

Abstract
Congenital heart disease (CHD) patients have increased prevalence of extra-cardiac congenital anomalies (CA) and risk of neurodevelopmental disabilities (NDD). Exome sequencing of 1,213 CHD parent-offspring trios identified an excess of protein-damaging de novo mutations, especially in genes highly expressed in developing heart and brain. These mutations accounted for 20% of patients with CHD, NDD and CA but only 2% with isolated CHD. Mutations altered genes involved in morphogenesis, chromatin modification, and transcriptional regulation, including multiple mutations in RBFOX2, an mRNA splice regulator. Genes mutated in other cohorts ascertained for NDD were enriched in CHD cases, particularly those with coexisting NDD. These findings reveal shared genetic contributions to CHD, NDD, and CA and provide opportunities for improved prognostic assessment and early therapeutic intervention in CHD patients.
Extra-cardiac congenital anomalies (CA, structural or functional anomalies that arise in utero) occur in approximately 13% of newborns with congenital heart disease (CHD), including 2% with a genetic syndrome, almost twice the prevalence observed in infants without CHD (1). Newborns with CHD are also at risk for the emergence of neurodevelopmental disorders (NDD), including cognitive, motor, social and language impairments. NDD occurs in 10% of all children with CHD and in 50% with severe CHD (2). Explanations to account for the high frequency of CA and NDD in CHD patients include embryonic circulatory deficits and stresses associated with post-natal therapeutic interventions (3), but these hypotheses remain unproven.
We sequenced exomes in 1,213 CHD trios, (probands and their unaffected parents) enrolled in the Pediatric Cardiac Genetics Consortium (PCGC) (4) or the Pediatric Heart Network (PHN) (5) after excluding CHD cases with clinically recognized genetic syndromes. Analyses included 353 previously reported CHD trios (6). We compared de novo mutations identified in CHD that occurred in isolation, or accompanied by CA, NDD, or both (phenotypes in Tables S1 and Database S1). Previously sequenced trios (n=900) from the Simons Foundation Autism Research Initiative Simplex Collection, each consisting of the unaffected parents and sibling of a child with autism spectrum disorder, served as control trios (7–9).
CHD and control probands were analyzed for de novo mutations (Databases S2–3). To evaluate the significance of mutation frequencies, we adapted a recently reported de novo expectation model (10) to assess mutation rates by variant class (synonymous, loss of function [LoF; e.g., nonsense, frameshift, canonical splice disruptions], or missense). We derived gene-based rates of de novo mutation from local sequence context, and adjusted by per-base coverage separately in case and control cohorts (Databases S4–5). We extended the model by merging all possible transcripts to obtain transcript-independent probabilities and by adding rates for deleterious missense variants predicted by the Meta-SVM score (D-Mis) (11). This yielded an overall mean expected mutation rate of 1.1 de novo variant per proband.
The expected and observed numbers of de novo mutations in each variant class in all CHD and control subjects (Table 1) were compared using a Poisson distribution. De novo mutation rates per variant class were accurately predicted in controls, replicating previous model validations (10). However, among all CHD trios, we detected significant enrichment (i.e., observed divided by expected frequencies) of LoF and D-Mis variants of 1.3 (p=0.0016) and 1.6 (p=1.8×10−10), respectively, across all genes. The combination of LoF+D-Mis variants (hereafter denoted as “damaging”) was 1.4-fold enriched in CHD cases compared to expectation, similar to the observed case vs. control comparison (Table S2). This burden persisted after excluding 353 previously studied CHD trios (Table S3) and was found in each CHD category (conotruncal defects, left ventricular outflow tract obstruction and “other”), except for heterotaxy, which showed no excess (Table S4).
Damaging de novo mutations were markedly increased in CHD cases (enrichment=2.4, p=5.1×10−24) among 4,420 genes in the top quartile of expression during heart development (high heart expression, HHE (6)) (Table 1). Conversely, controls had no significant enrichment in de novo mutations in HHE genes. Neither cases nor controls were enriched in de novo mutations among genes within the lower three quartiles of developing heart expression (LHE) (Table 1). From the observed and expected values, we estimated that 58% of these damaging de novo mutations contributed to CHD.
Twenty-one genes had multiple damaging de novo mutations only in cases, an unlikely chance occurrence (Fig. 1A; median expected 7; p=1.1×10−5 by permutation), suggesting that these genes are likely to be pathogenic for CHD (Fig. 1B). Indeed, this list includes seven genes previously implicated in CHD (PTPN11, KMT2D, CHD7, MYH6, JAG1, NOTCH1, ZEB2). Enrichments were not observed among genes with multiple de novo synonymous variants in CHD cases or across any variant class among controls (Fig. 1A and Table S5). A variety of cardiac malformations were associated with mutations in each of these 21 genes (Fig 1B and Table S6). From simulations based on these data (12), we estimate that de novo mutations in ~392 HHE genes contribute to CHD pathogenesis (Fig. S1).
Fig. 1.
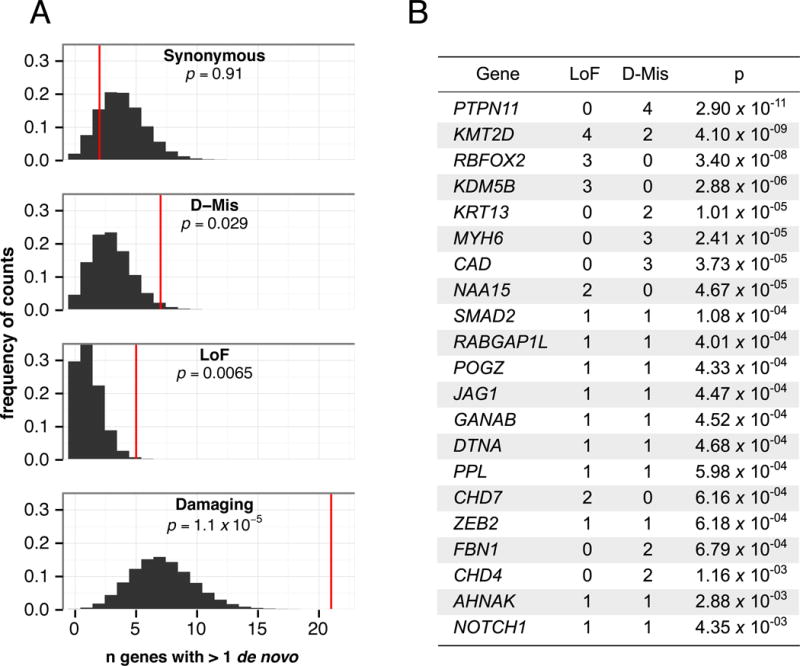
Genes with multiple de novo mutations are candidate CHD risk genes. A: Histograms show the expected distribution of the number of genes containing multiple de novo mutations (empirically derived using 1M permutations, black), and the observed number of genes with multiple mutations in cases (red line) for each class. P-values were calculated by permutation. B: Twenty-one genes with multiple damaging de novo mutations in cases. P-values are from Poisson test against expectation, with significance threshold < 9×10−7. Further details are shown in Table S6.
Within this 21-gene set, PTPN11, KMT2D (MLL2), and RBFOX2, each had significantly more damaging de novo mutations than expected (Bonferroni corrected threshold for genome-wide significance = p<9×10−7; Fig. 1B). RBFOX2, an RNA binding protein that regulates alternative splicing has not been previously implicated in CHD. RBFOX2 harbored three distinct de novo LoF mutations, a highly significant finding (Fig. 1B; p=3.4×10−8). Additionally, we previously identified a de novo copy number loss that encompasses RBFOX2 in another CHD proband (13). Remarkably, these four probands all have hypoplastic left heart syndrome (HLHS). RBFOX2 is critical for zebrafish heart development (14), and regulates epithelial-mesenchymal transitions (EMT) (15). Disruption of EMT is felt to underlie HLHS pathogenesis (16). Notably, we observed significant enrichment of damaging mutations in RBFOX2 target genes (17) in CHD cases (1.9-fold, p=6.6×10−8) but not controls (Table S7).
Gene ontology (GO) analysis revealed enrichment of damaging de novo mutations in genes involved in anatomic structure morphogenesis (GO:0009653; 2.4-fold; Bonferroni p=3.4×10−14), cardiovascular system development (GO:0072358; 3.2-fold; Bonferroni p=7.5×10−9), neurodevelopmental abnormality (HP:0012759; 2.6-fold; Bonferroni p=1.8×10−6), and others (Database S6). We replicated the reported excess of de novo LoF mutations affecting chromatin modification (6), even after including only newly studied cases (GO:0016568, 5.1-fold enrichment, p-value=7.2×10−5; Database S7). In the full CHD cohort, there were 25 de novo LoF mutations in chromatin modifying genes, a 5.3-fold enrichment over expectation (p=5.7×10−11, Table S8; Fig. S2), strongly supporting the conclusion that these damaging de novo mutations impart large effects on CHD risk.
We examined the prevalence of damaging de novo mutations in CHD with or without NDD and/or CA (Fig 2A–C) after excluding 19 subjects found to have de novo mutations in known syndromic CHD genes and 279 subjects with uncertain NDD/CA status. Damaging de novo mutations in HHE genes were not significantly enriched in 356 subjects with isolated CHD nor in controls, but were ~3-fold enriched in 559 CHD cases with CA and/or NDD (CHD + Extra, p=1.1×10−18) including 97 probands diagnosed with either NDD or CA but unknown for the other phenotype. Excluding these 97 probands, we observed a 4.7-fold enrichment of damaging de novo mutations in HHE genes among 138 CHD cases with both NDD and CA (p=5.6×10−15), a 2-fold enrichment in CHD cases with only NDD (252 probands, p=3.8×10−4) and a 2.9-fold enrichment in CHD cases with only CA (72 probands, p=7.4×10−4). By comparing de novo rates in cases against expectation, we estimate that damaging de novo mutations in HHE genes contributed to 20% of CHD with both NDD and CA (95% confidence interval 12–30%), 10% (7–14%) of CHD with CA and/or NDD, 10% (2.5–23%) and 6% (2–11%) of CHD with CA or NDD only, respectively, and only 2% (0.5–5%) of isolated CHD (Fig. 2B). These results implied frequent pleiotropic effects of de novo mutations in CHD and raise the possibility that mutations in these same genes might also contribute to non-syndromic NDD and/or other CA. Indeed, we find that genes mutated in CHD are not only enriched for high expression in developing heart, they are also enriched for high expression in developing brain (Table S9).
Fig. 2.
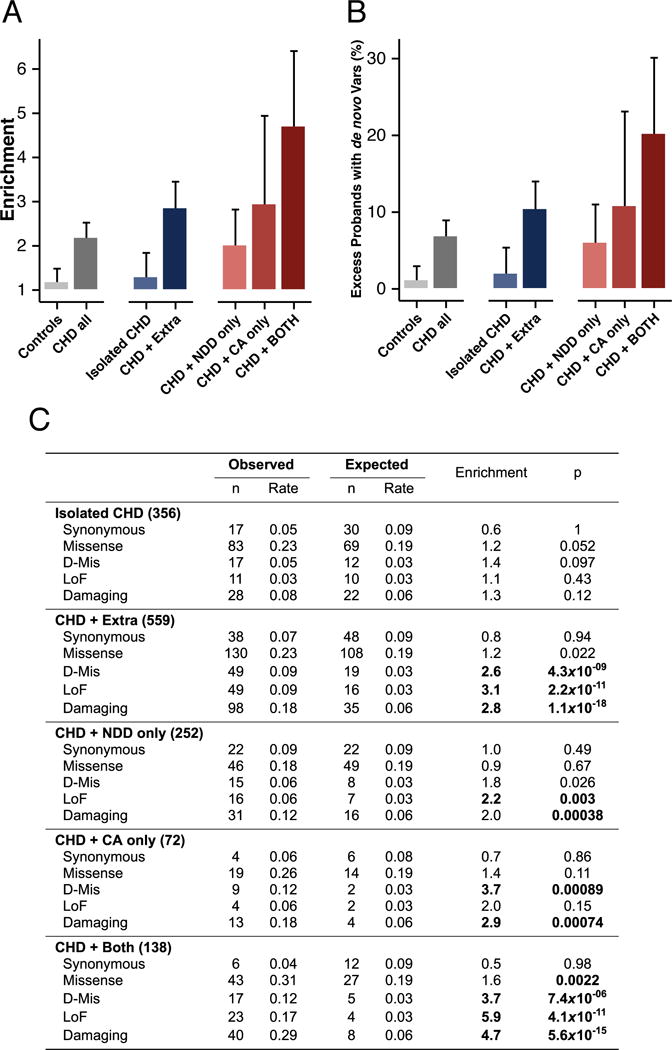
Burden of damaging de novo mutations in HHE genes among CHD cases with extra-cardiac phenotypes. A: The enrichment (ratio of observed/expected) of damaging de novo mutations in HHE genes is shown for each phenotype (± 95% confidence interval). Case probands were excluded if they carried de novo mutations in known CHD syndrome genes (n=19), had unknown extracardiac phenotype for both NDD and CA (n=6), or had one unknown phenotype and were negative for the other (n=273). Cases with either CA or NDD and unknown status for the other phenotype (n=97) were included in the “Extra” category but excluded from the “only” categories. B: Percent excess of individuals carrying damaging de novo mutations in HHE genes by indicated phenotype (± 95% confidence interval). Explanation of calculation is provided (12). C: Table of observed and expected de novo rates for the indicated variant classes by phenotype.
To further explore these pleiotropic effects, we considered whether genes with damaging de novo mutations in CHD with NDD overlapped with 1,161 genes (Database S8) found to contain damaging de novo mutations in seven cohorts ascertained for NDD phenotypes excluding CHD (published NDD, P-NDD gene set) (7, 18–23). Sixty-nine genes (Table S10) with damaging de novo mutations (n=85 mutations) were shared in CHD and P-NDD cohorts, far more than expected by chance (expected = 32 mutations; 2.6-fold enrichment; p=8.9×10−15, Fig. 3A and Table S19). HHE genes were particularly enriched among P-NDD genes that were mutated in CHD (4.4-fold for all CHD cases, p=1.2×10−23; Fig. 3A and Table S11). Moreover, genes mutated both in P-NDD and CHD cohorts are in the top quartile of both developmental heart and brain expression far more than expected by chance (observed = 38, expected = 11, p=6.1×10−11, binomial test, Fig. 3B). Input of these 69 overlapping genes into GO ontology analysis revealed significant terms that were broadly involved in the regulation of developmental transcription programs. These included 19 chromatin modifiers (GO:0016568 9.3-fold, p=8.5×10−10; Database S9 and Fig. S2) including genes responsible for altering the methylation, acetylation or ubiquitination status of numerous regulatory lysine residues on the nucleosome. Additionally, there were 32 transcriptional regulators (GO:0006355 2.8-fold p=1.5×10−4; Database S9), including genes involved in the Wnt (CTNNB1, DVL3, LRP5) and Notch (NOTCH1, EP300) signaling, important pathways in cardiac development. These findings demonstrate shared genetic etiologies for CHD and NDD patients, and confirm pleiotropic effects of mutations in these genes. Because it is unlikely that many P-NDD-ascertained patients with these mutations had clinically important CHD, and not all CHD patients with these mutations have NDD (see below), our findings also indicate that these mutations have variable expressivity, including isolated CHD, isolated NDD, or both.
Fig. 3.
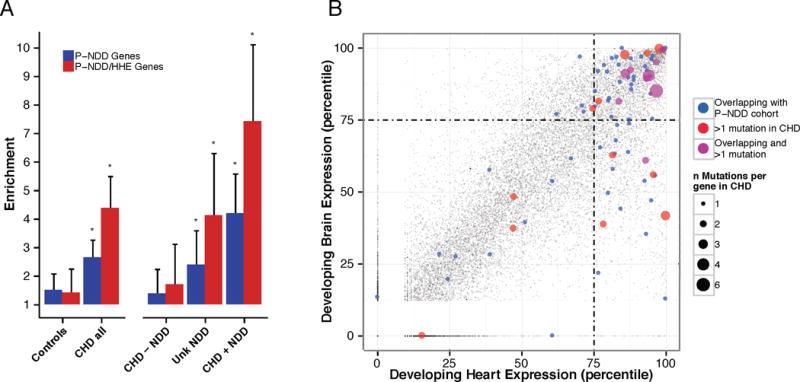
Genes containing de novo mutations in CHD cases show pleotropic developmental effects. A: Individuals with CHD carry an excess of damaging de novo mutations among 1,161 genes identified by containing damaging de novo mutations in 7 published studies of NDD (P-NDD cohort) (7, 18–23). All CHD cases were subdivided by NDD status (CHD + NDD, n=417 subjects, CHD – NDD n=440, Unknown NDD, n=363). P-values ≤ 0.005 as indicated (stars) were calculated from Poisson test against model-derived distribution (values in Table S11). P-NDD gene set (blue) was further filtered for HHE genes (P-NDD/HHE, red, 564 genes). Enrichments are shown ± 95% confidence intervals. B: Percentile gene expression ranks (100=high) are shown for all genes (grey) in the developing brain and heart, highlighting 69 genes with damaging de novo mutations in both CHD cases and the P-NDD cohort (blue or purple). Genes with multiple de novo mutations in CHD (red or purple) are shown. Point size represents numbers of de novo events.
Consistent with these observations, CHD subjects with damaging de novo mutations in these 69 overlapping genes (Fig. 3A) had a significantly increased risk of NDD (absolute risk of 73%, odds ratio 3.1, p=7.9×10−5, Fisher exact test). Damaging mutations (28 mutations in 27 subjects) in chromatin modifiers showed the highest risk of NDD (19 subjects with NDD, 8 with unknown NDD status due to age <1 year at evaluation). Moreover, the marked enrichments in damaging de novo mutations among P-NDD genes with HHE (Fig. 3A and Table S11), 7.4-fold in 413 CHD cases with NDD (p=3.9×10−22), 4.1-fold in 362 CHD infants with unknown NDD status (p=2.2×10−7) and no significant enrichment in 438 CHD cases without NDD (p=0.075), strongly implies a future risk of NDD among CHD infants with these variants. These observations suggest that genotype is a strong predictor for future development of NDD in CHD infants. Despite these highly significant findings, our estimates are based purely on statistical grounds and limited to in silico predictions of damaging variants, a caveat that should be considered when extrapolating these results to identify causative/predictive mutations in individual patients.
Contemporary therapeutic interventions have substantially improved survival among newborns with serious CHD. Despite these advances, many life-long medical issues remain. The demonstration that damaging de novo gene mutations cause CHD, particularly when associate with NDD and other birth defects has both clinical and research implications. First, clinical genotyping may help stratify CHD patients and identify those at high risk for NDD, enabling surveillance and early interventions to improve school performance, employability and quality of life. Second, the pleiotropic consequence of these mutations implies that further study of these genes may uncover critical regulation of broad developmental programs. Finally, the high frequency of mutation in transcriptional regulators suggests that mutations in regulatory elements (promoters and enhancers) may be additional causes of CHD, particularly isolated CHD.
Supplementary Material
Acknowledgments
The authors are grateful to the patients and families who participated in this research and team members who supported subject recruitment and sequencing: Danielle Awad, Carlos Breton, Katrina Celia, Charina Duarte, Davina Etwaru, Nathan Fishman, Mereurt Kaspakova, Jennie Kline, Rosalind Korsin, Alyssa Lanz, Emma Marquez, Dawn Queen, Ashley Rodriguez, Janine Rose, Jaswinder K. Sond, Dorothy Warburton, Abigail Wilpers, and Roslyn Yee (Columbia Medical School), B. McDonough, A. Monafo, J. Stryker (Harvard Medical School); N. Cross (Yale School of Medicine); S. M. Edman, J. L. Garbarini, J. E. Tusi, S. H. Woyciechowski (Children’s Hospital of Philadelphia); J. Ellashek, N. Tran (Children’s Hospital of Los Angeles); K. Flack L. Panesar, N. Taylor (University College London); D.Gruber, N. Stellato (Steve and Alexandra Cohen Children’s Medical Center of New York); D. Guevara, A. Julian, M.Mac Neal, C. Mintz (Icahn School of Medicine at Mount Sinai); E. Taillie (University of Rochester School of Medicine and Dentistry). We thank P. Candrea, E. Mazaika, K. Pavlik, V. Spotlow, and M. Sotiropoulos for production exome sequences and variant confirmation.
This work was supported by grants from the National Heart, Lung, and Blood Institute (Pediatric Cardiac Genomics Consortium, Pediatric Heart Network and Cardiovascular Development Consortium) and the National Human Genome Research Institute of the National Institutes of Health, Howard Hughes Medical Institute, Simons Foundation for Autism Research, John S. LaDue Fellowship at Harvard Medical School, Medical Scientist Training Program and National Research Science Award, Academy of Medical Sciences, British Heart Foundation, Wellcome Trust, Arthritis Research UK and the NIHR Cardiovascular Biomedical Research Unit at Royal Brompton and Harefield NHS Foundation Trust and Imperial College London, Leducq Foundation, Heart and Stroke Foundation of Ontario, Ted Roger Centre for Heart Research, Kostin Family Innovation Fund, Aaron Stern Professorship at the University of Michigan and Braylon’s Gift of Hope Fund. The views expressed are those of the authors and do not necessarily reflect those of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Footnotes
Editor correspondence to: cseidman@genetics.med.harvard.edu
References and Notes
- 1.Egbe A, Lee S, Ho D, Uppu S, Srivastava S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann Pediatr Cardiol. 2014;7:86–91. doi: 10.4103/0974-2069.132474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marino BS, et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126:1143–1172. doi: 10.1161/CIR.0b013e318265ee8a. [DOI] [PubMed] [Google Scholar]
- 3.Gaynor JW, et al. Neurodevelopmental outcomes after cardiac surgery in infancy. Pediatrics. 2015;135:816–825. doi: 10.1542/peds.2014-3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pediatric Cardiac Genomics Consortium et al. The Congenital Heart Disease Genetic Network Study: rationale, design, and early results. Circulation research. 2013;112:698–706. doi: 10.1161/CIRCRESAHA.111.300297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohye RG, et al. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med. 2010;362:1980–1992. doi: 10.1056/NEJMoa0912461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaidi S, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iossifov I, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samocha KE, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong C, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Materials and methods are available as supplementary materials on Science Online.
- 13.Glessner JT, et al. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circulation research. 2014;115:884–896. doi: 10.1161/CIRCRESAHA.115.304458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallagher TL, et al. Rbfox-regulated alternative splicing is critical for zebrafish cardiac and skeletal muscle functions. Developmental biology. 2011;359:251–261. doi: 10.1016/j.ydbio.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braeutigam C, et al. The RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene. 2014;33:1082–1092. doi: 10.1038/onc.2013.50. [DOI] [PubMed] [Google Scholar]
- 16.Hickey EJ, Caldarone CA, McCrindle BW. Left ventricular hypoplasia: a spectrum of disease involving the left ventricular outflow tract, aortic valve, and aorta. J Am Coll Cardiol. 2012;59:S43–54. doi: 10.1016/j.jacc.2011.04.046. [DOI] [PubMed] [Google Scholar]
- 17.Yeo GW, et al. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. American journal of human genetics. 2014;95:360–370. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 20.Rauch A, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 21.van der Bom T, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu B, et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet. 2011;43:864–868. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldberg CS, et al. Factors associated with neurodevelopment for children with single ventricle lesions. J Pediatr. 2014;165:490–496.e8. doi: 10.1016/j.jpeds.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human nonsynonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ware JS, Samocha KE, Homsy J, Daly M. Interpreting de novo variation in human disease using denovolyzeR. Curr Protoc Hum Genet. 2015;87:1–15. doi: 10.1002/0471142905.hg0725s87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reimand J, Arak T, Vilo J. g:Profiler–a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–15. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.