

Summary
A major goal in understanding autoimmune diseases is to define the antigens that elicit a self-destructive immune response, but this is a difficult endeavor. In an effort to discover autoantigens associated with type 1 diabetes (T1D), we used epitope surrogate technology that screens combinatorial libraries of synthetic molecules for compounds that could recognize disease-linked autoantibodies and enrich them from serum. Autoantibodies from one patient revealed a highly phosphorylated form of peripherin, a neuroendocrine filament protein, as a candidate T1D antigen. Peripherin antibodies were detected in 67% of donor patient sera. Further analysis revealed that the T1D-associated antibodies only recognized a dimeric conformation of peripherin. These data explain why peripherin was dismissed as an important T1D antigen previously. The discovery of this novel autoantigen would not have been possible using standard methods, such as hybridizing serum antibodies to recombinant protein arrays, highlighting the power of epitope surrogate technology for probing the mechanism of autoimmune diseases.
eTOC Blurb
Characterizing the autoantigens that trigger adaptive immune responses is challenging, especially when epitopes are non-linear or post-translationally modified. Using “epitope surrogate” small molecules to purify disease-linked antibodies from serum, Doran et al demonstrate that phosphorylated peripherin is a major autoantigen in patients with type 1 diabetes.
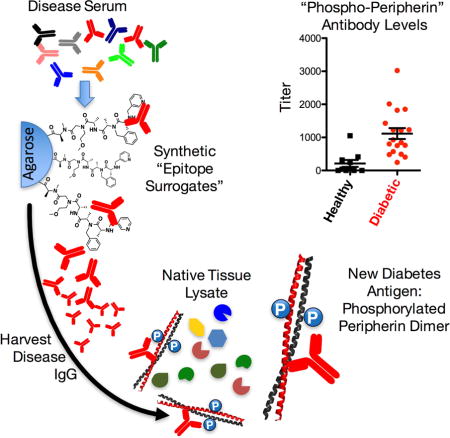
Introduction
A central goal in the study of autoimmune diseases is to characterize autoantigens involved in the process. In addition to potentially shedding light on the biology of the disease, autoantigens can often be used in the development of diagnostically useful blood tests based on capture of the corresponding autoantibody from serum. Unfortunately, identifying autoantigens is challenging, once the candidate approach has been exhausted and some type of unbiased search must be conducted. The most common approach is to screen case and control serum samples against large collections of potential autoantigens, for example using protein (Balboni et al., 2006), lipid (Kanter et al., 2006), peptide or oligonucleotide arrays (Larman et al., 2011). However, it is always possible that these collections will not contain the native autoantigen. For example protein arrays generally do not display molecules with post-translational modifications that might be important for antibody recognition. As a result, many screening efforts of this type have been unsuccessful.
A good example is the hunt for autoantigens within pancreatic islet cells that are targeted in type 1 diabetes (T1D). It has been known since the 1970s that there exists a humoral immune response against islet cell antigens (ICAs) that occurs years before onset of symptoms (Atkinson and Leiter, 1999). Yet, despite many efforts to uncover potential ICAs, progress in this area has been slow. To date, only four major antigens (insulin, GAD65, IA-2 and ZnT8) are known to elicit a measurable humoral response in patients who have, or will develop, T1D (Bonifacio et al., 2000; Bonifacio et al., 1995; Lan et al., 1996; Roep and Peakman, 2012; Velloso et al., 1993; Verge et al., 1996; Yu et al., 2000). High levels of antibodies to two or more of these antigens can be detected in ≈80% of individuals who later go on to develop T1D (Ziegler et al., 2013). The remaining ≈20% of T1D patients only possess high antibody titers to a single antigen. Unfortunately, an antibody response to just one antigen is of low diagnostic utility because it is also present in 80–85% of genetically at-risk individuals who never develop the disease. Moreover, a small fraction of T1D patients never possess elevated levels of antibodies towards any these antigens. Given that the detection of antibodies to a combination of the four known autoantigens can only accurately predict T1D with 80% sensitivity, the discovery of other, perhaps more primary, autoantigens would greatly improve the accuracy of multiplexed tests used to diagnose the disease.
Towards this end, we report here the application of “epitope surrogate” technology (Kodadek, 2014) to human T1D. This involves screening case and control serum antibodies against a large library of synthetic, unnatural molecules, with the intent of identifying compounds that retain far more antibodies from the case than the control samples. These epitope surrogates can then be used as affinity reagents to purify the antibodies they recognize. These antibodies, in turn, can be employed to “fish” for the native antigen in tissue extracts (Doran et al., 2015). Using this methodology, we report here the discovery that antibodies highly enriched in the serum of many T1D patients recognize a dimeric, phosphorylated form of the intermediate filament protein peripherin (Aletta et al., 1989).
Results
A synthetic epitope surrogate for T1D-related antibodies
A one bead one compound (OBOC) library of synthetic oligomers comprised of N-substituted peptide and peptoid units (Alluri et al., 2003; Figliozzi et al., 1996; Gao and Kodadek, 2013) (Supporting Information Fig. S1) was subjected to comparative screening using case and control serum samples to identify ligands that recognized IgG antibodies from the sera of patients with T1D, but not from control donors who were non-diabetic. Several copies of the library (i.e., each compound was displayed on more than one bead) were incubated first with a pool of three different control serum samples. Beads that retained significant amounts of IgG antibodies were collected by addition of a biotinylated anti-human IgG antibody, streptavidin-coated magnetic particles and exposure to a strong magnet. These beads were discarded since they display ligands to antibodies that are not associated with T1D (Fig. 1). The remainder of the library was screened against pooled T1D serum samples. Again, beads that retained significant amounts of IgG antibodies were collected magnetically.
Figure 1. Compounds 1 and 2 are epitope surrogates for diabetes-related antibodies.
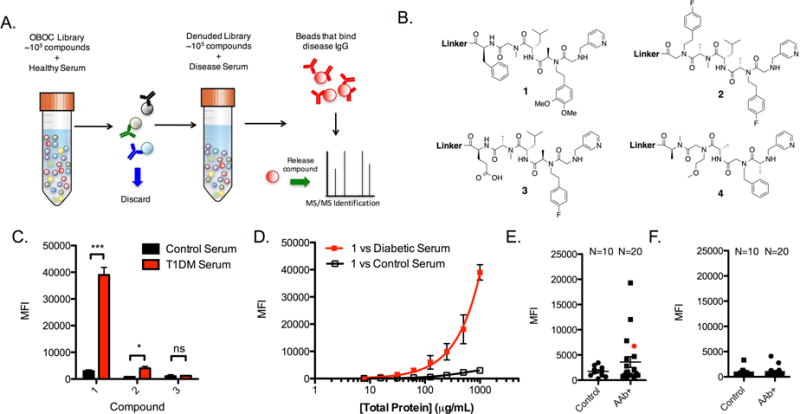
A) Illustration of the general comparative screening procedure. A OBOC library is mixed with healthy serum and beads that bind high levels of antibody are removed with an appropriately tagged secondary antibody. The remaining library is incubated with disease serum and beads that bind significant antibody are isolated. The compound is released from the bead and identified by tandem mass spectrometry. See also Figure S1. B) Chemical structures of compounds used throughout this study. See also Figure S2. C) Binding experiment using the liquid array platform wherein compounds 1–3 are immobilized onto the 10 μm TentaGel bead liquid array. The beads were mixed with diluted serum and fluorescent secondary antibody and binding of serum IgG antibodies was measured as the mean fluorescence intensity (MFI) of the beads using a flow cytometer. D) Binding isotherm generated for 1 using serially diluted concentrations of diabetic or healthy control serum and read using the cytometry assay. E) Analysis of 10 control serum samples and 20 AAb+ serum samples for binding to compound 1 using the cytometry binding assay. Serum was collected from 10 of the diabetes patients who were positive for classic T1D antibodies, but had not yet experienced hyperglycemia at the time of blood collection. F) Antibody binding to control compound 3 using 10 control serum samples and 20 AAb+ serum samples using the cytometry assay. Conditions for binding experiments: 500 μg mL−1 diluted serum in binding buffer for 1.5 h at room temperature. Binding experiment data reported in bar and line graphs represent the mean fluorescence intensity ± s.d. from three experiments. Statistical significance was determined using an unpaired t-test: *P < 0.05; ***P < 0.001; ns = not significant. Each dot plot data point is the average of two independent measurements for each serum sample tested.
Since this protocol is known to have a high false positive rate (Doran et al., 2014; Lian et al., 2013), a second differential screen was conducted. The ≈ 2500 beads collected magnetically were stripped of bound protein using a denaturing buffer. They were then incubated with control serum and washed. Secondary antibodies linked to red quantum dots (QDot655) were hybridized to the beads and those that displayed a red halo were collected manually with a micropipette under a low-power fluorescence microscope and discarded. The remaining beads were then incubated with a pool of three different sera from patients with confirmed T1D (Supplemental Table S1), washed, and probed with a QDot655-conjugated secondary antibody. Antibody-binding beads at this stage were collected, placed individually into wells of a microtiter plate, stripped of bound protein and the displayed molecules were released from the bead by CNBr-mediated cleavage at a methionine residue in the linker. The sequence of each “hit” was determined by tandem mass spectrometry. Of the 211 hits obtained from the quantum dot screen, two compounds (1 and 2, Fig. 1B) were isolated on more than one bead from the library. Isolation of the same compound multiple times indicates that the ligand likely binds with high affinity or specificity, whereas “singletons” are likely to be false positives (Doran et al., 2014). Therefore, we focused our efforts on testing compounds 1 and 2 as epitope surrogates.
These compounds and a control molecule, 3 (Fig. 1B), were synthesized with a C-terminal cysteine-containing linker for facile coupling to solid supports. The C-terminal linker excluded methionine and bromobenzene, which were monomers from the library that facilitated identification by mass spectrometry. The furfuryl-containing monomer was retained as a handle for future conjugation reactions (Astle et al., 2010), while the remaining monomers were retained to aid in compound solubilization. After HPLC purification, the compounds were linked to 10 μm TentaGel microspheres displaying a terminal bromoacetamide group and encoded in the protein-inaccessible domain of the bead with blue and orange dyes to distinguish distinct bead populations (Doran and Kodadek, 2013). Upon mixing the beads and blocking with BSA, pooled control and case serum samples (1 mg mL−1) used in the discovery set were incubated with the beads for 1.5 h at room temperature. After washing, a red fluorescent secondary antibody was hybridized to the bead, and antibody binding was measured for each bead population using a flow cytometer. Fig 1C shows that compound 1 retained significantly more IgG antibodies from T1D serum than from the control donor serum (an ≈12-fold difference). Though 2 appeared to distinguish case from control antibodies, the difference was only ≈5-fold. Thus, compound 1 was selected for further characterization.
The diagnostic utility of ligand 1 was evaluated in the context of a cohort of donor sera consisting of 10 control samples from healthy individuals, and 20 samples from patients who were autoantibody positive for major T1D-related antigens at the time of blood draw (herein referred to as AAb+, Table S1). Binding to 1 was assessed for each patient using the cytometer-based assay. Fig. 1E summarizes the results, where each serum sample is represented by a dot. No appreciable binding of antibodies to 1 was observed for the control samples; four of the diabetic or pre-diabetic serum samples had antibodies that were bound by 1 at a level above the background defined by the average of the control sample plus three standard deviations. These data translate into a 20% diagnostic sensitivity for compound 1. Conversely, when beads containing 3 were probed with serum, neither control nor diabetic serum antibodies were retained on the beads (Fig 1F).
Ligand optimization efforts provide a new epitope surrogate
The diagnostic sensitivity of 1 is modest. In an attempt to improve this, we carried out a library-driven affinity improvement experiment (Gao et al., 2015). Our hypothesis was that a higher affinity ligand for these antibodies might bind well enough to immunoglobulins in the serum of diabetic or pre-diabetic patients that fell below the 3σ line to increase the diagnostic sensitivity of the assay. To guide the design of the library, several derivatives of 1 were made in which the chirality of a single stereocenter was reversed or a side chain was replaced with a methyl group. These compounds were all tested for binding activity, revealing important features of the ligand-antibody interaction (Supporting Figures S3–S4). The new library was then designed to retain the elements critical for binding, but vary the non-essential structural features using building blocks not included in the original library in an attempt to pick up new productive interactions. This library was screened under more stringent conditions against a serum sample with modest-affinity antibodies for 1 (highlighted in red in Fig 1E). The screen resulted in the isolation of compound 4 (Fig. 1B) multiple times, so we proceeded to characterize antibody binding to 4.
Compound 4 was attached covalently to 10 μm TentaGel microspheres and the flow cytometer-based assay was again used to analyze binding to antibodies in serum samples (Fig. 2). The binding isotherms shown in Fig 2A using the serum sample employed in the screen revealed a ≈20-fold differential binding between the case and control sera at 500 μg mL−1 total protein concentration. However, none of the other 29 samples analyzed contained antibodies that recognize 4. Therefore, compound 4 recognizes a very specific subset of antibody paratopes from a single patient.
Figure 2. Epitope surrogate 4 is a highly specific capture agent for diabetes-linked antibodies.
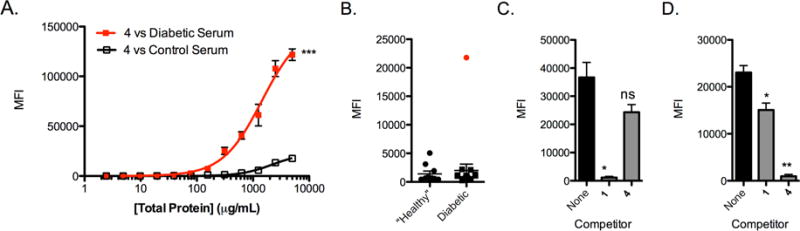
A) Binding isotherm for 4 generated by mounting 4 onto 10 μm TentaGel beads and incubating the beads with serially diluted concentrations of diabetic serum or healthy control serum. Antibody binding was measured using the cytometry assay. B) Analysis of 10 control serum samples and 20 AAb+ serum samples for binding to compound 4 using the cytometry binding assay. C) T1D antibody binding to beads containing 1 in the absence of a competitor molecule (black bar), or after incubation with 500 nM of soluble 1 or 3 (gray bars). D) T1D antibody binding to beads containing 3 in the absence of a competitor molecule (black bar), or after incubation with 500 nM of soluble 1 or 3 (gray bars). Conditions for binding experiments: 500 μg mL−1 diluted serum in binding buffer for 1.5 h at room temperature. Binding experiment data reported in bar and line graphs represent the mean fluorescence intensity ± s.d. from three experiments. Statistical significance was determined using an unpaired t-test: *P < 0.05; **P < 0.01; ***P < 0.001; ns = not significant. Each dot plot data point is the average of two independent measurements for each serum sample tested.
While unexpected, this result was not completely surprising. Serum antibody screening is inherently more complex than ligand discovery for more traditional protein targets. For example, antibodies from two patients that are raised against the same epitope may have different apparent affinities because of polyclonality or because they are present at varying levels in different patients. Alternatively, it could be that the interactions in one patient might represent off-target binding to antibodies that recognize completely different epitopes. We hypothesize that this was the case for binding of 1 to antibodies from the patient highlighted in red in Fig. 1E. If so, this means 4, which was derived by screening against that sample, binds completely different antibodies than 1, despite their structural similarity.
To test this, a competitive binding assay was carried out. T1D serum was pre-incubated with 500 nM soluble 1 or 4 (each at 500 nM) before adding the serum sample to beads displaying compound 1. Soluble 1 blocked antibody binding to immobilized 1, as expected, but soluble 4 did not (Fig. 2C). Moreover, soluble 4 competed antibody binding to immobilized 4, but soluble 1 did not (Fig. 2D). These data prove that compounds 1 and 4 recognize completely distinct antigen-binding sites.
Epitope surrogate 4 binds to anti-peripherin antibodies
Given the apparently strong and selective binding of compound 4 to antibodies in at least one T1D patient, we focused on attempting to identify the native antigen recognized by these antibodies. Serum from this patient was passed over a sepharose column displaying immobilized 4. After thorough washing, the bound antibody was released from the resin using mild elution conditions and buffer-exchanged. The eluted material (~ 75 μg total protein) was added to a human pancreas tissue lysate and immune complexes were captured using Protein A- and Protein G-conjugated sepharose beads. The bound proteins were denatured and analyzed by SDS-PAGE. Fig 3B shows the Coomassie-stained gel. A prominent band at ~60 kDa that was not present in a control lane (Fig. 3C) was excised. Trypsin digestion and LC/MS analysis of the proteins in this band revealed three candidate proteins that were not present in a corresponding area of a control gel (Supplemental Table S2 and Table S3), thereby ruling out the possibility that these proteins were isolated as an artifact of the immunoprecipitation experiment. One of these proteins was peripherin, a type III intermediate filament protein found mostly in peripheral neurons (Portier et al., 1983). Because peripherin has been implicated as a T1D in NOD mice (Boitard et al., 1992), we focused initially on pursuing this protein as the native antigen.
Figure 3. Peripherin is the antigen recognized by antibodies that bind 4.
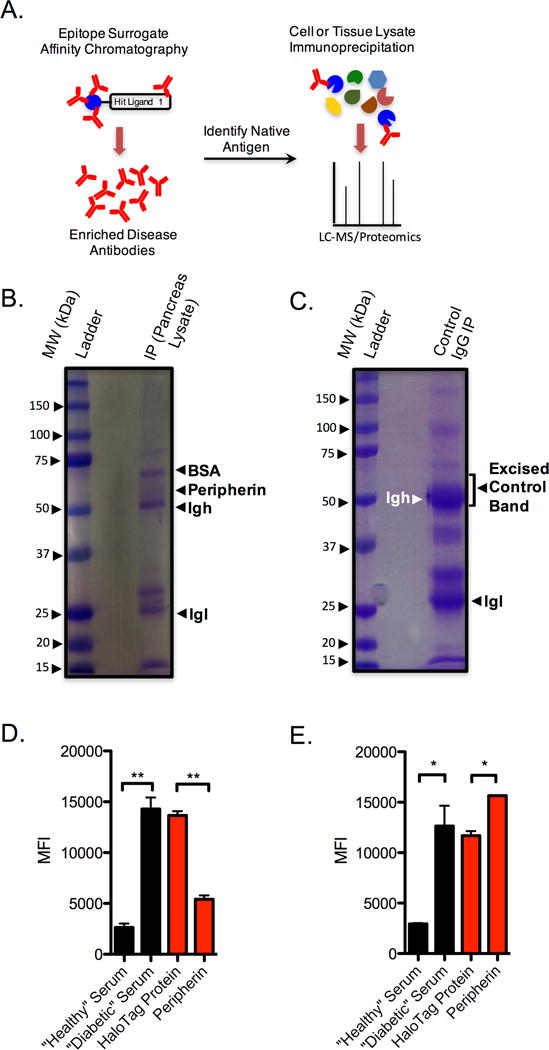
A) General experimental flow for antigen discovery using epitope surrogates. B) SDS-PAGE gel following immunoprecipitation of pancreas tissue lysate with serum-derived antibodies that were purified over a sepharose column containing 4. C) Control SDS-PAGE gel after immunoprecipitation of pancreas tissue lysate with control IgG. D) Assessment of serum antibody binding to 4 from healthy and control donors in the absence of competitor (black bars). Competition binding experiments (red bars) were performed using T1D serum in the presence of 500 nM nonspecific competitor (HaloTag protein) or 500 nM peripherin. E) Competition analyses performed on an epitope surrogate 1, that does not compete with 4 for antibody binding. Black bars represent binding in the absence of any competitor protein. Red bars indicate competition of T1D serum using 500 nM of the competitor indicated. In all cases data are reported as the mean ± s.d. from three experiments. Statistical significance was determined using an unpaired t-test: **P < 0.01; ***P < 0.001; ns = not significant.
A competitive binding experiment was performed to determine if peripherin is indeed the antigen recognized by antibodies that engage 4. Human peripherin was expressed in HEK 293T cells with a C-terminal His6-tag and purified over Ni-NTA agarose. As shown in Fig. 3D, incubation of soluble peripherin with serum from the patient containing antibodies that bind to 4 blocks the association of these antibodies with immobilized 4. In contrast, soluble peripherin had no effect on binding of antibodies to immobilized 1 (Fig. 3E), which binds antibodies distinct from those that recognize compound 4 (Fig. 2 C and D). We conclude that the antibodies that bind to compound 4 are indeed anti-peripherin antibodies.
Anti-peripherin antibodies are present in multiple T1D patients
To determine if anti-peripherin antibodies are common in T1D patients, or if they are restricted to the single patient with antibodies that bind compound 4, peripherin was immobilized and employed in a flow cytometry-based serological competition assay. To do so, a HaloTag-peripherin fusion protein purified from HEK 293 cells was immobilized on 10 μm TentaGel beads displaying the HaloTag ligand (Los et al., 2008). The beads were then incubated with a serum sample in the presence of 100 nM free peripherin or, as a control, 100 nM HaloTag-GST fusion protein. After washing, the amount of captured antibody was quantified with a fluorescently labeled secondary antibody and passing the beads through a flow cytometer. Fig. 4C shows the results plotted as the mean fluorescence intensity difference between the experiments using the two soluble competitor proteins (ΔMFI = MFI (HaloTag competitor) – MFI (peripherin competitor)). Two autoantibody positive (AAb+) serum samples were completely consumed during our preliminary analyses, so a total of 18 AAb+ and 10 control serum samples were tested in this and all subsequent competition analyses. Of these remaining samples, 67% of AAb+ relatives were positive for anti-peripherin antibodies. Only a single control sample exhibited binding beyond a threshold defined by three standard deviations above the mean of the control samples (“3-sigma” in Fig. 4C).
Figure 4. Antibodies derived from AAb+ donor sera recognize peripherin.
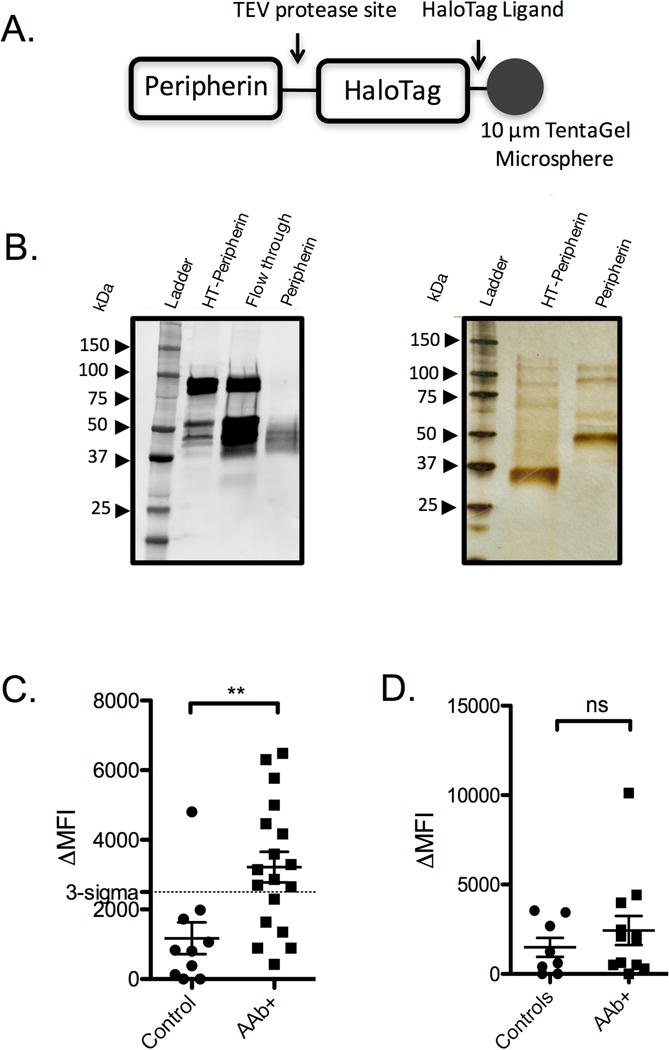
A) Illustration of the HaloTag-peripherin construct after mounting onto 10 μm TentaGel microspheres. B) Immunoblot and silver stain gel of HaloTag-peripherin fusion protein following nickel column affinity purification before and after TEV protease cleavage. C) Competition binding experiment using peripherin-mounted microspheres and serum derived from control donors and AAb+ donors. Binding was measured by flow cytometry after probing with a fluorescent secondary antibody. D) Competition binding experiment using vimentin-mounted microspheres and serum derived from control donors and AAb+ donors. Data points are reported as the mean ± s.d. from two independent serum competition experiments. Statistical significance was determined using an unpaired t-test: **P < 0.01.
Though unlikely, it is reasonable to suspect that peripherin and vimentin were pulled down in the immunoprecipitation experiment because they are relatively insoluble filament proteins, and may bind nonspecifically to antibodies from T1D patients. Indeed, vimentin shares 58% sequence identity with peripherin. Therefore, before moving forward with the validation of peripherin as a T1D-specific antigen, we evaluated vimentin as a diagnostic capture agent for antibodies in T1D patients. A HaloTag-vimentin contruct was expressed in HEK293T cells and mounted onto 10 μm TentaGel beads that displayed a HaloTag ligand. 8 control patient sera and 12 AAb+ serum samples were subjected to a competitive binding experiment that was performed identically to the experiment described for the assay shown in Fig. 4C, with the exception that soluble vimentin was used as the specific competitor (see also Fig. S5). Fig 4D shows that vimentin did not bind significant levels of serum antibodies from the T1D patient cohort. Thus, binding of antibodies from T1D patient sera cannot be attributed solely on the physical properties of filamentous proteins, leading us to pursue the detailed nature of the peripherin-autoantibody interaction.
The central rod domain of peripherin is recognized by T1D antibodies
Peripherin was separated into individual domains to locate regional epitopes recognized by T1D- associated antibodies. The known domains in peripherin were separated into two constructs, one of which contained the helical rod domain and the other the non-helical head and tail domains (Fig. 5A). The domains were expressed with GST fusion tags and mounted onto 10 μm TentaGel beads that displayed covalently-bound glutathione. The beads were exposed to sera from 10 control patients and 18 AAb+ patients in a blinded fashion. These were the same samples used in the Fig. 4C experiment. Antibody binding to the beads was measured using the serological cytometry assay format (Doran and Kodadek, 2013). The results, plotted in Fig. 5B, reveal that the rod domain is sufficient to recognize diabetes serum antibodies in 30% of the autoantibody-positive (AAb+) subjects tested and none of the control patients. Conversely, the head and tail domain failed to differentiate between case and control subjects (Fig. 5C). Note that not as many of the AAb+ samples were scored as T1D cases in this experiment as was the case when full-length peripherin was used (see Fig. 4C), indicating that the anti-peripherin autoantibodies are polyclonal and some require epitopes outside of the rod domain to bind tightly, while some do not.
Figure 5. The rod domain of peripherin is recognized by autoantibodies in peripherin-reactive sera.
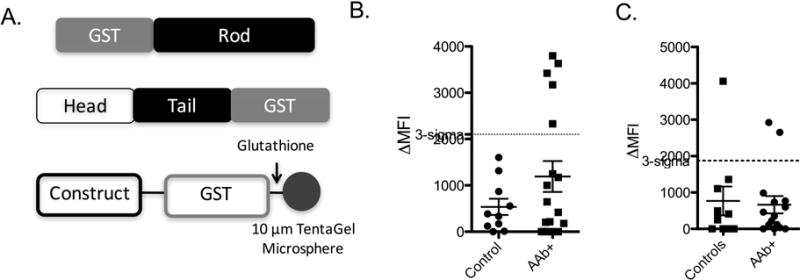
A) Schematic representation of the GST fusion proteins used to determine the antibody-binding domain. Each construct was mounted onto the surface of 10 μm TentaGel microspheres via glutathione, which was covalently attached to the microspheres to perform binding competition assays using the flow cytometry assay. See also Figure S6 and Table S5. B) Serum competition binding analysis using the coiled coil domain-GST fusion protein as a capture agent. C) Serum competition binding analysis using the head/tail domain-fusion protein as a capture agent.
Dimeric, phosphorylated peripherin is the key determinant for T1D antibody recognition
The rod domain of peripherin self-associates into coiled coil dimers and tetramers (Fuchs and Weber, 1994), which are the building blocks of intermediate filament assembly (Fig 6A). We asked whether anti-peripherin antibodies recognized one of these specific assembly states of peripherin. We tested this by cross-linking peripherin-His6 using Ru(bpy)3Cl2 and visible light (Fancy and Kodadek, 1999). Analysis of the products by SDS-PAGE and immunoblotting showed the presence of monomer, dimer and trace amounts of tetramer (Fig. 6B). The solution was passed through a gel filtration column to remove excess crosslinking reagent, then added to 30 μL of pooled T1D sera. Immunocomplexes containing peripherin bound to IgG antibodies were separated from other serum proteins via tandem nickel and Protein G affinity chromatography. Peripherin that was not associated with IgG was collected in the flow through of the protein G column. The two samples were analyzed by SDS-PAGE and immunoblotting. As shown in Fig. 6C, the antibody-associated peripherin (IP-pull down in Fig. 6C) is enriched for the dimer. Conversely, this band is markedly depleted in the non-antibody-associated fraction (Flow Through in Fig. 6C). The band intensities were determined by densitometry and plotted in Fig. 6D. Therefore, we conclude that antibodies from T1D sera primarily recognize a dimeric conformation of peripherin.
Figure 6. Phosphorylation mediates T1D antibody recognition.
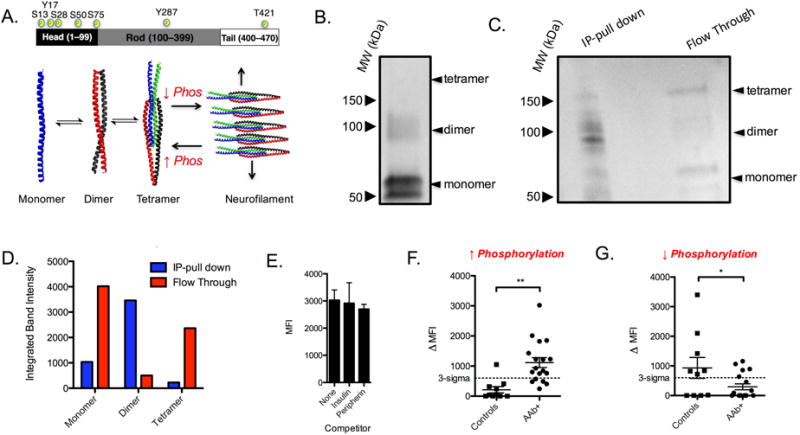
A) Top panel: Schematic representation of the three domains that comprise peripherin. Known phosphorylation sites are labeled with yellow circles. Bottom panel: the effect of peripherin self-association into dimer and tetramer is illustrated (bottom panel). When the phosphorylation state is high, type III intermediate filaments remain in a low oligomerization states such as dimer and tetramer. Dephosphorylation favors filament formation, and a reduction in low-order oligomers. B) Immunoblot of crosslinked peripherin. C) Immunoprecipitation of crosslinked peripherin using serum antibodies from T1D donors. D) Densitometry plot showing the integrated signal for monomer, dimer and tetramer for the IP and supernatant lanes. E) Competitive binding analysis of diabetes serum antibodies to mounted HaloTag-peripherin in the absence of competitor or presence of 500 nM insulin or 500 nM peripherin expressed in E. coli. Binding was measured using the cytometry assay. F) Competition serum binding analysis using phosphoprotein enriched peripherin as the specific competitor. G) Competition binding immunoassay on 10 normal control and 18 AAb+ subjects using HaloTag-peripherin that was treated with alkaline phosphatase to remove phosphate groups. For competition assays, the specific competitor was 100 nM of peripherin and the nonspecific competitor was 100 nM HaloTag-GST fusion protein. Dot plot data points are reported as the mean ± s.d. from two independent serum competition experiments. Statistical significance was determined using an unpaired t-test: **P < 0.01.
Since peripherin dimer and tetramer formation is regulated by phosphorylation (Aletta et al., 1989; Giasson and Mushynski, 1998) we asked whether phosphorylation had any affect on antibody recognition. To test this, we attached the HaloTag-peripherin fusion protein to HaloTag ligand-primed TentaGel beads and attempted to block binding of serum antibodies to these beads using soluble peripherin that was purified from E. coli, which would not be phosphorylated. As a control, binding of serum antibodies to the beads was also tested in the presence of insulin or in the absence of any competitor altogether. The results shown in Fig. 6E demonstrate that peripherin prepared in bacteria failed to block antibody binding to the beads displaying peripherin purified from human cells. These data support a role for post-translational modifications in peripherin recognition by T1D autoantibodies.
We next examined whether phosphorylation was the specific post-translational modification required for peripherin antigenicity. To do so, we set out to rigorously purify phospho-peripherin from human cells. As before, we expressed the peripherin-HaloTag conjugate (with a Tobacco Etch Virus protease cleavage site between the proteins) in HEK 293T cells and enriched for phosphoprotein using a TiO2 affinity column. The phospho-protein-enriched fraction was then mixed with TentaGel beads displaying the HaloTag ligand to capture the HaloTag protein-peripherin conjugate. The beads were washed and about half of them were subjected to TEV protease cleavage. This protocol thus provides preparations of immobilized HaloTag protein-peripherin and soluble peripherin highly enriched for the phosphorylated protein.
18 AAb+ and 10 control serum samples were evaluated using the competitive binding assay. The net signal (amount of fluorescent signal competed by soluble phospho-peripherin-signal competed by a control protein) for each of the 28 subjects is shown in Fig. 6F as a dot plot, which shows that 65% of the AAb+ subjects exhibited levels of antibody binding above the “three sigma” threshold compared to only one control patient.
This experiment was then repeated, but using as the capture agent and soluble competitor a HaloTag protein-peripherin preparation and a TEV-cleaved soluble peripherin protein that were incubated with alkaline phosphatase overnight to remove accessible phosphate groups. In stark contrast to the experiment using phospho-peripherin, in this case the AAb+ samples did not exhibit a higher signal than the control group (Fig. 6G). These data argue that peripherin phosphorylation is important for antibody recognition.
Discussion
The characterization of autoantigens involved in T1D is important to fully understand disease etiology and to develop simple antibody-based blood tests. While several autoantigens are already known, including insulin, IA-2, ZnT8 and GAD65, none are present in all T1D patients, leading to the assumption that more autoantigens remain to be discovered, possibly including molecules involved in the earliest, disease-initiating immune responses. To hunt for new T1D autoantigens in an unbiased fashion, we employed the “epitope surrogate” technology developed in our laboratory (Doran et al., 2015; Kodadek, 2014; Raveendra et al., 2013; Reddy et al., 2011). This involves screening a large library of synthetic molecules for compounds that retain far more antibodies from the serum of diabetic patients than non-diabetic controls. A screening hit is then used as an affinity reagent to enrich the antibodies that recognize it. Those T1D-associated antibodies, in turn, are used to “fish out” the native proteins that they recognize in a pancreatic tissue lysate.
The first screen for epitope surrogates resulted in the isolation of compound 1 that recognized antibodies in 20% of T1D patients. A second screen with a library that explored chemical space around the structure of 1 resulted in the isolation of 4, which was highly selective for antibodies from a single patient (Fig. 2). Surprisingly, 4 and 1 proved to bind different antibodies, as demonstrated by the fact that they do not compete with one another for IgG binding (Fig. 2C and D). Nonetheless, given the apparently high selectivity of binding of 4 to serum antibodies from that single patient, we decided to proceed forward to attempt to determine the native antigen recognized by 4-binding IgGs. Indeed, an affinity column comprised of immobilized 4 allowed us to enrich enough antibody to carry out an immunoprecipitation experiment using a non-denatured pancreatic lysate. The immunocomplex, (Fig. 3B), was found to contain peripherin. Peripherin competed for binding to antibodies that recognize 4, arguing that these antibodies were indeed raised against peripherin in this patient.
Given that 4 likely represents a surrogate for a single epitope, we tested the prevalence of anti-peripherin antibodies in the T1D donors using peripherin expressed in human cells mounted onto a liquid array platform. A competitive binding format for antibody detection was employed, in order to control for non-specific antibody binding to the immobilized protein. Our initial results indicated that peripherin antibodies were present in ≈67% of AAb+ patients and only one control patient (Fig. 4C).
It is important to point out that while there has been some mention of peripherin as a potential humoral autoantigen in T1D, the findings reported in this study are unique. Boitard, et al. used a combination of immunoblotting and immunoprecipitation to show that serum from non-obese diabetic (NOD) mice, the pre-eminent animal model for T1D, contained antibodies that recognize peripherin (Boitard et al., 1992). Though the binding epitope was not investigated, the use of denaturing conditions during Western blot analyses in this study argues strongly against a conformational epitope. Indeed, later studies aimed at more thoroughly mapping the peripherin epitope that is recognized by islet-infiltrating NOD mouse B cells revealed a single linear epitope located within the C-terminal domain (Garabatos et al., 2014; Puertas et al., 2007). This is obviously different than the antigen described here in human T1D patients.
Based on these mouse data, Strom, et al. used an electrochemiluminescence (ECL) assay and recombinant peripherin expressed in rat cells to carry out a focused search for anti-peripherin antibodies in human T1D patients. (Strom et al., 2010). Their results showed that a minority if people contain varying levels of anti-peripherin IgG, but that this is independent of T1D status, arguing against a linkage between anti-peripherin antibodies and T1D in humans. This is in contrast to the results reported here. The data reported by the Strom, et al paper are similar to what we see when de-phosphorylated peripherin is used as a probe (Fig. 6G). Thus, it is possible that Strom, et al. used a peripherin preparation that did not have the appropriate post-translational modifications that are required for specific recognition by the T1D-associated anti-peripherin antibodies that are linked to human T1D. Recall that our peripherin preparation was from human cells and even in this case, a few preparations failed to show significant reactivity with patient IgGs.
Finally, Lennon and co-workers reported that some patients with neurological conditions, particularly various neuropathies, have antibodies against peripherin (Chamberlain et al., 2010). However, when they analyzed a number of diabetes patients for these antibodies, almost none of them tested positive. Thus, these are different antibodies than the ones reported here. This study represents the first clear demonstration of the widespread presence of anti-peripherin antibodies in T1D patients.
Because of this confusing literature, when we first identified peripherin as the likely autoantigen, the importance of better characterizing the nature of the autoantigen-autoantibody interactions was clear. As shown in Fig. 5, about 30% of T1D patients and none of the controls tested, have antibodies that recognize the central rod domain of peripherin. The fact that about twice as many T1D patients have autoantibodies that recognize the full-length protein argues that many anti-peripherin recognize epitopes antibodies either partially or entirely outside this domain. Nonetheless, this is an interesting result from a practical point of view, since the GST-rod domain protein is much better behaved biochemically than full-length, relatively insoluble peripherin and could be used as a capture agent in some type of multiplexed assay in the future.
Even more importantly, we demonstrated that the T1D-associated anti-peripherin antibodies recognize a dimeric, phosphorylated form of the protein (Fig. 6). After cross-linking native peripherin, we found that the antibodies preferentially removed the dimeric product from the mixture. When peripherin was treated with a phosphatase, selective recognition by the T1D-associated antibodies was abolished. These results provide a striking demonstration that it would have been impossible to identify these interesting T1D-related autoantibodies by probing serum samples with an array of recombinant proteins.
Significance
Characterizing disease-linked antigens is challenging because the ideal antigen capture agents—cognate antibodies that bind antigens with high affinity and selectivity— are equally elusive and are likely present at low levels in serum. We have shown previously that synthetic molecules can be identified from combinatorial screens that bind to the antigen-binding site of disease-linked antibodies with sufficient affinity to retain them from serum. We call these ligands “epitope surrogates” because they occupy a “chemical descriptor” space shared by the native antigen, but due to their abiological composition, do not mimic antigen-antibody binding interactions completely. Importantly, the chemical diversity of synthetic combinatorial libraries does not limit discovery to antibodies against simple linear or unmodified epitopes, but instead, may contain compounds that recognize antibodies to more complex antigens. For example, some compounds may bind modestly well to antibodies raised against epitopes that are uniquely modified, discontinuous or conformational. In this study, we show that epitope surrogates are indeed capable of binding to such complex epitopes in the context of type 1 diabetes (T1D). An epitope surrogate that binds selectively to diabetes patient antibodies was used to enrich the antibodies on an affinity column. When exposed to pancreas lysates, these antibodies precipitated a dimeric conformation of phosphorylated peripherin, a type III intermediate filament found in neuroendocrine cells. These antibodies were found in 67% of the diabetic patients tested, suggesting that it is a major disease antigen. Most notably, the complexity of the antigenic recognition element would have prevented its discovery using conventional recombinant protein arrays, thus underscoring the advantage of using our chemical approach for unbiased antigen discovery.
Experimental Procedures
Please also see refer to the detailed procedures in the Supplemental Information.
Blood Collection
Peripheral blood was drawn into a 2.5 mL SST tube, inverted 5 times, then allowed to clot for 20–30 minutes at room temperature. The tube was centrifuged at 800 × g for 10 minutes to separate the serum, which was aliquoted into 1.8 mL cryovials and then frozen until further use. Blood was drawn from patients after informed consent was obtained. All human studies were approved by the Scripps Florida IRB.
Immunoprecipitation of native antigen from tissue lysates
A mixture containing 500 μg pancreas lysate (Novus Biologicals, catalogue # NB82059244) and 250 μg pancreas membrane fraction (Novus Biologicals, catalogue # NB82060604), both containing 1% NP-40 and a cocktail of protease inhibitors, were diluted 1:1 in immunoprecipitation buffer (IP buffer, 25 mM Tris, 150 mM NaCl, pH 7.2) and pre-cleared of IgG antibody using 100 μL of Protein A-sepharose resin (Thermo Fisher Scientific) and 100 μL Protein G-PLUS agarose resin (Pierce). The unbound supernatant was removed and to this was added 500 μL of ≈135 μg mL enriched disease antibody containing BSA. The mixture was incubated overnight at 4 °C with gentle rotation. The solution was added to a 1.5 mL microcentrifuge tube containing 25 μL Protein A and 25 μL Protein G and the suspension was mixed for 2 h at room temperature. The beads were washed with 1 mL immunoprecipitation buffer four times. The resin was washed with 500 μL water. The beads were centrifuged at 500 × g, and ≈400 μL of water was discarded. The remaining pellet was suspended in 100 μL 2X Laemmli loading buffer containing 1 M DTT, and the resin was heated to 95 °C for five min. The beads were spun at 5,000 × g and 100 μL was loaded onto the lanes of a 10% TGX gel (Bio-rad) for Coomassie staining, and 20 μL was added to a separate gel for silver staining. A control immunoprecipitation was performed by following the steps outlined above with the exception that enriched disease antibody was replaced with Pierce Human IgG, Whole Molecule (Thermo Scientific, Product # 31154) at the same total protein concentration as the enriched IgG eluted from the affinity column.
Preparation of peripherin-labeled microspheres
30 mg of 10 μm TentaGel® microspheres (0.25 mmol/g loading, 6.25 μmol) was suspended in anhydrous DMF for 20 min. The beads were pelleted on low-speed benchtop centrifuge and the DMF was decanted. HaloTag® Succidimidyl Ester (O2) Ligand (2.5 mg, 9.5 μm) was dissolved in DMF to a concentration of 5 mg/mL. To this solution was added 2 eq diisopropylethylamine (DIEA), and this solution was added to the pelleted beads. The bead mixture was mixed vigorously and allowed to react overnight with gentle agitation. The beads were washed in DMF (3 × 400 μL) and transferred to a Solvinert MultiScreen filter plate in equal aliquots of 7.5 mg to facilitate washing. The beads were washed with water (10 × 300 μL), and the final water wash proceeded for 6 h. The beads were equilibrated in PBS containing 0.05% tween 20 for 1 h and transferred back into a microcentrifuge tube, where they were pelleted. The appropriate peripherin construct was added to the pellet was added 1 mL of ~300 μg/mL peripherin-HaloTag® fusion protein in PBS solution. The suspension was rotated overnight at 4 °C. The beads were pelleted and the supernatant was removed. A small aliquot of ~3 mg beads was transferred to a filter plate and washed three times with TBS-T and then blocked with 0.5% BSA in TBS-T. The beads were used for binding assays within 2 hours of washing.
Preparation of human peripherin for competition binding
TentaGel beads containing the peripherin-HaloTag® fusion construct were subjected to TEV protease cleavage to release peripherin from the beads. 20 mg of TentaGel resin containing the construct was suspended in 400 μL of a solution containing 2 μg His-tagged rTEV (R&D Systems) in Tris buffer containing 1 mM DTT and the cleavage was performed overnight at 4 °C. The microspheres were pelleted and the supernatant was diluted to 5 mL in PBS and subjected to spin filtration in a 5 mL 10K MWCO filter to reduce the volume to 500 μL. After an additional round of spin filtration to reduce the DTT concentration, the sample was added to Ni-NTA resin (100 μL) that had been pre-equilibrated with 5 resin volumes of PBS. The beads with the Ni-NTA column for 30 min at 4 °C, Added 0.2% tween 20 and peripherin was collected by removing the supernatant by spin filtration at 500 × g. The peripherin concentration was adjusted to 15 μg mL−1, (~250 nM) and the protein was used immediately. Occassionaly, peripherin aggregated while concentrating, but was resolubilized by incubating at room temperature in the presence of 0.2% Tween 20. The insoluble fraction was pelleted at 10,000 × g and the supernatant was collected.
Phosphoprotein enrichment of peripherin
The HaloTag-peripherin solution was diluted to 100 μg/mL and phosphoproteins were enriched using a phosphoenrichment kit (Cell Biolabs, Inc.). After eluting the phosphoprotein with elution buffer, the eluent was collected by removing the beads through a 10 μm filter (Mo Bi Tec) using a low-speed centrifuge. The eluted phosphoproteins were dialyzed into PBS overnight at 4 °C and then added to 10 μm TentaGel beads displaying a HaloTag ligand (prepared using the methods described above) overnight at 4 °C with constant rotation. An aliquot containing approximately 2 mg of beads was removed and incubated an additional 3 h at room temperature.
Dephosphorylation of peripherin
To ~1 mL of the Peripherin-HT-His6 fusion protein solution was added 100 μL of 10X alkaline phosphatase buffer (Promega) and 100 U of alkaline phosphatase from calf intestine (Promega). The solution was incubated at 37 °C for 1 h and then an additional 16 h at 4 °C with gentle mixing on a rotator.
Binding assays
Dye-encoded 10 μm TentaGel beads exterior-linked ligands were blocked as a suspension of TBS-T containing 0.5% BSA at a concentration of ~10 mg/mL for at least 1 h at room temperature with constant rotation. The beads were added to 500 μg mL−1 of serum diluted into PBS containing 0.5% BSA and 0.05% tween 20 (herein referred to as “binding buffer”). The suspension of beads in serum was incubated at room temperature for 1.5 h and transferred to a filter plate. The beads were washed with TBS-T (2 × 500 μL). For blinded experiments, serum samples were diluted to 1 mg mL−1 in PBS containing 1.5% BSA and de-identified by S. S. For binding experiments, the serum samples were diluted to 500 μg mL−1 and the binding assay was performed by T. M. D. as described above.
Competition binding assays
Recombinant proteins and epitope surrogates were spun at 10,000 × g for 5 min and diluted to 200 nM in “binding buffer” for full-length peripherin, or 100 nM for GST-tagged head-tail domain or GST-tagged rod domain. The competitor solution was diluted 1:1 into 20 μL of serum (1 mg/mL total protein) containing 1% BSA (final serum protein concentration = 500 μg mL−1). The serum and competitor mixture was incubated at room temperature for 15 minutes. The beads containing bound antigens or epitope surrogates were suspended in the diluted serum and competitor mixture. The suspension was incubated with constant rotation for 1.5 h at room temperature. The beads from each sample were transferred to individual wells of a MultiScreen Solvinert filter plate and washed with TBS-T (2 × 500 μL). The beads were incubated for 1 h with 150 μL of Alexa Fluor 647-conjugated goat anti-human secondary antibody diluted 1:200 in TBS-T containing 0.5% BSA. Binding was analyzed on an LSRII flow cytometry. ΔMFI = MFI (HaloTag competitor) − MFI (Peripherin competitor). Negative values were constrained to 0. For 3-sigma threshold calculations, an F-test was used to remove outliers, which applied in the case of a single false positive in Fig. 4C and Fig. 6F.
Photo-induced cross-linking of unmodified protein (PICUP)
Proteins solutions in PBS-T were diluted to the desired concentration. 1 μL of 1 mM tris(bipyridine)ruthenium (II) chloride followed by 1 μL 20 mM ammonium persulfate (APS) were added for every 18 μL of peripherin solution. The solution was mixed and irradiated with a short-wave UV lamp for 10 s. Samples being analyzed by SDS-PAGE were immediately quenched with 5SDS-PAGE loading buffer containing 500 mM DTT. The samples were heated to 95 °C for 5 min, centrifuged at 500 × g and loaded onto 4–15% gradient TGX gels (Bio-Rad). For crosslinked samples that were used for downstream binding assays, the crosslinking reaction was quenched with 20 μL 100 mM DTT and DTT was removed via spin filtration and then overnight dialysis in a MINI cassette.
Immunoprecipitation of crosslinked peripherin
Pooled T1D sera (30 μL) was added to crosslinked peripherin and incubated for 4 h at 4 °C in a 1.5 mL microcentrifuge tube. The mixture was purified over ≈100 μL HisPur Ni-NTA resin that had been pre-washed with Ni-NTA buffer. The suspension was incubated at 4 °C for 30 min. The resin was washed four times with 1 mL wash buffer. The bound complexes were removed with 250 mM imidazole, which was removed in two passes of spin filtration using a 30 kDa MWCO spin filter and concentrated to 1 mL. Immunoprecipitation was performed by adding the eluted peripherin-antibody complexes to 100 μL pre-washed Protein G-PLUS agarose (Pierce) for 1 h at room temperature. The supernatant was saved and concentrated to 40 μg mL−1 in a 10 kDa MWCO filter Amicon Ultra 0.5 mL spin filter. The protein G agarose beads were washed four times with 1 mL IP buffer followed by 1 mL water. 100 μL 2X Laemmli sample buffer containing 1 M DTT was added to the beads and they were heated at 95 °C for 5 min. The beads were centrifuged at 5,000 × g for 5 min and the supernatant was collected. The solution was centrifuged at 500 × g for 1 min and separated by SDS-PAGE. The separated proteins were blotted onto nitrocellulose, blocked with TBS-T containing 2% BSA and probed at 4 °C overnight with chicken anti-peripherin polyclonal antibody (Pierce) diluted 1:500 in TBS-T containing 1% BSA. The blot was washed with TBS-T (4 × 5 min). Anti-chicken HRP (1:25,000) in TBS-T containing 1% BSA was incubated with the blot for 1 h at room temperature. The blot was washed with TBS-T (4 × 5 min) and developed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo). Chemiluminescence was visualized using a Typhoon 9410 Variable Mode Imager.
Supplementary Material
Highlights.
Chemical “epitope surrogates” are selective affinity probes for T1D-related IgG.
Affinity-purified antibodies bind to the neuroendocrine protein, peripherin.
Serum antibodies from T1D patients recognize dimeric peripherin.
Phosphorylation of peripherin is critical for T1D antibody recognition.
Acknowledgments
We would like to thank K. Lowe and B. Torres of the Scripps Florida Flow Cytometry Core. We are grateful to R. Flefil and P. Martinez of the Scripps Florida Proteomics Core. This work was supported by a DP3 award from the NIH/NIDDK (DK-094309).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
T. K. conceived the project. T. M. D. and T. K. designed experiments, analyzed results and wrote the manuscript; T. M. D. performed experiments; J. M. made the primary epitope surrogate library; A. P. and J. S. supervised blood collection and serum preparation, characterized subjects in this study for T1D and autoantibody status, and edited the manuscript; S. S., E. J. K. and L. F. C. cloned plasmids. L. F. C. purified proteins and edited the manuscript. K. P. and S. L. S. resynthesized ligands.
References
- Aletta JM, Shelanski ML, Greene LA. Phosphorylation of the peripherin 58-kDa neuronal intermediate filament protein. Regulation by nerve growth factor and other agents. J Biol Chem. 1989;264:4619–4627. [PubMed] [Google Scholar]
- Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. Isolation of Protein Ligands from Large Peptoid Libraries. J Am Chem Soc. 2003;125:13995–14004. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- Astle JM, Simpson LS, Huang Y, Reddy MM, Wilson R, Connell S, Wilson J, Kodadek T. Seamless Bead to Microarray Screening: Rapid Identification of the Highest Affinity Protein Ligands from Large Combinatorial Libraries. Chem Biol. 2010;17:38–45. doi: 10.1016/j.chembiol.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: As good as it gets? Nature medicine. 1999;6:601–604. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- Balboni I, Chan SM, Kattah M, Tenenbaum JD, Butte AJ, Utz PJ. Multiplexed protein array platforms for analysis of autoimmune diseases. Annu Rev Immunol. 2006;24:391–418. doi: 10.1146/annurev.immunol.24.021605.090709. [DOI] [PubMed] [Google Scholar]
- Boitard C, Villa MC, Becourt C, Gia HP, Huc C, Sempe P, Portier MM, Bach JF. Peripherin: an islet antigen that is cross-reactive with nonobese diabetic mouse class II gene products. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:172–176. doi: 10.1073/pnas.89.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacio E, Lampasona V, Bernasconi L, Ziegler AG. Maturation of the humoral autoimmune response to epitopes of GAD in preclinical childhood type 1 diabetes. Diabetes. 2000;49:202–208. doi: 10.2337/diabetes.49.2.202. [DOI] [PubMed] [Google Scholar]
- Bonifacio E, Lampasona V, Genovese S, Ferrari M, Bosi E. Identification of protein tyrosine phosphatase-like IA2 (islet cell antigen 512) as the insulin-dependent diabetes-related 37/40K autoantigen and a target of islet-cell antibodies. Journal of immunology. 1995;155:5419–5426. [PubMed] [Google Scholar]
- Chamberlain JL, Pittock SJ, Oprescu AM, Dege C, Apiwattanakul M, Kryzer TJ, Lennon VA. Peripherin-IgG association with neurologic and endocrine autoimmunity. J Autoimmun. 2010;34:469–477. doi: 10.1016/j.jaut.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran TM, Gao Y, Mendes K, Dean S, Simanski S, Kodadek T. The utility of redundant combinatorial libraries in distinguishing high and low quality screening hits. ACS Comb Sci. 2014;16:259–270. doi: 10.1021/co500030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran TM, Kodadek T. A Liquid Array Platform for the Multiplexed Analysis of Synthetic Molecule-Protein Interactions. ACS Chem Biol. 2013;9:339–346. doi: 10.1021/cb400806r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran TM, Simanski S, Kodadek T. Discovery of native autoantigens via antigen surrogate technology: Application to Type I diabetes. ACS Chem Biol. 2015;10:401–412. doi: 10.1021/cb5007618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy DA, Kodadek T. Chemistry for the analysis of protein-protein interactions: Rapid and efficient cross-linking triggered by long wavelength light. Proc Natl Acad Sci USA. 1999;96:6020–6024. doi: 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. Synthesis of N-substituted glycine peptoid libraries. Meth Enzymol. 1996;267:437–447. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- Gao Y, Amar S, Pahwa S, Fields G, Kodadek T. Rapid Lead Discovery Through Iterative Screening of One Bead One Compound Libraries. ACS Comb Sci. 2015;17:49–59. doi: 10.1021/co500154e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Kodadek T. Synthesis and screening of stereochemically diverse combinatorial libraries of peptide tertiary amides. Chem & Biol. 2013;20:360–369. doi: 10.1016/j.chembiol.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garabatos N, Alvarez R, Carrillo J, Carrascal J, Izquierdo C, Chapman HD, Presa M, Mora C, Serreze DV, Verdaguer J, Stratmann T. In Vivo Detection of Peripherin-Specific Autoreactive B Cells during Type 1 Diabetes Pathogenesis. J Immunol. 2014;192:3080–3090. doi: 10.4049/jimmunol.1301053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Mushynski WE. Intermediate filament disassembly in cultured dorsal root ganglion neurons is associated with amino-terminal head domain phosphorylation of specific subunits. J Neurochem. 1998;70:1869–1875. doi: 10.1046/j.1471-4159.1998.70051869.x. [DOI] [PubMed] [Google Scholar]
- Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, Steinman L, Robinson WH. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138–143. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- Kodadek T. Chemical tools to monitor and manipulate the adaptive immune system. Chem & Biol. 2014;21:1066–1074. doi: 10.1016/j.chembiol.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan MS, Wasserfall C, Maclaren NK, Notkins AL. IA-2, a transmembrane protein of the protein tyrosine phosphatase family, is a major autoantigen in insulin-depedent diabetes mellitus. Proc Natl Acad Sci USA. 1996;93:2307–2311. doi: 10.1073/pnas.93.13.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larman HB, Zhao Z, Laserson U, Li MZ, Ciccia A, Gakidis MAM, Church GM, Kesari S, LeProust EM, Solimini NL, Elledge SJ. Autoantigen discovery with a synthetic human peptidome. Nat Biotechnol. 2011;29:535–541. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian W, Upadhyaya P, Rhodes CA, Liu Y, Pei D. Screening Bicyclic Peptide Libraries for Protein-Protein Interaction Inhibitors: Discovery of a Tumor Necrosis Factor-alpha Antagonist. Journal of the American Chemical Society. 2013;135:11990–11995. doi: 10.1021/ja405106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Portier MM, de Nechaud B, Gros F. Peripherin, a new member of the intermediate filament protein family. Dev Neurosci. 1983;6:335–344. doi: 10.1159/000112360. [DOI] [PubMed] [Google Scholar]
- Puertas MC, Carrillo J, Pastor X, Ampudia RM, Planas R, Alba A, Bruno R, Pujol-Borrell R, Estanyol JM, Vives-Pi M, Verdaguer J. Peripherin Is a Relevant Neuroendocrine Autoantigen Recognized by Islet-Infiltrating B Lymphocytes. J Immunol. 2007;178:6533–6539. doi: 10.4049/jimmunol.178.10.6533. [DOI] [PubMed] [Google Scholar]
- Raveendra B, Hao W, Baccala R, Reddy MM, Schilke J, Bennett JL, Theofiliopolous AN, Kodadek T. Discovery of peptoid ligands for anti-Aquaporin 4 antibodies. Chem & Biol. 2013;20:350–359. doi: 10.1016/j.chembiol.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MM, Wilson R, Wilson J, Connell S, Gocke A, Hynan L, German D, Kodadek T. Identification of candidate IgG biomarkers for Alzheimer’s Disease via combinatorial library screening. Cell. 2011;144:132–142. doi: 10.1016/j.cell.2010.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harbor perspectives in medicine. 2012;2:a007781. doi: 10.1101/cshperspect.a007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom A, Sonier B, Chapman HD, Mojibian M, Wang GS, Slatculescu CR, Serreze DV, Scott FW. Peripherin-Reactive Antibodies in Mouse, Rabbit, and Human Blood. J Proteome Res. 2010;9:1203–1208. doi: 10.1021/pr900492y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velloso LA, Kampe O, Hallberg A, Christmanson L, Betsholtz C, Karlsson FA. Demonstration of GAD-65 as the main immunogenic isoform of glutamate decarboxylase in type 1 diabetes and determination of autoantibodies using a radioligand produced by eukaryotic expression. J Clin Invest. 1993;91:2084–2090. doi: 10.1172/JCI116431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996;45:926–933. doi: 10.2337/diab.45.7.926. [DOI] [PubMed] [Google Scholar]
- Yu L, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen K, Eisenbarth GS. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A. 2000;97:1701–1706. doi: 10.1073/pnas.040556697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. J Am Med Assoc. 2013;309:2473–2479. doi: 10.1001/jama.2013.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.