent from the hg19 human genome reference sequence but are present in the 1000 Genomes Phase III MEI data set (Sudmant et al. 2015). Upon comparing our sequenced elements to additional data sets, we learned that Beck et al. (2010) had previously identified and sequenced the Chr 17 source element. The Chr 17 source element from Beck et al. (2010) was cloned from a Yoruban individual (NA19129, Coriell) and is identical in sequence to the Chr 17 source element that we cloned from our patient (Supplemental Table S2). Beck et al. (2010) previously tested the Chr 17 source element in a cell culture-based retrotransposition assay (Moran et al. 1996) and reported that it is indeed a hot L1, with activity levels of 137% compared to the hot L1.3 control (GenBank ID L19088) (Beck et al. 2010). Therefore, since the Chr 17 source element sequences were identical in both studies, these data indicate that the somatic L1 insertion in our patient's APC gene was generated by an exceptionally hot L1 source element.
Expression of L1 source elements in normal versus tumor tissues
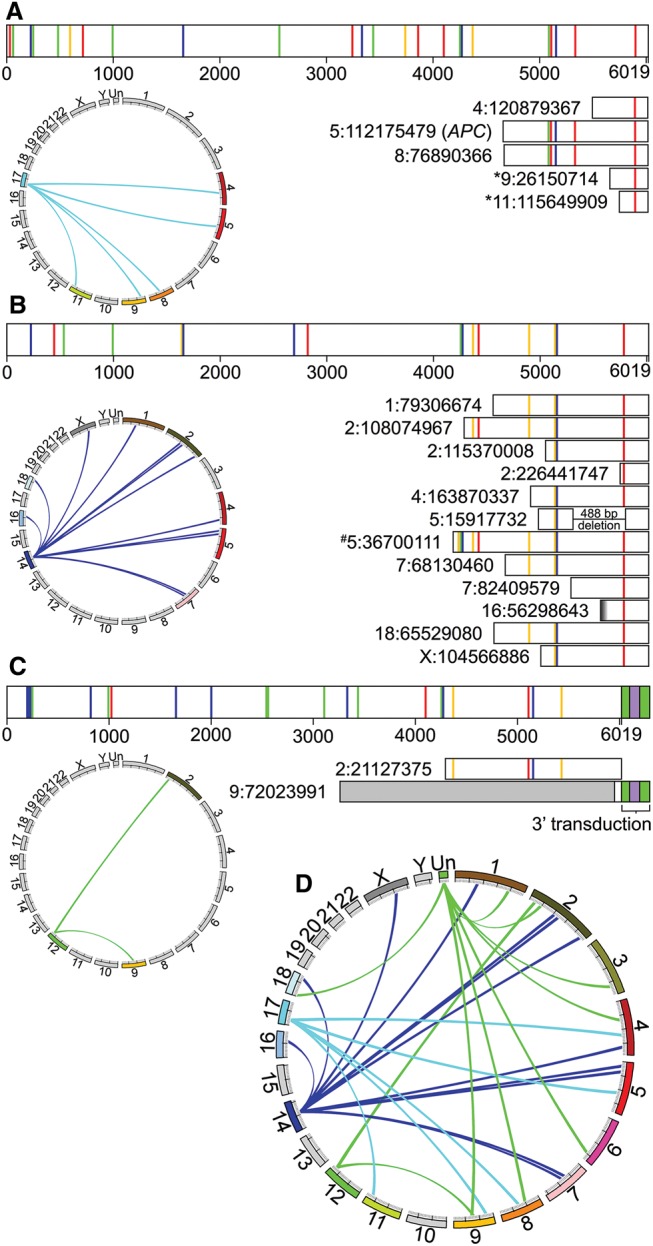
We next sought to better understand how the somatic L1 insertion in APC was generated in tissues that should have repressed the Chr 17 source element. The Chr 17 source element must have been active in normal cells at the earliest stages of tumorigenesis in order to generate the L1 insertion in APC sufficiently early to initiate tumorigenesis. This suggested the possibility that the Chr 17 source element might have been expressed in the normal somatic colon tissues of this patient. To determine whether this was the case, we performed strand-specific RNA-seq analysis and learned that the Chr 17 source element indeed was expressed in both the normal and tumor tissues of this patient (Fig. 4A; Supplemental Table S3; Methods). To perform this analysis, we leveraged the interior mutation profiles of the FL-L1Hs elements (Fig. 2; Supplemental Table S2) to identify source elements that were expressed in the patient's tissues. In each case, we identified multiple RNA-seq traces that exactly matched the unique mutation profiles of these source elements on the appropriate genomic DNA strand (Methods).
Figure 4.
FL-L1Hs source element expression in normal and tumor tissues. The unique interior mutation profiles of FL-L1Hs elements in the patient's genome (Fig. 2) were used to quantify expression of the 31 nonreference FL-L1Hs source elements including the Chr 17, Chr 14, and Chr 12 source elements (A) and the remaining 264 reference FL-L1Hs source elements in the patient's genome, using strand-specific RNA-seq (B) (Supplemental Table S3; Methods). Expression for each element is depicted in the normal and tumor tissues as the mean number of independent reads covering all mutations unique to a FL-L1Hs source element. Expression of FL-L1Hs source elements that could not be differentiated from the expression of the surrounding gene in the same orientation were excluded from this analysis (A, n = 4; B, n = 37) (Supplemental Table S3). The horizontal dotted lines represent a cutoff where the mean = two traces per unique site, and a total of 10 elements were expressed above this level. (C) DNA methylation analysis of the Chr 17 source element promoter. The top panel displays bisulfite sequencing results for the control 1000 Genomes Project (1KGP) sample (GWD sample HG02583, which is heterozygous for the Chr 17 element; Coriell). The two panels below show the results of bisulfite sequencing in the normal and tumor tissues of the CRC patient. Each circle represents a CpG site in the promoter (for a total of 29 CpGs at positions: 21, 37, 54, 60, 63, 72, 102, 137, 155, 160, 164, 166, 171, 181, 205, 231, 251, 255, 269, 284, 293, 305, 317, 320, 327, 351, 363, 369, 377 relative to the reference L1.3 sequence; GenBank ID L19088). White circles indicate no DNA methylation; black circles indicate DNA methylation. The red highlighting indicates the CpG at position 60 that previously was shown to be critical for repression of the L1 promoter by DNA methylation (Hata and Sakaki 1997). This CpG is mutated in the Chr 17 element (G61A). The three remaining CpG sites that are critical for repression of the L1 promoter by DNA methylation also are indicated (positions 54, 63, 72) (Hata and Sakaki 1997). The blue highlighting indicates a CpG to TpG mutation at position 37 that destroys an additional CpG in the promoter region.
In addition to the Chr 17 source element, the Chr 14 and Chr 12 source elements also were expressed in these tissues, although the Chr 12 element was expressed at very low levels and only in the normal tissues (Fig. 4A,B). Eight additional FL-L1Hs elements, including two nonreference and six reference elements, also evaded somatic repression and were expressed in the tissues of this patient (Fig. 4A,B). Consistent with the idea that FL-L1Hs elements are thought to be mostly repressed in normal colon tissues, we generally detected low levels (or no expression) of the remaining reference and nonreference FL-L1Hs elements (Fig. 4A,B; Supplemental Table S3).
To understand why the Chr 17 and Chr 14 source elements were inappropriately expressed in these tissues, we examined the interior sequences of these elements more closely. The Chr 17 element has a mutation (G61A) in its promoter region that maps to one of the four CpGs that previously were shown to be essential for suppression of the L1 promoter by DNA methylation (Fig. 4C; Hata and Sakaki 1997). We therefore sought to determine the DNA methylation status of the Chr 17 element in the affected patient. Bisulfite sequence analysis of the Chr 17 source element indicated that most of the CpG sites in the promoter region of this element were hypomethylated in both the normal and tumor tissues of this patient (Fig. 4C; Supplemental Methods). Therefore, it appears that the Chr 17 source element may have evaded somatic repression because its promoter was not sufficiently methylated at many CpG sites, including the critical CpG site at position 60 and three other critical sites that were identified in the Hata study (Fig. 4C; Hata and Sakaki 1997; Discussion).
The Chr 17 and Chr 14 source elements are restricted to African and African-derived populations
We next evaluated the population genetics of the Chr 17, Chr 14, and Chr 12 source elements in 26 diverse human populations using the MEI data that we generated for the 1000 Genomes Project (The 1000 Genomes Project Consortium 2015; Sudmant et al. 2015). Interestingly, we found that the Chr 17 and Chr 14 source elements are restricted to African and African-derived populations (Fig. 5; Supplemental Table S4). These data are consistent with the fact that our patient is African American (Methods) and that Beck et al. (2010) sequenced the same Chr 17 source element from a Yoruban individual. In contrast, the Chr 12 source element is found in all continental groups and populations (Fig. 5; Supplemental Table S4). Overall, these data indicate that the source FL-L1Hs content of an individual's genome is likely to vary considerably depending on the ancestry of the individual, and that differences in L1 content are likely to influence cancer risk. Our patient's genome had a population-specific hot L1 source element that is absent from most genomes and apparently increased her cancer risk considerably.
Figure 5.
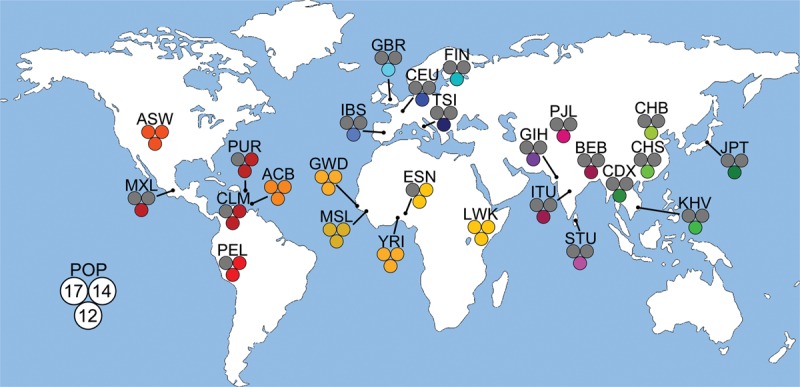
Population genetics of source elements in 26 diverse human populations. The 26 diverse human populations that were studied by the 1000 Genomes Project were examined to determine the frequencies of the Chr 17, Chr 14, and Chr 12 elements in global populations. The measurements are depicted by population on the world map as a set of three circles corresponding to the three FL-L1Hs source elements that gave rise to somatic offspring in this study with an accompanying population abbreviation (Supplemental Table S4): (Chr 17) upper left circle; (Chr 14) upper right circle; (Chr 12) bottom circle. Colored circles represent an allele frequency greater than 0 for that respective population, whereas gray circles represent an allele frequency of 0 (Supplemental Table S4). The Chr 17 and Chr 14 source elements are restricted to populations from Africa or African ancestry, whereas the Chr 12 element is found in all 26 of the diverse populations. World map provided by Vector Open Stock (www.vectoropenstock.com), under the Attribution Creative Commons 3.0 license.
Discussion
Next-generation sequencing has revolutionized somatic L1 discovery over the past 6 yr, leading to the identification of thousands of somatic L1 insertions in several types of human epithelial cancers (Iskow et al. 2010; Lee et al. 2012; Solyom et al. 2012; Shukla et al. 2013; Helman et al. 2014; Tubio et al. 2014; Doucet-O'Hare et al. 2015; Ewing et al. 2015; Rodic et al. 2015). The fact that L1 mobilization occurs frequently in human tumors raises the possibility that somatic L1 insertions could act as driver mutations during tumor initiation, progression, and metastasis. However, very few somatic L1 insertions have been recovered in known oncogenes and tumor suppressors, where a clear role in tumorigenesis could be established in the tumors in which they were discovered.
In addition to the Miki et al. (1992) insertion described above, one of the strongest driver candidates identified to date is a somatic L1 insertion in the ST18 gene that led to up-regulation of this gene in a case of hepatocellular carcinoma (Shukla et al. 2013). Interestingly, the insertion disrupted a repressor of transcription in an intron of the gene, which led to up-regulation of the gene in the tumor. In human breast and lung cancer, ST18 acts as a tumor suppressor (Jandrig et al. 2004; Job et al. 2010), but it appears to have acted as an oncogene in this case (Shukla et al. 2013). The precise role of ST18 in the development of hepatocellular carcinoma is unknown, and thus it is unclear whether this L1 insertion might have influenced tumor initiation, or instead, later stages of tumorigenesis. Another strong example of a potential L1 driver mutation was identified in the sixth exon of the PTEN tumor suppressor gene in a case of uterine corpus endometrial carcinoma (Helman et al. 2014). Because this insertion disrupted a coding exon, the tumor likely had lower levels of PTEN activity. However, once again, the precise role of this somatic insertion in this case of uterine cancer is unclear.
A number of other genes have been hit by somatic L1 insertions multiple times independently in tumors, suggesting that these insertions might also constitute driver mutations (e.g., Helman et al. 2014; Ewing et al. 2015; Rodic et al. 2015). However, most of these insertions map to sites within genes that are difficult to interpret (such as introns), or they map to genes with tenuous connections to the cancers in which they were discovered. Thus, even when L1 driver candidates are identified in a given tumor, it can be difficult to assign these mutations to clear roles in tumorigenesis, particularly in tissues for which the landscape of driver mutations has not been well established. In most tumor types, the temporal order whereby specific gene mutations influence tumorigenesis also has not been well established, making it even more difficult to determine whether L1 can initiate tumorigenesis.
Here, we present a clear example of a somatic L1 driver mutation initiating tumorigenesis through the classic route of CRC progression that has been mapped out by Vogelstein and colleagues (Fig. 6; Kinzler and Vogelstein 1996; Markowitz and Bertagnolli 2009; Fearon 2011; The Cancer Genome Atlas Network 2012). The identification of a second somatic L1 insertion in the APC gene >20 yr after the original Miki et al. (1992) insertion provided us with an opportunity to determine whether such insertions can actually initiate tumorigenesis. We show that both gatekeeper APC alleles are mutated in our patient's tumor, and this occurred in an MSS genetic background where such mutations would be expected to initiate CRC. A key factor in this study was the availability of relatively inexpensive whole-genome sequencing, which was not available at the time of the Miki et al. (1992) study. This allowed us to determine how the L1 insertion in APC was generated and then how it worked together with other mutations in APC, PIKC3A, KRAS, and ACVR1B to drive tumorigenesis (Fig. 6).
Figure 6.
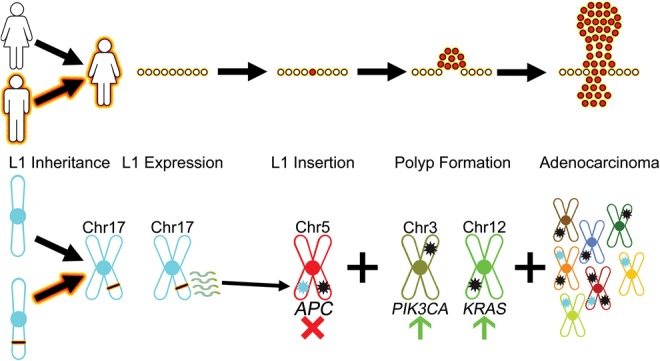
An oncogenic hot L1 evades somatic repression and initiates CRC. L1 Inheritance: Inheritance of a hot FL-L1Hs source element begins the process of L1-mediated cancer (this study). In this case, the patient inherited an African-specific hot FL-L1Hs source element on Chromosome 17 (black bar with fire outline, bottom) from one of her parents. L1 Expression: The inherited FL-L1Hs source element evades somatic repression and generates transcripts (squiggle lines, bottom) in normal colon tissues (this study). L1 Insertion: A somatic L1 offspring element is integrated into the sixteenth exon of the APC gene, thereby disrupting one APC allele (light blue star on Chr 5, bottom; this study) (Miki et al. 1992). The second APC allele is disrupted by the somatic mutation p.R1450* (black star on Chr 5, bottom; this study). Thus, both gatekeeper APC alleles are disrupted and the adenoma phase is initiated. Polyp Formation: Following loss of APC function, additional important driver mutations in the PIK3CA and KRAS genes (black stars on Chr 3 and Chr 12, respectively, bottom) result in progression to adenocarcinoma (cluster of red cells, top). Adenocarcinoma: Additional driver and passenger mutations occur to further drive progression of adenocarcinoma. These changes include new somatic L1 insertions (light blue stars, bottom), SNVs and indels (black stars, bottom), and perhaps other structural variants.
Our data support a model whereby an exceptionally hot L1 source element on Chromosome 17 of the patient's genome evaded somatic repression and produced an offspring insertion that disrupted the APC gene in a normal colon cell. This hot L1 source element has a mutation in one of the four CpGs that are necessary for repression of the L1 promoter by DNA methylation (Fig. 4C; Hata and Sakaki 1997). Indeed, our data indicate that this source element likely evaded somatic repression because its promoter was not sufficiently methylated (Fig. 4C). Moreover, all four of the CpGs that previously were shown to be essential for repression of the L1 promoter were either mutated or hypomethylated in the normal colon and tumor tissues of our patient (Fig. 4C). One intriguing possibility is that the G61A mutation that eliminated one of these critical CpGs also served as an “epimutation” that somehow caused the hypomethylation of the entire promoter region of the Chr 17 element. Similar mechanisms have been observed previously in several types of human cancers. For example, a recent report demonstrated that a 2-bp mutation altered the DNA methylation pattern of the entire RB1 gene promoter in a pedigree of human retinoblastomas (e.g., Quiñonez-Silva et al. 2016). If this was the case, then it appears that only a single mutation in L1 might have enabled the transposon to explore an entirely different niche of mutagenesis in somatic cells, thereby expanding upon its ability to mutagenize the germline.
Several previous studies have demonstrated that somatic L1 insertions occur relatively frequently in human CRC (Lee et al. 2012; Solyom et al. 2012; Tubio et al. 2014; Ewing et al. 2015). Somatic L1 insertions also are abundant in adenomatous polyps of the colon, suggesting that L1 mobilization can occur relatively early in the process of CRC (Tubio et al. 2014; Ewing et al. 2015). We now show that somatic L1 insertions can initiate CRC in normal colon cells, and thus, can act at the earliest stages of tumorigenesis that precede adenomatous polyp formation. Once adenomas are formed, it appears that additional somatic L1s can be generated (Tubio et al. 2014; Ewing et al. 2015). Although PIK3CA and KRAS mutations are thought to play key roles in driving the subsequent transition from adenoma to adenocarcinoma (Fearon 2011), it seems unlikely that L1 insertions could play a role in this transition, because such insertions probably have a limited capacity to activate oncogenes. In fact, in our patient's tumor, PIK3CA and KRAS were mutated by an activating somatic point mutation and a short duplication, respectively, underscoring the idea that L1 works together with other types of somatic mutations to drive human cancers. However, L1 could be envisioned to impact tumor suppressors such as PTEN, e.g., which has been implicated in later stages of CRC (Markowitz and Bertagnolli 2009; Fearon 2011).
Cancer risk from an African-specific hot L1
The hot FL-L1Hs Chr 17 source element, and a second source element on Chromosome 14 that generated most of the remaining somatic L1 insertions in our patient's tumor, are both restricted to African and African-derived individuals (Fig. 5). Such source elements appear to cause a novel form of ancestry-specific cancer risk. Remarkably, a wide range of epithelial cancers have been diagnosed in our patient's immediate family, suggesting the possibility that these two FL-L1Hs source elements might be responsible for increased levels of cancer risk in her family. In fact, the patient's father and seven of 12 siblings had a history of cancer (including vocal cord, breast, liver, prostate, lung, and esophageal cancers). These two FL-L1Hs source elements could potentially be active in a range of somatic tissues, where L1 is normally repressed and hence, may help to drive tumorigenesis in these other tissues. Such elements also may be active in somatic cells of the brain where they could impact neurological diseases such as Aicardi-Goutieres syndrome (Upton et al. 2015) and schizophrenia (Bundo et al. 2014) in an ancestry-specific manner. Finally, these elements would presumably remain active in the germline as well, where they could impact other human traits and diseases in an ancestry-specific manner.
As outlined above, several previous studies have suggested that L1 driver mutations may be rare in human tumors. However, a possible explanation for this observation is that the L1 insertions in these previous studies might have been generated by source elements that were derepressed only after tumorigenesis was well under way. As a consequence, such tumors might be enriched for passenger L1 insertions that played no clear role in tumorigenesis. Our study suggests that more emphasis needs to be placed on L1 source elements that evade somatic repression in normal tissues as a means to identify tumors that harbor L1 driver mutations. Follow-up studies with cancer-prone families like our patient's family, who carry source elements that can evade somatic repression, may help to establish a firmer link between somatic L1 mutagenesis and human cancers.
Impact of APC mutations
The somatic L1 insertion and the p.R1450* stop codon that we identified in APC both map to the mutation cluster region (MCR) of the gene, where the majority of disease-causing mutations have been discovered in sporadic CRCs (Fig. 1C). Most of the mutations that have been identified in sporadic CRCs introduce frameshifts or premature stop codons into the MCR and thus truncate the encoded APC protein within or near this region (Miyoshi et al. 1992). Both of the mutations discovered in our study cause these very same types of disruptions: The p.F1396L1 insertion causes a frameshift and premature termination of the encoded protein within the inserted L1, whereas the p.R1450* mutation introduces a stop codon that terminates the protein within the MCR. Moreover, the p.R1450* mutation occurred at one of the most commonly mutated APC codons in sporadic CRC (Polakis 1995; Fearon 2011). Thus, both of these terminations are consistent with previously identified disease-causing APC alleles in sporadic CRCs. Likewise, our RNA-seq data indicate that both of these mutant alleles were expressed in the tumor, suggesting that these variants escape nonsense-mediated mRNA decay (NMD) and likely produce truncated proteins (Supplemental Table S3).
Interestingly, the original Miki et al. (1992) L1 insertion lies slightly downstream from the MCR but is still very close to this region (codon 1526) (Fig. 1C). The Miki et al. (1992) insertion is in the same orientation as the APC gene, whereas our L1 is in the opposite orientation of APC. However, both L1 insertions cause frameshifts and terminate the protein within the inserted L1 sequences, suggesting that they effectively truncate APC in similar ways. Importantly, we now formally demonstrate that our somatic L1 insertion in APC initiated tumorigenesis. We cannot rule out that the Miki et al. (1992) insertion at codon 1526 also might have initiated CRC, but without knowing the status of the second allele and whether the tumor had stable microsatellites, it remains possible that the tumor was instead initiated by faulty DNA repair (Markowitz and Bertagnolli 2009; Fearon 2011). However, APC mutations are known to contribute to later stages of tumor progression in mutator-initiated CRCs, suggesting that both L1 insertions likely acted as driver mutations, perhaps at different stages of tumorigenesis. The fact that two independent L1 insertions have been identified in APC suggests that gateway tumor suppressors are sensitive targets for L1 mutagenesis, particularly when an inherited hot L1 evades somatic repression.
Methods
Genetic analysis of CRC patient tissue samples
Ten CRC samples with adjacent normal colon tissues were obtained from the Greenebaum Cancer Center Tumor Bank at the University of Maryland Medical Center under IRB protocol HP-00060447. The following samples were obtained: adenocarcinomas 19079, 19084, 19202, 20085, 20106, 20559; mucinous adenocarcinomas 19120 and 20444; tubular adenoma 20267; and carcinoid 20558. Patient 20444 (the major focus of this study) was a 62-yr-old female African American with a high-grade mucinous adenocarcinoma of the colon stage T4 N0. Genomic DNA was isolated from frozen tissue samples using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer's instructions. Total RNA for patient 20444 was isolated from both the normal and tumor tissues with the RNeasy Kit (Qiagen). Normal/tumor pairs were screened for somatic L1 insertions in CRC using the L1-seq method as described previously (Supplemental Methods; Iskow et al. 2010). WGS, RNA-seq, PCR validation of variants, and bisulfite sequencing were performed as described in the Supplemental Methods.
Microsatellite assessment using MSIsensor
Using MSIsensor (Niu et al. 2014), we determined the total number of somatically mutated microsatellites in our normal/tumor pair plus three control TCGA normal/tumor colorectal cancer pairs in which the microsatellite status was known (two MSS samples and one MSI sample) (The Cancer Genome Atlas Network 2012; data not shown). Cases with <3.5% of microsatellites somatically mutated are considered to have stable microsatellites (MSS), whereas cases with >3.5% of microsatellites somatically mutated are considered to have microsatellite instability (MSI).
MELT analysis
L1 discovery was performed on normal/tumor pairs using WGS Illumina data and the MELT algorithm with default parameters (http://melt.igs.umaryland.edu) (Supplemental Table S6; Supplemental Data S2; Sudmant et al. 2015). Sites were manually examined using the IGV (Thorvaldsdottir et al. 2013) and also were validated by PCR and/or Sanger sequencing (Supplemental Table S5; Supplemental Methods). Analysis of reference FL-L1Hs elements was conducted by genotyping all reference elements using the MELT-Deletion algorithm (http://melt.igs.umaryland.edu). The MELT-Deletion algorithm genotypes 4645 reference L1Hs elements using L1 sites provided by RepeatMasker (Smit et al. 1996–2010; Jurka et al. 2005). Of these, 19 FL-L1Hs source elements (L1Hs >5900 bp) were absent from the patient's genome and were excluded from further analysis (Supplemental Table S2; Supplemental Data S3).
Analysis of FL-L1 Hs source elements in the patient's genome
The Chr 14 and Chr 17 FL-L1Hs source elements were amplified with long-range PCR using LA Taq DNA Polymerase (Takara), cloned into plasmids with the TOPO XL PCR Cloning Kit (Invitrogen), and sequenced in triplicate with Sanger sequencing using 20 custom L1 primers and universal flanking primers (Supplemental Table S5). Thirty-one nonreference FL-L1Hs elements that were detected in the patient's genome with MELT were sequenced with Pacific Biosciences sequencing. Each element was amplified from either the normal sample DNA from patient 20444 or from a 1000 Genomes Project sample that also had the element (see Supplemental Table S5 for a listing of DNAs and primers). Amplification was performed using LA Taq DNA Polymerase (Takara) with the following reaction conditions (Supplemental Table S5): 90 sec at 94°C, followed by 32 cycles for 30 sec at 94°C, for 30 sec at 57°C, and for 8 min 30 sec at 68°C. A final elongation step was performed for 10 min at 68°C. All amplicons were subsequently pooled in approximately equimolar amounts and then purified with AMPure SizeSelect Beads (Beckman Coulter Genomics). The amplicons were prepared for PacBio sequencing using the DNA Template Prep Kit 1.0 (Pacific Biosciences) by following the manufacturer's protocol. Small fragments and extra adapters were removed from the sample using a BluePippin with a 0.75% agarose cassette (Sage Science). One SMRT cell was sequenced per amplicon pool, using P6C4 chemistry and a 240-min movie on the PacBio RS II.
Following PacBio sequencing, reads were aligned to their genomic locations using BLASR 1.3.1 (Chaisson and Tesler 2012). Reads were clustered based on genomic locations and assembled to form a consensus for each amplicon using version 2.3.0.140936 of ConsenseTools, which is available as part of the SMRTAnalysis software package from Pacific Biosciences (https://github.com/PacificBiosciences/SMRT-Analysis/wiki/ConsensusTools-v2.3.0-Documentation). SNVs and FL-L1Hs subfamilies were then identified using the LINEU tool found in the MELT analysis package (http://melt.igs.umaryland.edu). We used the Sanger sequencing and PacBio sequencing data for FL-L1Hs elements outlined above, together with the FL-L1Hs sequences available in hg19, to develop a profile of 295 potential source elements in the patient's genome (Fig. 2A,B; Supplemental Table S2). Equivalent results were obtained using FL-L1Hs sequences available in the GRCh38/hg38 build of the human genome reference sequence together with our nonreference FL-L1Hs elements.
Strand-specific RNA-seq analysis of FL-L1Hs source elements
Total RNA samples were prepared for strand-specific RNA-seq analysis by first treating the samples with DNase I (Invitrogen). Ribosomal RNA was reduced prior to library construction using the Ribo-Zero rRNA Removal Kit (Human/Mouse/Rat; Illumina). Illumina RNA-seq libraries were prepared from this material with the TruSeq RNA Sample Prep Kit (Illumina) according to the manufacturer's protocol. Between first and second strand cDNA synthesis, the primers and nucleotides were removed from the samples with NucAway spin columns (Ambion). The second strand was synthesized with a dNTP mix containing dUTP. Adapters containing indexes 6 nt in length were ligated to the double-stranded cDNA. After adapter ligation, the second strand cDNA was digested with 2 units of Uracil-N-Glycosylase (Applied Biosystems). The DNA was purified between enzymatic reactions, and size selection of the library was performed with AMPure XT beads (Beckman Coulter Genomics). Libraries were sequenced on the HiSeq4000 with paired end runs (one sample per lane) (Supplemental Table S6).
To evaluate FL-L1Hs RNA expression, raw RNA-seq FASTQ files that were generated by strand-specific RNA-seq (Supplemental Table S6) were aligned to the reference FL-L1Hs L1.3 element (GenBank ID L19088) (Dombroski et al. 1993) using Bowtie 2 version 2.2.4 (Langmead and Salzberg 2012). Reads with perfect matches to the FL-L1Hs element profiles outlined in Figure 2 were identified and quantified (Supplemental Table S3). To determine relative expression of all FL-L1Hs copies, the read coverages over element-specific SNV signatures were pooled to obtain a raw expression value for each element, and the mean coverage was reported (Fig. 4; Supplemental Table S3).
Data access
Sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under BioProject accession number PRJNA292328. The BioSample numbers are listed in Supplemental Table S6.
Acknowledgments
We thank Brandi Cantarel for help with informatics analysis; Naomi Sengamalay, Sandra Ott, Kelly Klega, Xuechu Zhao, Lisa Sadzewicz, and Luke Tallon for assistance with Illumina and PacBio sequencing; Doris Powell for advice on methylation experiments; Shari Corin for critical evaluation of the manuscript; and the three anonymous reviewers for helpful comments. This work was funded by the following NIH grants: National Cancer Institute (NCI) grant T32 CA154274 (E.C.S.), National Institute of Diabetes and Digestive and Kidney Diseases grant T32 DK067872 (N.T.C.), NCI grant R01 CA077337 (P.M.V.), NCI grant R01 CA166661 (S.E.D.), and National Human Genome Research Institute grant R01 HG002898 (S.E.D.).
Author contributions: E.C.S. performed L1-seq assays, PCR validations, sequencing of somatic L1 insertions, analysis of the two APC alleles, cloning and sequencing of the Chr 17 and Chr 14 FL-L1Hs elements with Sanger capillary sequencing, and bisulfite methylation experiments; E.J.G. aligned WGS raw sequencing data to the reference genome, generated BAM files, and performed MELT analysis, RNA-seq analysis, FL-L1Hs analysis, PCR validations, and bisulfite analysis; A.M. performed non-MEI somatic variant analysis and patient data evaluation; E.J.G. and N.T.C. performed PacBio FL-L1Hs experiments and analysis. E.C.S., P.M.V., and S.E.D. designed methylation experiments. E.C.S., E.J.G., and S.E.D. designed experiments, performed data analysis, prepared display items, and wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.201814.115.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, et al. 2011. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479: 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. 2010. LINE-1 retrotransposition activity in human genomes. Cell 141: 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Garcia-Perez JL, Badge RM, Moran JV. 2011. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet 12: 187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Chevret P, Furano AV. 2000. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol 17: 915–928. [DOI] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr. 2003. Hot L1s account for the bulk of retrotransposition activity in the human population. Proc Natl Acad Sci 100: 5280–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundo M, Toyoshima M, Okada Y, Akamatsu W, Ueda J, Nemoto-Miyauchi T, Sunaga F, Toritsuka M, Ikawa D, Kakita A, et al. 2014. Increased L1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81: 306–313. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH. 2009. L1 retrotransposition in human neural progenitor cells. Nature 460: 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombroski BA, Scott AF, Kazazian HH Jr. 1993. Two additional potential retrotransposons isolated from a human L1 subfamily that contains an active retrotransposable element. Proc Natl Acad Sci 90: 6513–6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet-O'Hare TT, Rodić N, Sharma R, Darbari I, Abril G, Choi JA, Young Ahn J, Cheng Y, Anders RA, Burns KH, et al. 2015. LINE-1 expression and retrotransposition in Barrett's esophagus and esophageal carcinoma. Proc Natl Acad Sci 112: E4894–E4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrony GD, Cai X, Lee E, Hills LB, Elhosary PC, Lehmann HS, Parker JJ, Atabay KD, Gilmore EC, Poduri A, et al. 2012. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151: 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Kazazian HH Jr. 2010. High throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res 20: 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Gacita A, Wood LD, Ma F, Xing D, Kim MS, Manda SS, Abril G, Pereira G, Makohon-Moore A, et al. 2015. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Res 25: 1536–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER. 2011. Molecular genetics of colorectal cancer. Annu Rev Pathol 6: 479–507. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al. 2014. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43: D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Lutz S, Morrish TA, Moran JV. 2005. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol 25: 7780–7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata K, Sakaki Y. 1997. Identification of critical CpG sites for repression of L1 transcription by DNA methylation. Gene 189: 227–234. [DOI] [PubMed] [Google Scholar]
- Helman E, Lawrence MS, Stewart C, Sougnez C, Getz G, Meyerson M. 2014. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res 24: 1053–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CR, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, et al. 2010. Mobile interspersed repeats are major structural variants in the human genome. Cell 141: 1171–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE. 2010. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 141: 1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandrig B, Seitz S, Hinzmann B, Arnold W, Micheel B, Koelble K, Siebert R, Schwartz A, Ruecker K, Schlag PM, et al. 2004. ST18 is a breast cancer tumor suppressor gene at human chromosome 8q11.2. Oncogene 23: 9295–9302. [DOI] [PubMed] [Google Scholar]
- Job B, Bernheim A, Beau-Faller M, Camilleri-Broët S, Girard P, Hofman P, Majières J, Toujani S, Lacroix L, Laffaire J, et al. 2010. Genomic aberrations in lung adenocarcinoma in never smokers. PLoS One 5: e15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. 1997. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci 94: 1872–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. 1996. Lessons from hereditary colorectal cancer. Cell 87: 159–170. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ III, Lohr JG, Harris CC, Ding L, Wilson RK, et al. 2012. Landscape of somatic retrotransposition in human cancers. Science 337: 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. 1993. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 72: 595–605. [DOI] [PubMed] [Google Scholar]
- Markowitz SD, Bertagnolli MM. 2009. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y. 1992. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res 52: 643–645. [PubMed] [Google Scholar]
- Miyoshi Y, Nagase H, Ando H, Hori A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. 1992. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1: 229–233. [DOI] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr. 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87: 917–927. [DOI] [PubMed] [Google Scholar]
- Moran JV, DeBerardinis RJ, Kazazian HH Jr. 1999. Exon shuffling by L1 retrotransposition. Science 283: 1530–1534. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. 2005. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435: 903–910. [DOI] [PubMed] [Google Scholar]
- Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, Wendl MC, Ding L. 2014. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 30: 1015–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EA, Kazazian HH Jr. 2001. Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res 11: 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. 1995. Mutations in the APC gene and their implications for protein structure and function. Curr Opin Genet Dev 5: 66–71. [DOI] [PubMed] [Google Scholar]
- Quiñonez-Silva G, Dávalos-Salas M, Recillas-Targa F, Ostrosky-Wegman P, Aranda D, Benítez-Bribiesca L. 2016. Monoallelic germline methylation and sequence variant in the promoter of the RB1 gene: a possible constitutive epimutation in hereditary retinoblastoma. Clin Epigenetics 8: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodić N, Steranka JP, Makohon-Moore A, Moyer A, Shen P, Sharma R, Kohutek ZA, Huang CR, Ahn D, Mita P, et al. 2015. Retrotransposon insertions in the clonal evolution of pancreatic ductal adenocarcinoma. Nat Med 21: 1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R, Upton KR, Muñoz-Lopez M, Gearhardt DJ, Fisher ME, Nguyen T, Brennan PM, Baillie JK, Collino A, Ghisletti S, et al. 2013. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 153: 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 1996–2010. RepeatMasker Open 3.0. http://www.repeatmasker.org.
- Solyom S, Ewing AD, Rahrmann EP, Doucet T, Nelson HH, Burns MB, Harris RS, Sigmon DF, Casella A, Erlanger B, et al. 2012. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res 22: 2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C, Kural D, Strömberg MP, Walker JA, Konkel MK, Stütz AM, Urban AE, Grubert F, Lam HY, Lee WP, et al. 2011. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet 7: e1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MHY, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio JM, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, Gundem G, Pipinikas CP, Zamora J, Raine K, et al. 2014. Mobile DNA in cancer: extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 345: 1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton KR, Gerhardt DJ, Jesuadian JS, Richardson SR, Sánchez-Luque FJ, Bodea GO, Ewing A, Salvador-Palomeque C, van der Knapp MS, Brennan PM, et al. 2015. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161: 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]