

Abstract
The maternal and paternal copies of the genome are both required for mammalian development, and this is primarily due to imprinted genes, those that are monoallelically expressed based on parent-of-origin. Typically, this pattern of expression is regulated by differentially methylated regions (DMRs) that are established in the germline and maintained after fertilization. There are a large number of germline DMRs that have not yet been associated with imprinting, and their function in development is unknown. In this study, we developed a genome-wide approach to identify novel imprinted DMRs in the human placenta and investigated the dynamics of these imprinted DMRs during development in somatic and extraembryonic tissues. DNA methylation was evaluated using the Illumina HumanMethylation450 array in 134 human tissue samples, publicly available reduced representation bisulfite sequencing in the human embryo and germ cells, and targeted bisulfite sequencing in term placentas. Forty-three known and 101 novel imprinted DMRs were identified in the human placenta by comparing methylation between diandric and digynic triploid conceptions in addition to female and male gametes. Seventy-two novel DMRs showed a pattern consistent with placental-specific imprinting, and this monoallelic methylation was entirely maternal in origin. Strikingly, these DMRs exhibited polymorphic imprinted methylation between placental samples. These data suggest that imprinting in human development is far more extensive and dynamic than previously reported and that the placenta preferentially maintains maternal germline-derived DNA methylation.
The two haploid copies of the genome are not interchangeable, as both a maternally and paternally inherited copy is required for mammalian development. This is primarily due to imprinted genes, those that are expressed from a single allele based on parent-of-origin. This class of genes is particularly important for fetal and placental development (Frost and Moore 2010).
The monoallelic expression of imprinted genes is controlled by differentially methylated regions (DMRs) that are established either as “primary” (germline) imprints in the female or male gametes or as “secondary” (somatic) imprints in embryonic development. Imprinted genes can occur in isolation but are often found in centrally regulated clusters. Imprinted DMRs are all CpG-rich regions; however, paternal DMRs tend to be intergenic while maternal DMRs are located at promoters or intragenic regions (Kobayashi et al. 2006; Schulz et al. 2010).
Primary imprinted DMRs are erased and reestablished during germline development of each generation dependent on the sex of the embryo (Kelsey and Feil 2013). Shortly after germ cell specification in the embryo, there is a genome-wide removal of DNA methylation, thereby erasing all prior parentally inherited imprints (Seisenberger et al. 2012). Methylation is then reacquired in spermatocytes around the time of birth in males, while in females, DNA methylation is not fully established until oocyte growth in adulthood (Lucifero et al. 2004; Henckel et al. 2012). Once established, imprints are then passed on to offspring upon formation of the zygote and are maintained through the dynamic epigenetic reprogramming in the early embryo. Our current knowledge about the mechanisms and dynamics of imprinting has been determined almost exclusively using mouse models; however, evidence from human studies suggests that these processes are largely conserved (Ishida and Moore 2013; Guo et al. 2014).
Currently, 151 imprinted genes have been identified in mouse (www.mousebook.org), while there appears to be less extensive imprinting in humans, with only 78 genes identified (http://igc.otago.ac.nz). Despite this difference, the majority of the well-studied imprinted clusters are conserved in their expression, methylation patterns, and synteny (Onyango et al. 2000). In both mice and humans, a large number of DMRs of unknown function have been reported between sperm and oocyte (Smallwood et al. 2011; Guo et al. 2014), which raises the question as to whether imprinting is more extensive in the genome than previously reported.
Over the past decade, multiple approaches to identify novel imprinted genes in the human genome have been utilized, including comparative methodologies for genomic features (Luedi et al. 2007), gene expression (Metsalu et al. 2014; Babak et al. 2015), and DNA methylation (Choufani et al. 2011; Yuen et al. 2011a; Das et al. 2013; Stelzer et al. 2013; Court et al. 2014; Docherty et al. 2014) in various tissues. Importantly, these studies have resulted in the discovery and validation of many imprinted genes; however, the candidate lists are often not replicated between studies. Until recently, the genome-wide pattern of DNA methylation in human oocytes was not known (Guo et al. 2014), preventing the definitive identification of novel primary imprints in humans. In this study, we describe the dynamics of known imprinted DMRs throughout human development in somatic and extraembryonic tissues and identify novel germline imprinted DMRs utilizing gametic DNA methylation in combination with a validated approach of comparing placental samples with genomic imbalances (Yuen et al. 2011a).
Results
Characterization of known imprinted DMRs in human development
To evaluate imprinting in the human placenta, we identified DMRs (≥3 CpGs within 500 bp) between five diandric (two paternal copies of the genome and one maternal) and five digynic (two maternal copies and one paternal) triploid placentas, measured by the Illumina HumanMethylation450 BeadChip array (450K array) (Fig. 1A). Eight hundred eighty-two candidate DMRs, encompassing 6807 probes (CpG sites), were identified (Supplemental Table 1), including 43 that overlapped previously reported human imprinted DMRs (Supplemental Table 2; Court et al. 2014).
Figure 1.

Characterization of known imprinted DMRs in human development. (A) Digynic triploid conceptions contain two maternal (white) and one paternal (gray) genome, while diandric triploid conceptions contain one maternal and two paternal genomes. By comparing digynic (N = 5) and diandric (N = 5) triploid placental villi, DMRs were identified, defined as ≥3 CpGs within 500 bp. An example plot shows a maternal DMR near PEG3, which is more highly methylated among digynic triploid samples (black line), compared to diandric triploid samples (red line) across 37 CpG sites over a ∼4-kb region. (B) DNA methylation through early human development is shown for 43 DMRs, identified between triploid samples that overlap previously reported imprinted DMRs (Court et al. 2014). DNA methylation for human germ cells, early embryonic stages (zygote, two-cell, four-cell, eight-cell, and morula stage embryos), inner cell mass, and trophectoderm is an average of CpG sites across each DMR measured by RRBS. DNA methylation for placental villi, fetal tissues (brain, kidney, muscle, and spinal cord), and whole blood was an average of 450K array probes across each DMR. Primary (germline) imprinted DMRs (light gray), placental-specific DMRs (black), and secondary DMRs (dark gray) are denoted. Parental origin of DNA methylation is designated as maternal (magenta) or paternal (blue). White boxes in the heat map indicate DMRs with no data.
DNA methylation at these 43 known DMRs was assessed using the 450K array in fetal and extraembryonic tissues and using publicly available reduced representation bisulfite sequencing (RRBS) in human gametes and embryos (Guo et al. 2014). Thirty-seven DMRs were informative for germline DNA methylation and, as expected, the majority showed >50% difference in DNA methylation between germ cells, including two paternal and 32 maternal germline DMRs (Fig. 1B). The remaining three DMRs (MEG3, ZNF597, and GNAS DMR1) were unmethylated both in germ cells and the early embryo, suggesting these are somatic DMRs (Fig. 1B). Among the known DMRs, 15 are placental-specific in humans (Court et al. 2014) and all showed differential methylation between oocytes and sperm, with a mean difference of 67.5% (Fig. 1B). These placental-specific DMRs are intermediately methylated in both the inner cell mass (ICM) (35.6%) and trophectoderm (TE) (34.1%) of the blastocyst, suggesting that they are transiently imprinted in embryo, not just extraembryonic TE (Fig. 1B).
Identification of novel imprinted placental DMRs
We sought to identify novel imprinted DMRs by identifying CpG islands (CGIs) that showed a profile consistent with imprinting in early human development: DMRs that were both differentially methylated in the germline and between triploid placental samples. First, CGIs that were differentially methylated in the germline and intermediately methylated in blastocysts were identified (specific criteria detailed in the Methods), which included 1222 oocyte-specific and 288 sperm-specific DMRs (Fig. 2A). Of these candidate CGIs, 101 novel regions overlapped with the 882 DMRs identified in triploid placental villous samples (Fig. 2B). All 101 novel DMRs derived methylation from the oocyte, and the majority (N = 72) showed a placental-specific pattern of imprinting (Fig. 2C).
Figure 2.

Identification of novel imprinted placental DMRs. (A) DNA methylation is shown for oocyte-specific (left panel) and sperm-specific (right panel) DMRs in human gametes and early embryonic stages (zygote, two-cell, four-cell, eight-cell, and morula stage embryos), ICM, and TE. DMRs were defined as CGIs that showed a >50% methylation difference between gametes, with intermediate methylation (15%–60%) in the ICM and TE. (B) The number of DMRs that overlap between those identified between human gametes and those identified between triploid placental samples is shown. (C) DNA methylation through early human development is shown for the 101 novel imprinted DMRs. DNA methylation for human gametes, early embryonic stages (zygote, two-cell, four-cell, eight-cell, and morula stage embryos), ICM, and TE is an average of CpG sites across each DMR, measured by RRBS. DNA methylation for placental villi, fetal tissues (brain, kidney, muscle, and spinal cord), and whole blood was an average of 450K array probes across each DMR. Seventy-two novel DMRs showed a placental-specific pattern of imprinting, defined as <25% or >75% methylation in somatic tissues and intermediately (25%–75%) methylated in placenta.
To confirm that the novel DMRs maintain imprinted allele-specific methylation throughout development, allelic methylation was evaluated in oocytes, sperm, ICM, and TE for known and novel DMRs. This reaffirmed that ∼50% of DNA strands in the ICM and TE were partially/fully methylated and the proportion of methylated alleles was not significantly different between known and novel DMRs (Fig. 3A). It can be inferred that these methylated alleles were almost entirely derived from the oocyte (Fig. 3A), because it is assumed that there is almost no de novo methylation in the preimplantation embryo (Seisenberger et al. 2013).
Figure 3.

Imprinted methylation of novel placental DMRs. (A) DNA methylation was averaged across reads with ≥5 CpGs from RRBS. The proportion of reads that was completely unmethylated (black), methylated (red), or had mixed methylation (gray) is shown for oocyte, sperm, ICM, and TE for germline known (N = 40) and novel (N = 101) DMRs. The proportion of methylated alleles did not differ between known and novel DMRs (P = 0.981, χ2 test). (B) DNA methylation was averaged across reads with ≥5 CpGs from multiplexed bisulfite sequencing in 16 placental villi and 12 matched trophoblast samples. Distribution of read methylation across DMRs (binned in 10% intervals) is shown in the density plot, with a density heat map in grayscale showing the distribution at each known (orange) and novel (blue) DMR below. DMRs with a minimum read depth of ≥5 were reported. (C) Average DNA methylation is shown for the methylated and unmethylated allele at known (orange) and novel (blue) DMRs containing a heterozygous SNP. Each row corresponds to one heterozygous SNP for one individual at one DMR; for loci with more than one informative SNP, the SNP number is designated after the sample name. Parental origin was assigned where informative, with maternal (red) and paternal (blue) monoallelic methylation, alleles with no differential methylation (gray), and uninformative alleles (white) denoted.
In placenta, allelic DNA methylation was assayed at known and novel DMRs using multiplexed bisulfite sequencing (Supplemental Table 5) in 16 term placental villi samples with matched isolated trophoblast (N = 12) and parental blood (N = 20). One hundred two DMRs (32 known and 70 novel) were successfully assayed, revealing a bimodal distribution of read methylation in term placenta and trophoblast at these loci (Fig. 3B), consistent with that in the ICM and TE (Supplemental Fig. 1). This bimodal methylation pattern is indicative of monoallelic methylation, as opposed to heterogeneous CpG methylation expected at nonimprinted, intermediately methylated loci. DMRs that contained heterozygous SNPs showed that indeed this bimodal methylation pattern was attributable to monoallelic methylation (Fig. 3C). Furthermore, DMRs with informative parental origin showed that monoallelic methylation was entirely maternally derived, with the exception of paternally methylated H19 (Fig. 3C).
Polymorphic imprinted methylation in the human placenta
Strikingly, methylation across the novel DMRs was highly polymorphic, with 50% of heterozygous samples demonstrating monoallelic methylation and the remaining showing a complete loss of imprinting at these loci (Fig. 3C). The frequency of polymorphic imprinting was similar in placental villi (50.0%) and trophoblast (49.4%) (Supplemental Table 3), and allelic methylation was highly correlated between matched trophoblast and whole villi from the same individual (N = 34, R2 = 0.73, P < 0.0001, Spearman rank correlation). This suggests that the pattern of polymorphic imprinting is not attributable to differences in cell composition. Polymorphic methylation was confirmed among the 63 placental samples run on the 450K array (Fig. 4A), which included four samples assayed by multiplexed bisulfite sequencing. Using hierarchical clustering, the samples did not cluster by gestational age, suggesting polymorphic methylation across these loci is also not gestational age-dependent. Furthermore, polymorphic imprinting at these DMRs was placental-specific, as somatic tissues showed significantly less variability than placenta (P < 2.2 × 10−16) (Fig. 4B). These data suggest that polymorphic imprinting is pervasive in the human placenta.
Figure 4.

Polymorphic imprinted methylation in the human placenta. (A) DNA methylation shows a polymorphic pattern across known (orange) and novel (blue) DMRs among 63 placental samples from 1st (N = 5, black), 2nd (N = 9, light gray), and 3rd (N = 49, dark gray) trimester samples. The color scale was set to show those samples that deviate from the intermediate range of methylation. (B) Mean standard deviation across all DMRs (N = 144) is shown for placenta, fetal somatic tissues, and adult whole blood (P < 2.2 × 10−16, Kruskal–Wallis). Y-axis is on a logarithmic scale.
The placental-specific imprinted genes reported to date in human and mouse are not well-conserved (Frost and Moore 2010); therefore, we evaluated the DNA methylation of orthologous regions to placental-specific DMRs in mouse development using publicly available data (Smith et al. 2012, 2014; Hon et al. 2013). Only six DMRs showed differential germline DNA methylation in mouse gametes, but only one locus, orthologous to NAV2, showed a pattern consistent with placental-specific imprinting (Supplemental Fig. 2).
Timing of loss of imprinting in human embryonic lineages
To assess the developmental timing of loss of imprinting at placental-specific imprinted DMRs in somatic lineages, we took advantage of the methylation patterns observed in isolated extraembryonic cell types. The various cells contributing to the human placenta vary in the timing of differentiation and also the degree to which they comprise cells from embryonic origins. As extraembryonic tissues progressively originate later in development, they are more similar to the embryo (Fig. 5A). We found that the trophoblast and mesenchymal core of the placental villi showed the expected intermediate methylation at known placental-specific DMRs (51.3% and 46.5%, respectively). This supports that these DMRs are still imprinted in the TE and ICM after the blastocyst stage; while trophoblast originates entirely from the TE, mesenchyme is a mix of cell types largely derived from the ICM-derived extraembryonic mesoderm (Fig. 5A). Contrastingly, the average methylation was lower in chorion (33.0%) and amnion (8.3%) (Fig. 5B), and both mostly originate later from the extraembryonic mesoderm (Fig. 5A).
Figure 5.
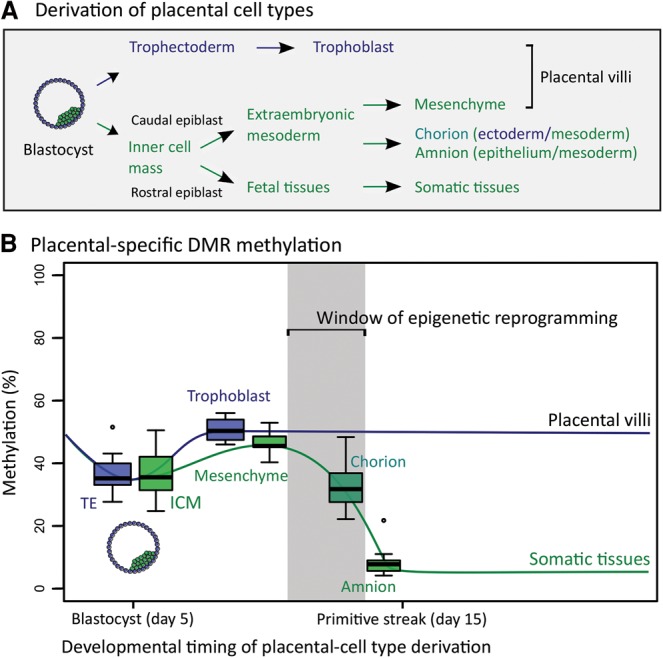
Timing of loss of imprinting in human embryonic lineages. (A) Schematic representation of the developmental origins of the extraembryonic cell types is shown, with TE-derived cells (dark blue), ICM-derived cells (green), and cells with mixed origin (turquoise) denoted. (B) The distribution of DNA methylation for known placental-specific DMRs that lose DNA methylation in somatic tissues (N = 14) is shown for extraembryonic cell types isolated from term placenta. Methylation for trophoblast and mesenchyme is an average from 1st, 2nd, and 3rd trimester samples. TE-derived cells (dark blue), ICM-derived cells (green), and cells with mixed origin (turquoise) are denoted. Extraembryonic cell types are ordered based on the approximate timing in development when each cell type is derived. The window of epigenetic reprogramming highlights the period in which DNA methylation at placental-specific DMRs is likely being erased in the ICM-derived lineage.
The exact developmental timing and origin of amnion and chorion are not firmly established. In mouse and, likely, in human, chorion is derived from both the TE and extraembryonic mesoderm (Downs 2011; Pereira et al. 2011), while molecular data in human suggest that there may also be a later contribution from epiblast, prior to primitive streak formation at day 15, to both chorion and amnion (Robinson et al. 2002; Penaherrera et al. 2012). The relative loss of DNA methylation observed in chorion and amnion compared to placental villi suggests that imprinting at placental-specific DMRs in the embryonic lineages has already begun to be lost at the time of primitive streak formation (Fig. 5B). Thus, it appears that these loci are either passively or actively losing methylation from the maternal allele, while much of the genome is acquiring de novo DNA methylation concomitant with somatic differentiation (Smith et al. 2012).
Characteristics and function of placental-specific DMRs
The monoallelic methylation of placental-specific DMRs (16 known and 72 novel) may be attributable to targeted protection of these regions or to the fact that these regions are a vestige of embryonic methylation patterns. To evaluate this, we first assessed whether there was evidence of de novo methylation in the post-implantation placenta. One thousand three hundred fifty-two CGIs were fully methylated in both oocyte and sperm, and are therefore not imprinted, and retained intermediate DNA methylation in the ICM and TE. In somatic tissues, almost all of these CGIs undergo de novo methylation to become fully methylated (83.2%), and this was similarly observed in the placenta (70.1%) (Supplemental Fig. 3A), suggesting the placenta exhibits a phase of post-implantation de novo DNA methylation. In addition, the majority of placental-specific DMRs (80.7%) were not associated with previously described large partially methylated domains characteristic of the placenta (Supplemental Fig. 3B; Schroeder et al. 2013). Together, these findings suggest that placental-specific DMRs may be specifically protected from loss or gain of methylation.
Zinc finger protein 57 (ZFP57) and di-/tri-methylation of histone 3 lysine 9 (H3K9me2/3) have been shown to protect imprinted DMRs from demethylation during embryonic reprogramming in mouse (Li et al. 2008; Nakamura et al. 2012) and thus may be important to facilitate the protection of placental-specific imprinting in humans. We observed a slightly higher proportion of placental-specific DMRs (62.9%) with at least one ZFP57 consensus sequence (TGCCGC) (Quenneville et al. 2011) than CGIs genome-wide (51.5%; P = 0.04), which was comparable to known imprinted DMRs (70.8%; P = 0.63) (Fig. 6A). Placental-specific DMRs were also enriched for H3K9me3 in placenta compared to somatic tissues (muscle, P = 2.2 × 10−16 and amnion, P = 2.2 × 10−16) (Fig. 6B; Supplemental Fig. 4). However, this enrichment was not a unique feature, as all CGIs were enriched for H3K9me3 in placenta compared to somatic tissue (muscle, P = 2.2 × 10−16 and amnion, P = 3.1 × 10−14) (Fig. 6B; Supplemental Fig. 4). These findings suggest that ZFP57 and H3K9me3 may, at least partially, be important for the maintenance of monoallelic DNA methylation at placental-specific DMRs; however, this is likely not a specific mechanism.
Figure 6.

Characteristics of placental-specific DMRs. (A) The proportion of known imprinted germline DMRs, placental-specific DMRs, and CGIs with one or more ZFP57 binding site consensus sequences is shown. (B) Enrichment for H3K9me3, using 1000-bp sliding windows with a 1000-bp step, was compared between human placenta and fetal muscle. Using the absolute difference for each probe, placental-specific DMRs (P = 2.2 × 10−16, t-test) and CGIs (P = 2.2 × 10−16, t-test) showed a significant enrichment compared to all other probes, in placenta. Placental-specific DMRs were not significantly different from CGIs (P = 0.70, t-test). (C) Expression of genes associated with placental-specific DMRs based on closest transcription start site (Supplemental Table 6) is shown in TE, and 2nd and 3rd trimester placental villi.
To investigate the potential role for placental-specific imprinting, gene expression in placenta was assessed using publicly available RNA-seq data sets (Bernstein et al. 2010; Yan et al. 2013; Metsalu et al. 2014). Fifty percent of the placental-specific DMRs had an associated gene that was expressed in at least one developmental stage (RPKM > 0.1) (Fig. 6C; Supplemental Table 6). However, gene ontology analysis revealed no enrichment for pathways typically associated with imprinted genes, such as fetal growth or placentation. The top gene ontology terms included alternative splicing, expression in the brain, splice variants, and the extracellular region (Supplemental Table 7).
Discussion
In this study, DNA methylation in human gametes and triploid placental villi was used to identify germline imprinted DMRs. We found 43 imprinted DMRs in placenta that overlapped with previously reported imprinted DMRs initially identified in somatic tissues (Court et al. 2014), and 34 of these were confirmed to be germline imprints. Fifteen DMRs reported by Court et al were not identified in this study; notably, 10 of these did not show germline differential methylation and therefore may be secondary DMRs that are not fully established in placenta. Fifteen known placental-specific DMRs did show differential germline methylation. This is supported by a similar finding in an independent study of oocyte DNA methylation (Okae et al. 2014); however, these findings contradict a previous report suggesting that these regions are unmethylated in human gametes (Court et al. 2014). This discrepancy may be explained by the use of parthenogenetic embryonic stem cells as a proxy for the oocyte methylome in that study (Court et al. 2014).
Recently, it was shown that many regions of the genome are differentially methylated between oocyte and sperm beyond the widely studied imprinted DMRs (Smallwood et al. 2011; Kobayashi et al. 2012; Guo et al. 2014), and the importance of this DNA methylation during development has been unclear to date. We have determined that some of the additional differential methylation in the human germline results in more widespread imprinting, specifically in the placenta. We identified 101 novel maternal germline imprinted DMRs in human placenta and found that 72 were placenta-specific. The monoallelic DNA methylation at these DMRs was validated by targeted multiplexed bisulfite sequencing in placenta and trophoblast, confirming maternally derived methylation in all informative cases.
While the importance of imprinting in placental development has been well-established (Varmuza and Miri 2015), the role of placental-specific imprinting has not been fully explored. Consistent with previously reported placental-specific imprinted genes (Frost and Moore 2010), the human placental-specific DMRs we identified were not conserved in mouse. This is somewhat unsurprising, because although imprinting is quite robustly conserved across eutherian mammals (Renfree et al. 2013), many of the additional germline DMRs identified thus far have not been conserved in methylation state across species (Smith et al. 2014). There has not been a genome-wide investigation of placental-specific imprinting in many other species, and therefore the extent and role of placental-specific imprinting in eutherian mammals is unclear.
All 101 novel DMRs we identified had maternally inherited DNA methylation, raising the number of maternal germline imprinted DMRs to at least 140 loci in placenta. With only two paternally derived germline imprinted DMRs identified, it suggests that the maternal genome is almost solely responsible for inter-generational epigenetic inheritance, in the form of germline imprinted DMRs. In the assessment of methylation at all CGIs in germ cells and embryos, we observed that oocyte-derived DNA methylation is more likely to be retained in the blastocyst; this is consistent with the majority of imprinted domains being maternal in origin and with previous observations made in the mouse (Smallwood et al. 2011). The maternally inherited DNA in the embryo is primarily passively demethylated after fertilization, as opposed to the active demethylation that occurs on paternal DNA (Santos et al. 2002; Inoue and Zhang 2011); this passive demethylation may more readily allow oocyte-derived methylation to be maintained. In addition, maternal DMRs are CG-rich (Kobayashi et al. 2006) and may therefore be more likely to contain a CG-rich domain like the ZFP57 binding site and preferentially acquire H3K9me3 in the placenta. The placenta may be able to uniquely maintain much of this monoallelic DNA methylation throughout development, perhaps through recruitment of protective complexes. Intriguingly, unlike mice, ZFP57 is not expressed in human oocytes but is expressed in the blastocyst (Yan et al. 2013), suggesting that it may have differing roles in the protection of imprints in human and mouse development.
The polymorphic imprinted methylation among the novel DMRs in the human placenta may be explained by underlying genetic variants affecting promoter usage or genomic variation affecting the surrounding epigenetic profile. Alternatively, it may be a reflection of epigenetic stochasticity present in early development, when only a small number of cells will contribute to the developed tissue, and that such methylation states are fixed in the placenta and not overwritten by global remethylation as occurs in somatic lineages. The future study of genetic and genomic variations in conjunction with placental methylation will help elucidate this.
While many genes associated with placental-specific DMRs are expressed in placenta, the role of the monoallelic methylation in regulating transcription remains unclear and is likely not consistent across loci. Placental-specific DMRs have been shown to regulate monoallelic expression, including DNMT1 (Yuen et al. 2011a; Das et al. 2013) and the Chromosome 19 microRNA cluster (C19MC) (Noguer-Dance et al. 2010). There are also examples of loci where biallelic expression has been documented despite a nearby imprinted DMR, such as at RASGRF1 and C3orf62 (Yuen et al. 2011a). This suggests that additional factors may be important for imprinted gene regulation, supported by the observation in mouse that monoallelic methylation of Rasgrf1 is widespread across tissues but only mediates monoallelic expression in a subset (Dockery et al. 2009). Furthermore, polymorphic imprinted expression has been described for IGF2R and SLC22A2, despite no difference in DNA methylation (Monk et al. 2006). Therefore, it is likely that at least some of the placental-specific imprinted DMRs are functionally important; however, the placenta may also present a unique epigenetic context that allows for the acquisition of imprints, regardless of function. To understand this fully, both allelic expression and methylation will need to be interrogated across all loci in a large study population.
There are several hypotheses proposed to explain the evolution of imprinting. The most commonly discussed is the parental conflict hypothesis (Haig and Westoby 1989; Moore and Haig 1991), which suggests imprinted genes arose through competing interests of the maternal and paternal genetic contributions to the developing fetus. While some of the novel DMRs identified may fit this model, there are several aspects that do not appear to, such as the solely maternal origin of DNA methylation, the relatively low expression of associated genes, and the lack of enrichment for genes involved in growth and development (Haig 1997). There are, however, several additional theories that may provide insight, including the parthenogenetic protection (Solter 1988; Hall 1990) and coadaptation (Keverne et al. 1996; Keverne and Curley 2008) hypotheses.
The parthenogenetic protection hypothesis suggests that maternally silenced genes important for placentation and implantation would prevent progression of spurious parthenogenetic pregnancies. This is consistent with an entirely maternal origin of methylation and perhaps with the placental-specificity; however, imprinting of only a few genes may be sufficient for this to be effective (Spencer and Clark 2014). The coadaptation hypothesis suggests that there is a need to coordinate fetal development and maternal resource management (Keverne et al. 1996; Keverne and Curley 2008). This supports the notion that the majority of imprinted DMRs would be maternally inherited and that, as the site for nutrient exchange, the placenta may be enriched for imprinted loci. Furthermore, imprinting of placental expressed genes that may regulate maternal behavior and metabolism through secreted protein products, such as NPY, which regulates appetite (Stanley et al. 1986), or RASGRF1, which has been implicated in pancreatic function (Manyes et al. 2014), may fit this hypothesis. Our data support that there may be multiple factors contributing to the evolution of imprinting, particularly in the human placenta, as no one theory appears to explain all aspects.
This study has shown that differentially methylated CGIs in the oocyte can uniquely maintain imprinted DNA methylation throughout pregnancy in the human placenta. The novel placental-specific imprinted DMRs identified are not conserved in methylation status between mouse and human and appear to be highly polymorphic in maintenance of methylation in the human population. Further study will be needed to understand the underlying factors contributing to this variability in human extraembryonic tissues and its importance in pregnancy and development.
Methods
Samples
All placenta, fetal tissue, and blood samples were collected through hospital postings with full informed consent at the BC Children's and Women's Health Centre, Vancouver, BC, or anonymously through the Embryopathology Laboratory at British Columbia Children's and Women's Hospital, Vancouver, BC. These samples have been previously described and utilized for studies of DNA methylation (GSE44667, GSE69502) (Yuen et al. 2011a,b; Price et al. 2012). Collection of samples was approved by the University of British Columbia Clinical Ethics Review Board and the ethics committee of the Children's and Women's Health Centre of British Columbia (H04-70488, H06-70085).
Control placental chorionic villous samples (N = 75) included 40 pregnancies delivered at term and 35 delivered preterm or miscarried. The range of gestational ages was 6.0–41.6 wk and included 37 male and 38 female conceptions. The preterm births were associated with one or more of the following: preterm labor, premature rupture of membranes, chorioamnionitis, placental abruption, and incompetent cervix. All samples were chromosomally normal, assessed by standard karyotyping or comparative genome hybridization, as previously described (Robinson et al. 2010). Two to four independent sites were taken from each placenta, from which DNA was pooled after extraction from chorionic villous. Maternal contamination was excluded using microsatellite markers (Robinson et al. 2010). To identify imprinted DMRs in the placenta, chorionic villous samples from five diandric and five digynic triploids pregnancies were assayed. The average (range) gestational ages for diandric and digynic triploids were 11.2 (8–13) wk and 9.4 (8–12) wk (P = 0.18), respectively.
Thirty-two fetal tissues, including brain (N = 7), spinal cord (N = 7), muscle (N = 9), and kidney (N = 9) were collected from second trimester fetuses, as previously described (Price et al. 2012). Adult female whole-blood samples (N = 10) were collected from control women. Extraembryonic cell types (N = 31), including cord blood, cord, amniotic membrane, chorionic membrane, 1st, 2nd, and 3rd trimester trophoblast and mesenchyme, and decidua (maternal), were isolated from control placental samples. Trophoblast and mesenchyme were enzymatically separated from whole villi, as previously described (Henderson et al. 1996; Robinson et al. 2010); trophoblast included both cytotrophoblast and syncytiotrophoblast but are referred to as trophoblast throughout. Isolated mesenchyme often contains a small proportion of contaminating trophoblast cells; therefore, data from mesenchyme should be interpreted with caution.
DNA methylation
Illumina HumanMethylation450 array
Using the DNeasy Blood and Tissue Kit (Qiagen), DNA was extracted and purified for all samples. For each sample, 750 ng of DNA was bisulfite-converted using the EZ DNA Methylation Kit (Zymo Research Corporation). DNA from five diandric triploids, five digynic triploids, 63 control placental villi, 32 fetal tissues, 10 adult blood, and 19 extraembryonic cell types were run on the Infinium HumanMethylation450 BeadChip array (Illumina). The arrays were processed as per the manufacturers’ guidelines, and data were collected on the HiScan System (Illumina).
Using GenomeStudio software (Illumina), background intensity levels were subtracted, and corrected values were exported into R version 2.14.0 (R Development Core Team 2011). Probes that met the following criteria were omitted before analysis, using additional annotation (Price et al. 2013): X and Y Chromosome, detection P-value > 0.01 in at least one sample, no average B-value in more than one sample, XY cross-hybridizing (Chen et al. 2013), and a SNP in the target C or G. The B-values for the remaining probes were then converted to M-values and normalized using subset-quantile within array normalization (SWAN) (Maksimovic et al. 2012).
Reduced representation bisulfite sequencing
DNA methylation for human oocytes (N = 5), sperm (N = 4), zygotes (N = 2), two-cell embryos (N = 2), four-cell embryos (N = 2), eight-cell embryos (N = 3), morulae (N = 3), ICM (N = 3), and TE (N = 3) from blastocysts was obtained using publicly available low-input RRBS data (GSE49828) (Guo et al. 2014). All bisulfite sequencing data described in this study were processed in the same manner, unless otherwise specified: Quality was evaluated using FastQC Version 0.11.2, adapters and quality trimming using Trim Galore! version 0.3.7, reads were aligned and methylation calls performed in Bismark version 0.14.0 (Krueger and Andrews 2011), and data were analyzed in SeqMonk version 0.29.0. All software can be found at http://www.bioinformatics.babraham.ac.uk/projects/. Reads were combined from each biological replicate to represent a DNA methylation profile for each development stage and gamete. Probes were generated using positions from the CpG island track from UCSC (https://genome.ucsc.edu/) with ≥5 CpGs with at least one methylation call. The library quality measures and coverage are detailed in Supplemental Table 4.
For comparison to mouse, DNA methylation for oocytes, sperm, ICM, TE, E6.5 epiblast, E6.5 extraembryonic ectoderm, placenta, heart, liver, and brain was obtained from publicly available data sets (GSE51239, GSE42836, GSE34864) (Smith et al. 2012, 2014; Hon et al. 2013). For the placental data (GSE42836), published methylation calls were utilized (Hon et al. 2013). Orthologous regions for the known and novel placental-specific DMRs (N = 88) were determined using the liftOver tool in UCSC (https://genome.ucsc.edu/cgi-bin/hgLiftOver) to convert coordinates from the human genome (hg19) to the mouse genome (mm10). Of placental-specific DMRs, 88.6% had a mapped orthologous region in the mouse genome, and a total of 58 loci had informative methylation calls.
Multiplex bisulfite sequencing
Isolation of DNA and bisulfite conversion for 48 samples (16 placental villi, 12 trophoblast, 16 maternal blood, and four paternal blood) were performed as described above. One hundred fifty-one PCR assays were designed for the 43 known and 101 novel DMRs (Supplemental Table 5). Primers were designed to contain no SNPs or CpGs within the primer sequences and each assay to interrogate at least one SNP. Parallel PCR reactions, with a final concentration of 1×HotStar PCR buffer, 0.2 mM dNTPs (Life Technologies), 1 U HotStar Taq (Qiagen), 0.4 µM each primer pair, and 2 ng DNA, were performed in a 15-µL reaction volume in 384-well plates. Plates were prepared on the Agilent Bravo Workstation (Agilent Technologies). Cycling conditions included a 15-min, 95°C denaturation, followed by 30 cycles of 95°C for 30 sec, 50°C for 30 sec, 72°C for 90 sec, with a 5-min final extension at 72°C, and reactions were performed on the C1000 Touch Thermal Cycler (Bio-Rad).
PCR products were pooled for each sample and SPRI-purified using Sera-Mag carboxylate-modified Magnetic SpeedBeads (Fisher Scientific) as per the manufacturer's recommendations. A-overhang addition was done using 1×NEB buffer 2, 0.2 mM dATP, 12.5 U Klenow fragment (3′->5′ exo-) (New England Biolabs) in a 25-µL reaction incubated at 37°C for 30 min. Following SPRI purification, Illumina adapters were ligated using the Quick ligation kit (New England Biolabs) in a 40-µL reaction. Following another SPRI purification, libraries were amplified (11 cycles) and barcoded using the Sanger eight-base index in a 50-µL reaction containing: 1×High Fidelity buffer, 1 U Phusion High Fidelity Taq (New England Biolabs), 0.2 mM dNTPs, and 0.1 µM each primer. Final libraries were SPRI-purified, and concentration and quality were assessed by the 2100 Bioanalyzer system (Agilent Technologies) and Kapa library quantification (Kapa Biosystems). The 48 barcoded libraries were pooled and run on the Illumina MiSeq System (Illumina) using 150-bp paired-end reads. Allelic methylation levels and distributions were extracted from the Bismark BAM files for reads with a linear match to the genome using a custom script (Supplemental File 1), which compared the bases in the reads to the expected SNPs designed into the amplicons.
Gene expression
RNA-seq
Expression of genes associated with placental-specific DMRs in early development was assessed using three publicly available RNA-seq data sets (Bernstein et al. 2010; Yan et al. 2013; Metsalu et al. 2014). Single-cell RNA-seq was utilized for TE (GSE36552), mRNA-seq for term placenta (GSE56524), and total RNA-seq for mid-gestation placenta (GSE18927). The single-cell RNA-seq data were trimmed using Trim Galore! version 0.3.7 and mapped using TopHat 2.0.12 guided by gene models from Ensembl release 61. Quality assessment was performed using the RNA-Seq QC module of SeqMonk. The mRNA-seq for term placenta was aligned using TopHat (Trapnell et al. 2009) with Bowtie 1, and --color --quals options (since the data are in color space). Aligned BAM files for the total RNA-seq in mid-gestation placenta were obtained directly from Gene Expression Omnibus (GSE18927).
Gene expression as reads per kilobase per million mapped reads (RPKM) was quantitated over exons with merged isoforms, correcting for transcript length using the RNA-seq pipeline quantitation in SeqMonk version 0.29.0. Placental-specific DMRs were assessed for associated gene expression, based on the closest TSS, using the UCSC Genome Browser gene annotation (Supplemental Table 6). Two genes were missed due to gene annotation differences between UCSC and Ensembl, the gene annotation track in SeqMonk version 0.29.0, resulting in 86/88 DMRs being reported. The data are presented using the natural log transformed RPKM+1.
Histone modifications
ChIP-seq
Publicly available ChIP-seq data for H3K9me3 in placental villi, amniotic membrane, and fetal muscle (GSE18927) (Bernstein et al. 2010) were assessed in SeqMonk version 0.29.0. Relative enrichment was assessed by calculating the difference (Δ) between placenta and somatic tissue (muscle or amnion) for each 1000-bp probe. Values of Δ enrichment for placental-specific DMRs and CGIs were compared to all other genome-wide probes using a t-test.
Statistical analyses
All statistical analyses were done using RStudio version 0.97.551 (RStudio Team 2015) and R version 3.0.3 (R Core Team 2014), unless otherwise specified.
Identifying imprinted DMRs
Using the DMRFinder tool in the CHARM package (Aryee et al. 2011), candidate DMRs were identified between diandric (N = 5) and digynic (N = 5) triploid samples (Supplemental Table 1). Known imprinted DMRs were those that overlapped a previously reported human imprinted DMR with adequate coverage on the 450K array (N = 57) (Supplemental Table 2; Court et al. 2014). Germline novel DMRs were those that were CGIs (defined by UCSC Genome Browser [https://genome.ucsc.edu/]) that (1) were >50% differentially methylated between sperm and oocytes, (2) were intermediately methylated (15%–60%, based on the 90th percentile observed at known imprinted DMRs) in the blastocyst (average of ICM and TE), (3) showed a >5% difference in methylation between diandric and digynic triploids, and (4) showed matching parental origin of DNA methylation between triploid villi and gametes. Placental-specific DMRs were defined as those with >25% and <75% DNA methylation in placenta and <25% in somatic tissues.
Allelic DNA methylation
Reads from the RRBS data for oocyte, sperm, ICM, and TE (Guo et al. 2014) were remapped based on phase of DNA methylation within a given read. Reads were categorized into fully methylated, fully unmethylated, or mixed methylation, and the proportion of each across a given DMR was determined.
For the RRBS and multiplex bisulfite sequencing data, DNA methylation was averaged across reads with ≥5 CpGs with a read depth of ≥5. Reads across DMRs were binned into 10% intervals to generate density plots of the distribution of read methylation. For DMRs with heterozygous SNPs, percent methylation was calculated for each allele separately for each informative individual sample. Where possible, parental origin was assigned based on parental genotype information.
Features of placental-specific DMRs
The frequency of ZFP57 consensus motifs (TGCCGC) (Nakamura et al. 2007; Quenneville et al. 2011) among known imprinted DMRs, placental-specific DMRs, and CGIs was determined using customized Perl scripts and compared using a χ2 test.
Gene ontology
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) gene ontology program was used on genes associated with placental-specific DMRs (Supplemental Table 6; Huang da et al. 2009a,b).
Data access
Illumina Infinium HumanMethylation450 array data and multiplexed targeted bisulfite sequencing from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE74738 and GSE76273.
Acknowledgments
We thank all the participants of this study. We also thank Kristina Tabbada at the Babraham Institute Sequencing Facility, Felix Krueger at the Babraham Institute Bioinformatics Programme, Magda Price at the Child and Family Research Institute, and Wendy Dean at the Babraham Institute for assistance and advice. This work was supported by Canadian Institutes of Health Research grant 49520 (W.P.R.) and UK Medical Research Council grant MR/K011332/1 (G.K.).
Author contributions: C.W.H. generated the research objectives, produced array and bisulfite sequencing data, performed analyses, and prepared the figures and manuscript. M.S.P. generated array data and helped with experimental design and interpretation. H.S. performed computational analyses. S.A. processed and analyzed allelic bisulfite sequencing data. D.E.M. collected and sampled human fetal tissues. G.K. and W.P.R. supported the study, assisted with experimental design, and helped with manuscript preparation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.196139.115.
Freely available online through the Genome Research Open Access option.
References
- Aryee MJ, Wu Z, Ladd-Acosta C, Herb B, Feinberg AP, Yegnasubramanian S, Irizarry RA. 2011. Accurate genome-scale percentage DNA methylation estimates from microarray data. Biostatistics 12: 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babak T, DeVeale B, Tsang EK, Zhou Y, Li X, Smith KS, Kukurba KR, Zhang R, Li JB, van der Kooy D, et al. 2015. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat Genet 47: 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28: 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. 2013. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choufani S, Shapiro JS, Susiarjo M, Butcher DT, Grafodatskaya D, Lou Y, Ferreira JC, Pinto D, Scherer SW, Shaffer LG, et al. 2011. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res 21: 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court F, Tayama C, Romanelli V, Martin Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simon C, Moore H, Harness JV, et al. 2014. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res 24: 554–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Lee YK, Strogantsev R, Jin S, Lim YC, Ng PY, Lin XM, Chng K, Yeo GS, Ferguson-Smith AC, et al. 2013. DNMT1 and AIM1 imprinting in human placenta revealed through a genome-wide screen for allele-specific DNA methylation. BMC Genomics 14: 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty LE, Rezwan FI, Poole RL, Jagoe H, Lake H, Lockett GA, Arshad H, Wilson DI, Holloway JW, Temple IK, et al. 2014. Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J Med Genet 51: 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockery L, Gerfen J, Harview C, Rahn-Lee C, Horton R, Park Y, Davis TL. 2009. Differential methylation persists at the mouse Rasgrf1 DMR in tissues displaying monoallelic and biallelic expression. Epigenetics 4: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs KM. 2011. Lineage commitments: emphasis on embryonic–extraembryonic interfaces. EMBO Rep 12: 987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JM, Moore GE. 2010. The importance of imprinting in the human placenta. PLoS Genet 6: e1001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y, Yan J, Ren X, Lin S, Li J, et al. 2014. The DNA methylation landscape of human early embryos. Nature 511: 606–610. [DOI] [PubMed] [Google Scholar]
- Haig D. 1997. Parental antagonism, relatedness asymmetries, and genomic imprinting. Proc Biol Sci 264: 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haig D, Westoby M. 1989. Parent-specific gene-expression and the triploid endosperm. Am Nat 134: 147–155. [Google Scholar]
- Hall JG. 1990. Genomic imprinting: review and relevance to human diseases. Am J Hum Genet 46: 857–873. [PMC free article] [PubMed] [Google Scholar]
- Henckel A, Chebli K, Kota SK, Arnaud P, Feil R. 2012. Transcription and histone methylation changes correlate with imprint acquisition in male germ cells. EMBO J 31: 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson KG, Shaw TE, Barrett IJ, Telenius AH, Wilson RD, Kalousek DK. 1996. Distribution of mosaicism in human placentae. Hum Genet 97: 650–654. [DOI] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B. 2013. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet 45: 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Inoue A, Zhang Y. 2011. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 334: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida M, Moore GE. 2013. The role of imprinted genes in humans. Mol Aspects Med 34: 826–840. [DOI] [PubMed] [Google Scholar]
- Kelsey G, Feil R. 2013. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos Trans R Soc Lond B Biol Sci 368: 20110336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keverne EB, Curley JP. 2008. Epigenetics, brain evolution and behaviour. Front Neuroendocrinol 29: 398–412. [DOI] [PubMed] [Google Scholar]
- Keverne EB, Fundele R, Narasimha M, Barton SC, Surani MA. 1996. Genomic imprinting and the differential roles of parental genomes in brain development. Brain Res Dev Brain Res 92: 91–100. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Suda C, Abe T, Kohara Y, Ikemura T, Sasaki H. 2006. Bisulfite sequencing and dinucleotide content analysis of 15 imprinted mouse differentially methylated regions (DMRs): Paternally methylated DMRs contain less CpGs than maternally methylated DMRs. Cytogenet Genome Res 113: 130–137. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. 2012. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet 8: e1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. 2008. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 15: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifero D, Mann MR, Bartolomei MS, Trasler JM. 2004. Gene-specific timing and epigenetic memory in oocyte imprinting. Hum Mol Genet 13: 839–849. [DOI] [PubMed] [Google Scholar]
- Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. 2007. Computational and experimental identification of novel human imprinted genes. Genome Res 17: 1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic J, Gordon L, Oshlack A. 2012. SWAN: Subset-quantile Within Array Normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol 13: R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manyes L, Arribas M, Gomez C, Calzada N, Fernandez-Medarde A, Santos E. 2014. Transcriptional profiling reveals functional links between RasGrf1 and Pttg1 in pancreatic β cells. BMC Genomics 15: 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsalu T, Viltrop T, Tiirats A, Rajashekar B, Reimann E, Koks S, Rull K, Milani L, Acharya G, Basnet P, et al. 2014. Using RNA sequencing for identifying gene imprinting and random monoallelic expression in human placenta. Epigenetics 9: 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk D, Arnaud P, Apostolidou S, Hills FA, Kelsey G, Stanier P, Feil R, Moore GE. 2006. Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci 103: 6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T, Haig D. 1991. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet 7: 45–49. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, Taniguchi H, Sekimoto T, Ikawa M, Yoneda Y, Okabe M, et al. 2007. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol 9: 64–71. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, Tachibana M, Ogura A, Shinkai Y, Nakano T. 2012. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 486: 415–419. [DOI] [PubMed] [Google Scholar]
- Noguer-Dance M, Abu-Amero S, Al-Khtib M, Lefevre A, Coullin P, Moore GE, Cavaille J. 2010. The primate-specific microRNA gene cluster (C19MC) is imprinted in the placenta. Hum Mol Genet 19: 3566–3582. [DOI] [PubMed] [Google Scholar]
- Okae H, Chiba H, Hiura H, Hamada H, Sato A, Utsunomiya T, Kikuchi H, Yoshida H, Tanaka A, Suyama M, et al. 2014. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet 10: e1004868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango P, Miller W, Lehoczky J, Leung CT, Birren B, Wheelan S, Dewar K, Feinberg AP. 2000. Sequence and comparative analysis of the mouse 1-megabase region orthologous to the human 11p15 imprinted domain. Genome Res 10: 1697–1710. [DOI] [PubMed] [Google Scholar]
- Penaherrera MS, Jiang R, Avila L, Yuen RK, Brown CJ, Robinson WP. 2012. Patterns of placental development evaluated by X chromosome inactivation profiling provide a basis to evaluate the origin of epigenetic variation. Hum Reprod 27: 1745–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira PN, Dobreva MP, Graham L, Huylebroeck D, Lawson KA, Zwijsen AN. 2011. Amnion formation in the mouse embryo: the single amniochorionic fold model. BMC Dev Biol 11: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price EM, Cotton AM, Penaherrera MS, McFadden DE, Kobor MS, Robinson W. 2012. Different measures of “genome-wide” DNA methylation exhibit unique properties in placental and somatic tissues. Epigenetics 7: 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ME, Cotton AM, Lam LL, Farre P, Emberly E, Brown CJ, Robinson WP, Kobor MS. 2013. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, et al. 2011. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell 44: 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- R Development Core Team. 2011. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Renfree MB, Suzuki S, Kaneko-Ishino T. 2013. The origin and evolution of genomic imprinting and viviparity in mammals. Philos Trans R Soc Lond B Biol Sci 368: 20120151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson WP, McFadden DE, Barrett IJ, Kuchinka B, Penaherrera MS, Bruyere H, Best RG, Pedreira DA, Langlois S, Kalousek DK. 2002. Origin of amnion and implications for evaluation of the fetal genotype in cases of mosaicism. Prenat Diagn 22: 1076–1085. [DOI] [PubMed] [Google Scholar]
- Robinson WP, Penaherrera MS, Jiang R, Avila L, Sloan J, McFadden DE, Langlois S, von Dadelszen P. 2010. Assessing the role of placental trisomy in preeclampsia and intrauterine growth restriction. Prenat Diagn 30: 1–8. [DOI] [PubMed] [Google Scholar]
- RStudio Team. 2015. RStudio: Integrated Development for R. RStudio, Inc, Boston, MA. [Google Scholar]
- Santos F, Hendrich B, Reik W, Dean W. 2002. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol 241: 172–182. [DOI] [PubMed] [Google Scholar]
- Schroeder DI, Blair JD, Lott P, Yu HO, Hong D, Crary F, Ashwood P, Walker C, Korf I, Robinson WP, et al. 2013. The human placenta methylome. Proc Natl Acad Sci 110: 6037–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R, Proudhon C, Bestor TH, Woodfine K, Lin CS, Lin SP, Prissette M, Oakey RJ, Bourc'his D. 2010. The parental non-equivalence of imprinting control regions during mammalian development and evolution. PLoS Genet 6: e1001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W. 2012. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell 48: 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S, Peat JR, Hore TA, Santos F, Dean W, Reik W. 2013. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos Trans R Soc Lond B Biol Sci 368: 20110330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G. 2011. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43: 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484: 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Humm KC, Karnik R, Mekhoubad S, Regev A, Eggan K, Meissner A. 2014. DNA methylation dynamics of the human preimplantation embryo. Nature 511: 611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solter D. 1988. Differential imprinting and expression of maternal and paternal genomes. Annu Rev Genet 22: 127–146. [DOI] [PubMed] [Google Scholar]
- Spencer HG, Clark AG. 2014. Non-conflict theories for the evolution of genomic imprinting. Heredity (Edinb) 113: 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley BG, Kyrkouli SE, Lampert S, Leibowitz SF. 1986. Neuropeptide Y chronically injected into the hypothalamus: a powerful neurochemical inducer of hyperphagia and obesity. Peptides 7: 1189–1192. [DOI] [PubMed] [Google Scholar]
- Stelzer Y, Ronen D, Bock C, Boyle P, Meissner A, Benvenisty N. 2013. Identification of novel imprinted differentially methylated regions by global analysis of human-parthenogenetic-induced pluripotent stem cells. Stem Cell Reports 1: 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varmuza S, Miri K. 2015. What does genetics tell us about imprinting and the placenta connection? Cell Mol Life Sci 72: 51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Yang M, Guo H, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng X, Yan J, et al. 2013. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol 20: 1131–1139. [DOI] [PubMed] [Google Scholar]
- Yuen RK, Jiang R, Penaherrera MS, McFadden DE, Robinson WP. 2011a. Genome-wide mapping of imprinted differentially methylated regions by DNA methylation profiling of human placentas from triploidies. Epigenetics Chromatin 4: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Neumann SM, Fok AK, Penaherrera MS, McFadden DE, Robinson WP, Kobor MS. 2011b. Extensive epigenetic reprogramming in human somatic tissues between fetus and adult. Epigenetics Chromatin 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]