

Abstract
α7 nicotinic acetylcholine receptor (α7nAChR, encoded by CHRNA7) is involved in dementia pathogenesis through cholinergic neurotransmission, neuroprotection and interactions with amyloid-β. Smoking promotes atherosclerosis and increases dementia risk, but nicotine exerts neuroprotective effect via α7nAChR in preclinical studies. No studies explored the gene-gene, gene-environment interactions between CHRNA7 polymorphism, apolipoprotein E (APOE) ε4 status and smoking on dementia risk. This case-control study recruited 254 late-onset Alzheimer’s disease (LOAD) and 115 vascular dementia (VaD) cases (age ≥65) from the neurology clinics of three teaching hospitals in Taiwan during 2007–2010. Controls (N = 435) were recruited from health checkup programs and volunteers during the same period. Nine CHRNA7 haplotype-tagging single nucleotide polymorphisms representative for Taiwanese were genotyped. Among APOE ε4 non-carriers, CHRNA7 rs7179008 variant carriers had significantly decreased LOAD risk after correction for multiple tests (GG + AG vs. AA: adjusted odds ratio = 0.29, 95% confidence interval = 0.13–0.64, P = 0.002). Similar findings were observed for carriers of GT haplotype in CHRNA7 block4. A significant interaction was found between rs7179008, GT haplotype in block4 and APOE ε4 on LOAD risk. rs7179008 variant also reduced the detrimental effect of smoking on LOAD risk. No significant association was found between CHRNA7 and VaD. These findings help to understand dementia pathogenesis.
The growing number of dementia patients has introduced a tremendous healthcare burden in the aging society. Alzheimer’s disease (AD) is the most common type of dementia, followed by vascular dementia (VaD). AD pathology is characterized by the selective loss of nicotinic acetylcholine receptors (nAChR)1 and elevated amyloid-β (Aβ) deposition in hippocampus and neocortex2. Reduced nAChR was also reported in the subcortical regions of VaD3.
α7nAChR is one of the most important nAChR subunits in the central nervous system and is often co-localized with Aβ deposition in the neuritic plaques of AD cortical neurons4. α7nAChR plays a pivotal role in dementia development through enhancing cholinergic neurotransmission5, inducing long-term potentiation6 and exerting neuroprotective effect7. However, as dementia progresses, elevated Aβ binds to α7nAChR with high affinity, inactivating α7nAChR and inhibiting its neuroprotective effect8. Another well-known risk factor of dementia is the cigarette smoking habit9, which increases dementia risk probably through accelerating atherosclerosis10. In contrast, the nicotine compound has a beneficial effect on cognition as it exerts neuroprotective action via α7nAChR in preclinical studies11. Taken together, complex interactions should exist among α7nAChR, Aβ, and cigarette smoking in the pathogenesis of dementia, which are currently unknown.
α7nAChR is encoded by CHRNA7 gene on chromosome 15q13–14, which is a region linked to several neuropsychiatric disorders, including bipolar affective disorder, schizophrenia, parkinsonism, several types of epilepsy, and autism12. CHRNA7 polymorphisms were also associated with decreased AD risk13 and slower progression from mild cognitive impairment to AD14, but other studies reported non-significant findings15,16,17,18,19,20. The inconsistence across studies may be attributable to differences in ethnicity and lack of information on gene–gene or gene–environment interactions. Apolipoprotein E (APOE) ε4, an essential genetic risk factor of late-onset AD (LOAD), is associated with increased Aβ deposition21. Despite prior in vitro evidence suggesting an important interaction between Aβ peptide and α7nAChR in the pathogenesis of dementia8, no studies explored the interaction between APOE ε4 and CHRNA7 polymorphisms on AD risk. Meanwhile, nicotine is an agonist of α7nAChR, but no studies examined whether the association of CHRNA7 polymorphisms with dementia varied depending on the smoking status.
This case-control study examined the association between CHRNA7 polymorphisms and dementia risk. Nine haplotype-tagging single nucleotide polymorphisms (htSNP) representative for Taiwanese were genotyped, capturing a majority of genetic information of CHRNA7. Interactions among CHRNA7 genotypes, APOE ε4, and dementia (LOAD, VaD) were explored. Stratified analyses were further performed by smoking history.
Materials and Methods
Study participants
Dementia patients were recruited from the neurology clinics of three teaching hospitals in northern Taiwan (National Taiwan University Hospital, En Chu Kong Hospital, and Cardinal Tien Hospital) from November 2007 to July 2010. Healthy controls were recruited from geriatric health checkup programs and from volunteers during the same period of time. All participants were Taiwanese (Han Chinese descents) who were 65 years and older. The exclusion criteria were participants with a history of depression, Parkinson’s disease, stroke, brain tumor, lack of blood sample, or poor DNA quality. After exclusion, a total of 254 LOAD cases, 115 small-vessel VaD cases, and 435 controls were included in the statistical analyses. All of the study protocols were approved by the Institutional Review Boards of National Taiwan University Hospital (200709031R, 200712102R), En Chu Kong Hospital (ECKIRB:98015), and Cardinal Tien Hospital (CTH-96-2-030). Informed consents were obtained from all subjects. Written consents were obtained from participants who were able to give consent by themselves, and from legal guardian/next of kin for those who couldn’t give consent themselves due to severe cognitive impairment. All of the experiments were carried out in accordance with the guidelines of the World Medical Association Declaration of Helsinki.
A detailed questionnaire was administered to all participants via a face-to-face interview with the assistance of informants. The collected information included data on demography, lifestyle, and comorbidity. Detailed smoking history (starting age of smoking habit, years of smoking, and years since quitting smoking) was obtained from the questionnaire. Ever-smokers were defined as those having smoked ≥100 cigarettes during their lifetime. Previous studies found good reliability between self-reported smoking status and elevated nicotine-related biomarkers in the body22. Blood samples were collected in EDTA tubes and genomic DNA was extracted from the buffy coat by using the QuickGene-Mini80 kit (Fujifilm, Tokyo, Japan) after centrifugation.
Dementia Evaluation
Probable LOAD was diagnosed by experienced neurologists as per the criteria defined by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association23. Brain images (computed tomography or magnetic resonance imaging) were performed to exclude organic brain lesions. VaD was diagnosed using the criteria of the National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences24. Because of different etiology between large- and small-vessel VaD, only patients with small-vessel VaD (e.g., lacunar infarction and leukoaraiosis) were included in this study in order to provide a more homogeneous outcome.
Mini-Mental State Examination (MMSE) was used to evaluate the cognitive performance of LOAD and VaD cases25. The controls were assessed by Short Portable Mental Status Questionnaire (SPMSQ) with the objective of excluding participants with possible cognitive impairment26. To further ensure that the controls were cognitively intact, only those without memory complaints and completely independent in performing activities of daily living and instrumental activities of daily living were included.
SNP Selection and Genotyping Assays
Common (frequency ≥5%) SNPs in CHRNA7 were selected from Han Chinese in Beijing (CHB) genotype data from the International HapMap Project (http://hapmap.ncbi.nlm.nih.gov/). Haplotype blocks were determined by the Haploview program (http://www.broadinstitute.org/haploview/haploview) using a modified Gabriel algorithm27,28. htSNPs were selected from each haplotype block using the tagSNP program with R2 > 0.7 in each haplotype block29. Genotypes of CHRNA7 SNPs were determined by TaqMan® Genomic Assays using the ABI 7900HT fast real-time PCR system (Applied Biosystems Inc., Foster City, CA, USA). APOE genotypes were determined by the assay developed by Chapman et al.30. The APOE diplotypes (ε2/ε2, ε2/ε3, ε3/ε3, ε2/ε4, ε3/ε4, and ε4/ε4) were determined by APOE112 (rs429358) and APOE158 (rs7412)31. APOE ε4 carriers were defined by participants carrying ε2/ε4, ε3/ε4, or ε4/ε4 diplotypes. Participants carrying other diplotypes (ε2/ε2, ε2/ε3, and ε3/ε3) were defined as APOE ε4 non-carriers. The genotyping call rate was greater than 95% for each SNP. The internal genotyping quality control obtained from 5% of samples in duplicates had a concordance rate of 100%.
Statistical Analyses
The Student’s t test (for normally-distributed continuous variables), Mann-Whitney U test (for non-normally distributed continuous variables), and χ2 test (for categorical variables) were used to compare the distribution of potential confounders by LOAD, VaD, and controls. The Hardy–Weinberg equilibrium (HWE) test in controls was performed for each SNP of CHRNA7 and APOE genes to examine possible genotyping errors or selection bias. The expectation-maximization algorithm was applied to estimate haplotype frequencies29. Participants were stratified by intervals of 5-years of age and cases were compared with controls within each age stratum in the multivariable analysis. Age (in years) was further adjusted in the multivariable analysis to control for residual confounding within each age stratum. Conditional logistic regression models were used to estimate the adjusted odds ratio (AOR) and 95% confidence interval (CI) for dementia (LOAD or VaD) in participants carrying 1 or 2 versus 0 copies of the minor allele of each SNP and each multilocus haplotype after adjustment for age, sex, APOE ε4, and education year.
Because APOE ε4 is an important risk factor for dementia and due to the in vitro evidence of interactions between Aβ and α7nAChR8, stratification analysis was performed by APOE ε4 status (carriers vs. non-carriers). The type I error resulting from multiple tests was controlled by false discovery rate (FDR)32.
To compare the joint effects of CHRNA7 polymorphisms and the smoking status on LOAD, four categories were created for each SNP and haplotype (non-variant carriers who ever smoked/never smoked and variant carriers who ever smoked/never smoked). Non-variant carriers who never smoked served as the reference group.
Because the numbers of homozygous variants were small and most of the included SNPs followed dominant mode of inheritance, all analyses were performed under dominant models. All statistical analyses were performed using SAS 9.4 (SAS Institute, Cary, NC, USA). A two-sided P < 0.05 was considered to be statistically significant.
Results
Population Characteristics
This study included 254 LOAD patients, 115 small-vessel VaD patients, and 435 healthy controls. Compared with controls separately, LOAD and VaD patients were significantly older (79.8 and 79.8 vs. 73.2 years-old, respectively), less educated (≥6 years of education: 49% and 40% vs. 88%, respectively), and more likely to be ever-smokers (24% and 28% vs. 17%, respectively). Compared with controls, LOAD patients showed a higher number of females (64% vs. 53%, respectively) and APOE ε4 carriers (39% vs. 15%, respectively), less hypertension rate (38% vs. 53%, respectively) and hypercholesterolemia (18% vs. 31%, respectively). VaD patients had more hypertension percentage (65% vs. 53%) and diabetes mellitus (35% vs. 14%) compared with the controls (Table 1).
Table 1. Characteristics of the study population.
| LOAD (N = 254) | VaD (N = 115 ) | Control (N = 435) | ||
|---|---|---|---|---|
| mean ± SD | ||||
| Age | 79.8 ± 6.3 | 79.8 ± 6.1 | 73.2 ± 5.8 | |
| MMSE score | 18.7 ± 5.1 | 15.2 ± 6.3 | NA | |
| SPMSQ (number of errors) | NA | NA | 0.1 ± 0.4 | |
| N (%) | ||||
| Female | 162 (64) | 66 (57) | 232 (53) | |
| Education | ≦6 years | 128 (51) | 69 (60) | 50 (12) |
| 6–12 years | 86 (34) | 34 (30) | 171 (39) | |
| >12 years | 37 (15) | 12 (10) | 212 (49) | |
| Ever smoking | 60 (24) | 32 (28) | 75 (17) | |
| Alcohol consumption | 28 (11) | 19 (17) | 48 (11) | |
| Hypertension | 96 (38) | 75 (65) | 232 (53) | |
| Diabetes mellitus | 48 (19) | 40 (35) | 62 (14) | |
| Hypercholesterolemia | 46 (18) | 25 (22) | 133 (31) | |
| APOE ε4 status | 99 (39) | 26 (23) | 65 (15) | |
Cases (LOAD or VaD) were compared to controls. Numbers in bold indicate significant findings (P < 0.05). Abbreviations: LOAD, late-onset Alzheimer’s disease; VaD, vascular dementia; SD, standard deviation; MMSE, Mini-Mental State Exmination; SPMSQ, Short Portable Mental Status Questionnaire; NA, not applicable; APOE, apolipoprotein E.
Haplotype-tagging SNPs in CHRNA7 gene
Nine common (frequency ≥5%) htSNPs, forming four haplotype blocks in the CHRNA7 gene, were selected and genotyped [Table 2, Fig. 1(a)]. Block1 contained two htSNPs (SNP1: rs885071, SNP2: rs8024987), block2 contained one htSNP (SNP3: rs4779565), block3 contained 4 htSNPs (SNP4: rs7402761, SNP5: rs904952, SNP6: rs4779978, SNP7: rs2651418), and block4 contained 2 htSNPs (SNP8: rs7179008, SNP9: rs2337980). The linkage disequilibrium (LD) structure is shown in Fig. 1(b). The minor allele frequencies (MAFs) for the nine htSNPs among controls ranged from 0.09 to 0.44 (Table 2), which were similar to those of the MAFs of Han Chinese from HapMap database (http://hapmap.ncbi.nlm.nih.gov/) and can reflect the genetic distribution among general Chinese population. None of the CHRNA7 SNPs were out of HWE after correction for multiple tests.
Table 2. Characteristics of CHRNA7 haplotype-tagging SNPs.
| Haplotype block | SNP name | rs no. | Nucleotide change | HapMap |
Control |
LOAD |
VaD |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CHB MAF | CEU MAF | MAF | HWE p | MAF | HWE p | MAF | HWE p | ||||
| 1 | SNP1 | rs885071 | T→G | 0.38 | 0.81 | 0.41 | 0.26 | 0.46 | 0.50 | 0.38 | 0.54 |
| 1 | SNP2 | rs8024987 | C→G | 0.11 | 0.24 | 0.11 | 0.05 | 0.15 | 0.14 | 0.11 | 0.79 |
| 2 | SNP3 | rs4779565 | G→T | 0.38 | 0.41 | 0.34 | 0.11 | 0.39 | 0.69 | 0.35 | 0.71 |
| 3 | SNP4 | rs7402761 | A→T | 0.45 | 0 | 0.44 | 0.37 | 0.44 | 0.79 | 0.44 | 0.32 |
| 3 | SNP5 | rs904952 | T→C | 0.29 | 0.53 | 0.27 | 0.16 | 0.34 | 0.16 | 0.33 | 0.13 |
| 3 | SNP6 | rs4779978 | C→T | 0.38 | 0.31 | 0.36 | 0.83 | 0.35 | 0.65 | 0.32 | 0.50 |
| 3 | SNP7 | rs2651418 | T→C | 0.42 | 0.53 | 0.39 | 0.62 | 0.42 | 0.41 | 0.44 | 0.89 |
| 4 | SNP8 | rs7179008 | A→G | 0.13 | 0.27 | 0.09 | 0.33 | 0.07 | 0.08 | 0.12 | 0.21 |
| 4 | SNP9 | rs2337980 | C→T | 0.26 | 0.50 | 0.24 | 0.03 | 0.25 | 0.69 | 0.23 | 0.96 |
All SNPs are intronic SNPs. Abbreviations: SNP, single nucleotide polymorphism; CHB, Han Chinese in Beijing, China;
CEU, Utah residents with ancestry from northern and western European; MAF, minor allele frequency; HWE, Hardy–Weinberg equilibrium test;
LOAD, late-onset Alzheimer’s disease; VaD, vascular dementia.
Figure 1. CHRNA7 genomic structure and linkage disequilibrium (LD) plot.
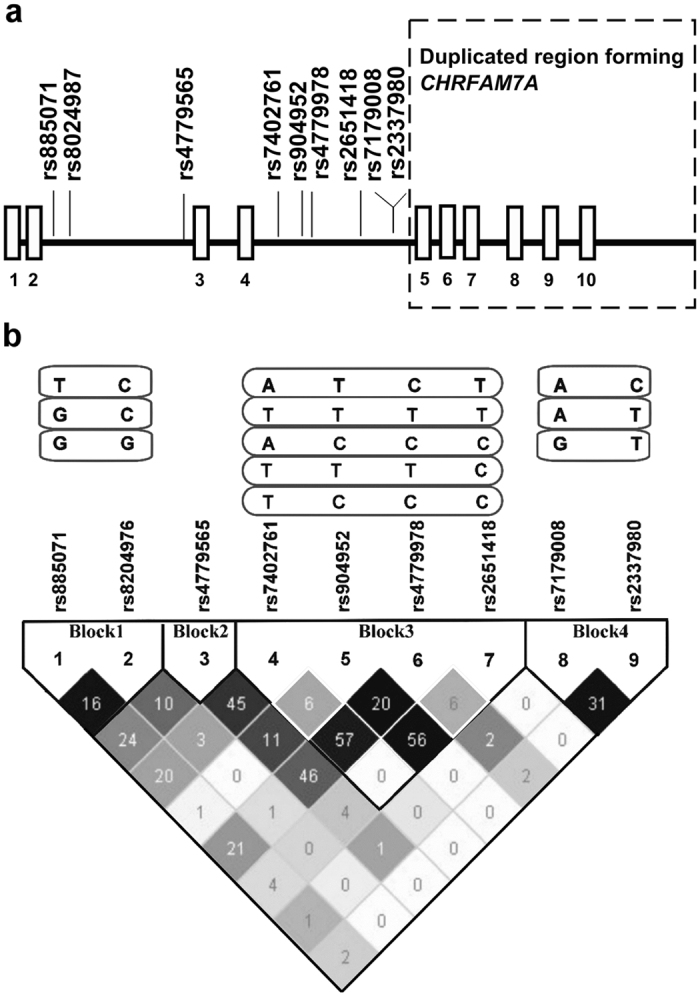
(a) shows the genomic structure of the CHRNA7 gene and the htSNPs in this study. The dashed rectangle indicates the region duplicated in the fusion gene, CHRFAM7A. Exons and introns are indicated by boxes and lines, respectively (not to scale). (b) shows the LD plot generated by Haploview program using genotype data from this study. Levels of pairwise D′, which indicate the degree of LD between two htSNPs, are shown in the LD structure in gray scale. Levels of pairwise r2, which indicate the degree of correlation between two SNPs, are shown as the number in each cell. Common (frequency ≥5%) haplotypes were identified in each haplotype block. A modified Gabriel et al. algorithm was used to define the haplotype block.
Association between CHRNA7 htSNPs or haplotypes and Dementia
The association between CHRNA7 polymorphisms and dementia was examined after adjustment for age, sex, APOE ε4 status, and education year. Three common (frequency ≥5% among controls) haplotypes were identified in CHRNA7 haplotype block1. Block2 included only one htSNP and was consequently excluded from the haplotype analysis. Five common haplotypes were identified in block3. Block4 included three common haplotypes. The effects of CHRNA7 SNPs and haplotypes in block4 on LOAD risk are shown in Table 3. Rs7179008 [htSNP in block4 (SNP8)] and GT haplotype in block4 [consisting of rs7179008 (SNP8) and rs2337980 (SNP9)] were associated with decreased LOAD risk (AOR = 0.50, 95%, CI = 0.28–0.92, P = 0.02; AOR = 0.49, 95% CI = 0.27–0.90, P = 0.02, Table 3). The effects of haplotypes in block1 and block3 on LOAD risk are shown in Supplementary Table S1. A protective effect against LOAD risk was also found among carriers of TC haplotype in block1 (AOR = 0.51, 95% CI = 0.30–0.86, Supplementary Table S1).
Table 3. Association between CHRNA7 SNPs and LOAD by APOE ε4 status.
| Haplotype block | SNP/Haplotype (frequency among controls) | 0 copies |
1 or 2 copies |
Pinteraction | |||
|---|---|---|---|---|---|---|---|
| Case/Control | AOR | Case/Control | AOR (95% CI) | ||||
| 1 | SNP1 | All | 76/145 | 1.00 | 178/290 | 1.48 (0.94–2.32) | 0.23 |
| APOE ε4 (−) | 44/121 | 1.00 | 110/247 | 1.25 (0.73–2.13) | |||
| APOE ε4 (+) | 31/24 | 1.00 | 68/41 | 2.10 (0.89–4.94) | |||
| 1 | SNP2 | All | 182/348 | 1.00 | 68/84 | 1.53 (0.93–2.52) | 0.69 |
| APOE ε4 (−) | 111/294 | 1.00 | 40/71 | 1.52 (0.84–2.75) | |||
| APOE ε4 (+) | 70/52 | 1.00 | 28/13 | 1.68 (0.63–4.42) | |||
| 2 | SNP3 | All | 95/179 | 1.00 | 157/255 | 1.16 (0.76–1.79) | 0.62 |
| APOE ε4 (−) | 59/155 | 1.00 | 94/212 | 1.26 (0.76–2.09) | |||
| APOE ε4 (+) | 35/23 | 1.00 | 63/42 | 0.84 (0.35–2.02) | |||
| 3 | SNP4 | All | 77/139 | 1.00 | 176/293 | 1.50 (0.73–1.82) | 0.59 |
| APOE ε4 (−) | 48/125 | 1.00 | 105/241 | 1.22 (0.72–2.09) | |||
| APOE ε4 (+) | 28/14 | 1.00 | 71/50 | 0.84 (0.32–2.21) | |||
| 3 | SNP5 | All | 115/225 | 1.00 | 134/205 | 1.34 (0.88–2.04) | 0.15 |
| APOE ε4 (−) | 64/189 | 1.00 | 51/35 | 1.59 (0.97–2.61) | |||
| APOE ε4 (+) | 87/175 | 1.00 | 46/29 | 0.79 (0.35–1.81) | |||
| 3 | SNP6 | All | 109/181 | 1.00 | 143/253 | 0.79 (0.51–1.21) | 0.82 |
| APOE ε4 (−) | 75/161 | 1.00 | 79/207 | 0.79 (0.48–1.29) | |||
| APOE ε4 (+) | 34/20 | 1.00 | 63/44 | 0.65 (0.26–1.62) | |||
| 3 | SNP7 | All | 88/160 | 1.00 | 165/274 | 1.10 (0.72–1.71) | 0.23 |
| APOE ε4 (−) | 48/135 | 1.00 | 105/232 | 1.30 (0.77–2.20) | |||
| APOE ε4 (+) | 40/24 | 1.00 | 59/41 | 0.73 (0.32–1.66) | |||
| 4 | SNP8 | All | 219/352 | 1.00 | 32/78 | 0.50 (0.28–0.92) | 0.03 |
| APOE ε4 (−) | 135/298 | 1.00 | 16/66 | 0.29(0.13–0.64)* | |||
| APOE ε4 (+) | 83/52 | 1.00 | 16/12 | 1.32 (0.45–3.87) | |||
| 4 | SNP9 | All | 143/260 | 1.00 | 110/174 | 1.07 (0.70–1.64) | 0.10 |
| APOE ε4 (−) | 93/221 | 1.00 | 60/146 | 0.87 (0.53–1.44) | |||
| APOE ε4 (+) | 50/39 | 1.00 | 49/26 | 1.79 (0.78–4.07) | |||
| 4 | Hap1: AC | All | 14/33 | 1.00 | 240/402 | 1.37 (0.59–3.23) | 0.61 |
| (76%) | APOE ε4 (−) | 7/27 | 1.00 | 147/341 | 1.57 (0.56–4.41) | ||
| APOE ε4 (+) | 7/6 | 1.00 | 92/59 | 0.99 (0.19–5.04) | |||
| 4 | Hap2: AT | All | 171/319 | 1.00 | 83/116 | 1.34 (0.84–2.12) | 0.66 |
| (15%) | APOE ε4 (−) | 109/271 | 1.00 | 45/97 | 1.28 (0.74–2.21) | ||
| APOE ε4 (+) | 62/47 | 1.00 | 37/18 | 1.52 (0.62–3.73) | |||
| 4 | Hap3: GT | All | 222/357 | 1.00 | 32/78 | 0.49 (0.27–0.90) | 0.03 |
| (9%) | APOE ε4 (−) | 138/302 | 1.00 | 16/66 | 0.28 (0.13–0.63)* | ||
| APOE ε4 (+) | 83/53 | 1.00 | 16/12 | 1.30 (0.44–3.83) | |||
All models were adjusted for age, sex, APOE ε4, and education year and conditional on 5-year age strata. Numbers in bold indicate significant findings (P < 0.05). *The association remained significant after correction for multiple tests by false discovery rate (FDR). The effects of CHRNA7 haplotypes in block1 and block3 on LOAD risk are shown in Supplementary Table S1 because the results were non-significant after correction for multiple tests. Block2 included only one htSNP and was excluded from the haplotype analysis. Abbreviations: AOR, adjusted odds ratio; CI, confidence interval; SNP, single nucleotide polymorphism; APOE, apolipoprotein E; Hap, haplotype.
The effects of CHRNA7 SNPs and haplotypes on VaD risk are shown in Table 4 and Supplementary Table S2 separately. Rs904952 (SNP5) was associated with increased VaD risk (AOR = 1.77, 95% CI = 1.01–3.09, P = 0.046, Table 4). TTTC haplotype in block3 was associated with decreased VaD risk (AOR = 0.33, 95% CI = 0.12–0.88, Supplementary Table S2). However, none of the above SNPs or haplotypes was significantly associated with LOAD or VaD after correction for multiple tests using FDR.
Table 4. Association between CHRNA7 SNPs and VaD by APOE ε4 status.
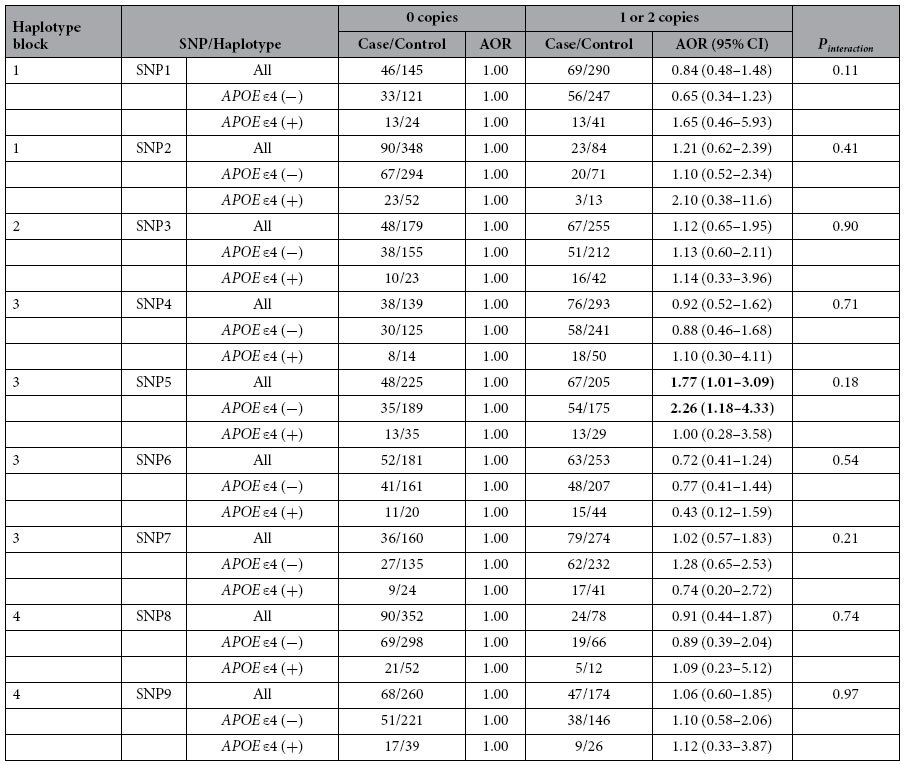
All models were adjusted for age, sex, APOE ε4, and education year and conditional on 5-year age strata. Numbers in bold indicate significant findings (P < 0.05). The results became non-significant after correction for multiple tests. Abbreviations: VaD, vascular dementia; AOR, adjusted odds ratio; CI, confidence interval; SNP, single nucleotide polymorphism; APOE, apolipoprotein E.
Modification Effect by APOE ε4 Status
A significant interaction was found between the variant rs7179008 (SNP8) and APOE ε4 on LOAD risk (Pinteraction = 0.03, Table 3). The effect of GT haplotype in block4 on LOAD risk was also significantly modified by APOE ε4 status (Pinteraction = 0.03, Table 3). Among APOE ε4 non-carriers, carriers of variant rs7179008 (AOR = 0.29, 95% CI = 0.13–0.64, P = 0.002 Table 3) and GT haplotype in block4 (AOR = 0.28, 95% CI = 0.13–0.63, P = 0.002, Table 3) demonstrated decreased LOAD risk compared with non-carriers. This decreased risk remained significant after correction for multiple tests using FDR based on 18 independent tests (9 SNPs by APOE ε4 status). No significant associations were observed between any of the CHRNA7 htSNPs, haplotypes, and LOAD among APOE ε4 carriers (Table 3).
The carriers of variant rs904952 (SNP5) were associated with increased VaD risk among APOE ε4 non-carriers (AOR = 2.26, 95% CI = 1.18–4.33, P = 0.01, Table 4)), but the result became non-significant after correction for multiple tests.
Modification Effect by Smoking
No significant interaction was observed between rs7179008 (SNP8) variant and smoking on LOAD risk (Pinteraction = 0.70). However, significant associations were found in a few subgroups. Ever-smokers had significantly increased LOAD risk after adjustment for age, sex, APOE ε4 status, and education year (AOR = 1.89, 95% CI = 1.01–3.52, P = 0.045). Among participants carrying rs7179008 wild-type, ever-smokers had increased LOAD risk compared with never-smokers (AOR = 2.00, 95% CI = 1.03–3.87, P = 0.008, Table 5). However, the detrimental effect of smoking was diminished if ever-smokers carried variant rs7179008 (AOR = 0.79, 95% CI = 0.21–2.96, P = 0.70, Table 5) compared with non-smokers with wild-type rs7179008.
Table 5. Polymorphisms of CHRNA7 rs7179008 and LOAD risk by smoking status.
| rs7179008 (SNP8) | Pinteraction | ||||
|---|---|---|---|---|---|
| 0 copies (AA) | 1 or 2 copies (AG+GG) | ||||
| Case/Control | AOR (95% CI) | Case/Control | AOR (95% CI) | ||
| Never-smokers | 164/291 | 1.00 | 27/63 | 0.54 (0.27–1.04) | 0.70 |
| Ever-smokers | 55/60 | 2.00 (1.03–3.87) | 5/15 | 0.79 (0.21–2.96) | |
All models were adjusted for age, sex, apolipoprotein E ε4, and education year and conditional on 5-year age strata. Numbers in bold indicate significant findings (P < 0.05). Abbreviations: AD, Alzheimer’s disease; SNP, single nucleotide polymorphism; AOR, adjusted odds ratio; CI, confidence interval.
Discussion
To the best of our knowledge, this is the first study exploring the gene–gene and gene–environment interactions of CHRNA7 polymorphisms on dementia risk. We found that the variants of CHRNA7 rs7179008 (SNP8) and GT haplotype in block4 significantly protect against LOAD among APOE ε4 non-carriers. The effects of rs7179008 and GT haplotype in block4 on LOAD risk were significantly modified by APOE ε4 status. The variant rs7179008 decreased the detrimental effect of smoking on LOAD risk. No significant associations were observed between CHRNA7 polymorphisms and VaD.
The analysis of CHRNA7 genome is complex due to its interaction with the highly polymorphic CHRFAM7A gene33. During evolution, exons 5–10 of CHRNA7 were duplicated and formed the fusion gene, CHRFAM7A33,34. The subunits encoded by CHRFAM7A lack part of the ligand binding site, and CHRFAM7A works as a dominant negative regulator on α7nAChR ion channel function35. Rs7179008 is located at intron 4 of CHRNA7 gene, which is a common breakpoint for gene rearrangement and may thus contribute to the polymorphisms of CHRFAM7A34,35. The variations in CHRFAM7A may in turn affect the expression of α7nAChR. Besides affecting α7nAChR expression by pre-mRNA alternative splicing36, it is also possible that this intronic SNP acts through affecting subsequent protein production36, or through LD with other functional genetic regions. GT haplotype in block4 consisted of rs7179008 and rs2337980, which explains the protective effect of this haplotype.
An Irish study previously found that TCC haplotype in CHRNA7 block1 was significantly associated with reduced AD risk13, which consisted of rs1514246, rs2337506, and rs8027814. Another European study found −86 C/T promoter polymorphism in CHRNA7 gene was associated with slower progression from mild cognitive impairment to AD14. The genome-wide association studies by Heinzen et al.16 and Swaminathan et al.15 found that the variants of CHRNA7 seem to contribute to AD risk and warrant further investigation. But the other candidate gene association studies19,20 and genome-wide association studies17,18 yielded non-significant results regarding the effect of CHRNA7 polymorphisms on dementia risk. These inconsistent findings may be related to different SNPs in Caucasians13,15,16,17,18,19 or lack of information on gene–gene and gene–environment interactions. Important genetic variants might be missed under gene-environment interactions, when the genetic association is opposite among different subgroups37. Because APOE ε4 and smoking are both well-established common risk factors for dementia and the abundant in vitro evidence suggesting their interactions with α7nAChR, our study takes the interactions into account. In addition, our study was adjusted for important risk factors for dementia (age, sex, APOE ε4, and education year), which was not considered in most of the previous studies13,16,19,20.
The following mechanisms may explain how CHRNA7 polymorphisms protect against LOAD risk through affecting the expression of α7nAChR. α7nAChR forms ligand-gated ion channels on neuron cell membranes, which are activated by the neurotransmitter acetylcholine or other agonists, e.g., nicotine5. When ligands bind to α7nAChR ion channel, the influx of sodium depolarizes cell membranes and increases cholinergic neurotransmission5. Presynaptic α7nAChR also modulates the release of other neurotransmitters38. Long-term potentiation is facilitated via α7nAChR6, which is important for memory consolidation. Furthermore, the stimulation of postsynaptic α7nAChR increases calcium influx and activates the intracellular signal transduction pathway, conferring neuroprotective effect by protecting neurons against Aβ toxicity7. In addition to disease prediction, our previous work has found that CHRNA7 polymorphisms may predict cognitive response to cholinesterase inhibitors and serve as a pharmacogenomic marker in LOAD treatment39.
The postulated mechanisms explaining the interactions among CHRNA7 polymorphisms, Aβ, and smoking on LOAD risk are shown in Fig. 2. In vitro evidence suggested an important interaction of Aβ and nAChR on AD pathogenesis8. As Aβ level increases pathologically with dementia progression, Aβ binds to α7nAChR with high affinity, which inactivates α7nAChR and decreases its neuroprotective effect8. Chen et al. also found Aβ impaired long term potentiation as a consequence of dysfunctional α7nAChR40. In addition, another in vitro study revealed that other APOE-derived peptides disrupt acetylcholine-mediated peak current response through the direct blockade of α7nAChR41. Compared with APOE ε4 non-carriers, APOE ε4 carriers showed increased Aβ deposits in brains21 and decreased nAChR binding sites42, which may further diminish the protection of α7nAChR. These facts corroborate our finding that the protective effect of the variant CHRNA7 rs7179008 was only observed among APOE ε4 non-carriers.
Figure 2. Postulated mechanism for the interaction between CHRNA7 polymorphisms, APOE ε4 and smoking.

CHRNA7 encodes α7nAChR and may affect the pathogenesis of LOAD through the following mechanisms: (1) modulation of neurotransmitter release in presynaptic neurons38, (2) memory enhancement via mediating cholinergic neurotransmission5 and long-term potentiation6, and (3) neuroprotection via α7nAChR7. In APOE ε4 carriers, high levels of Aβ bind to α7nAChR, inactivating the receptor and decreasing its neuroprotective effect8. The nicotine in cigarette smoking is an agonist to α7nAChR which potentiates the neuroprotective effect of the receptor in preclinical studies11. Upregulation of α7nAChR was reported among smokers44. Abbreviations: nAChR, nicotinic acetylcholine receptor; APOE, apolipoprotein E; Aβ, amyloid β.
The joint effect of smoking and CHRNA7 polymorphisms on LOAD risk had not been explored previously. We found that the detrimental effect of smoking was attenuated among carriers of variant CHRNA7 rs7179008. Accumulating evidence based on large prospective cohort studies revealed that smoking increases AD risk9, which was consistent with our findings. The increased risk may result from the pro-atherogenic effect of smoking that contributes to dementia progression10. α4β2nAChR, another common nAChR subunit in the central nervous system, binds to nicotine with high affinity. Many studies have demonstrated increased numbers of high-affinity nAChRs in the brains of smokers43,44. α7nAChR binds to nicotine with lower affinity. Despite preclinical studies found that nicotine enhances α7nAChR-mediated neuroprotection45, studies were inconsistent regarding whether the expression of α7nAChR is increased in the brains of smokers46. In a post-mortem brain biopsy, Mousavi et al. found a significantly increased α7nAChR protein levels in the temporal cortex of smokers compared with non-smokers44. Thus, nicotine might offset the harm of toxic compounds in cigarette smoke by increasing the protective effect of α7nAChR among CHRNA7 variant carriers. Future studies are required to determine the level of nicotine that stimulates α7nAChR, the duration of persisted receptor upregulation after smoking cessation, and the possible modifying effect by disease status and genetic variation.
However, we found that CHRNA7 polymorphisms were not associated with VaD risk, which was not previously reported as far as we know. Few studies examined the change of nAChRs during VaD pathogenesis. One study reported decreased α4β2nAChR expression in subcortical region of VaD patients3, but another study did not find the association47. Genetic susceptibility to VaD has been much less understood compared with AD. One possible cause is that different types of ischemic stroke and VaD (e.g., small-vessel occlusion and large artery atherosclerosis) are related to different genetic factors48,49. Thus, genetic analyses by VaD subtypes provide clearer explanation to different etiologies50. VaD subtypes were often neglected in previous genetic association studies. Therefore, in this study, we included only small-vessel VaD cases to minimize the heterogeneity.
This study presents a few strengths. To the best of our knowledge, for the first time, we explored the joint effects of CHRNA7 polymorphisms, APOE ε4 status, and smoking on LOAD and VaD risk. Exploring the gene–gene and gene–environment interactions may help us further understand the pathogenesis of dementia. CHRNA7 polymorphisms may be important genetic risk factors for APOE ε4 non-carriers. Second, in this study, we used a systematic approach to select htSNPs representative of Taiwanese, which captured abundant genetic information of CHRNA7 gene (R2 > 0.7) and were different from SNPs selected for Caucasians. Finally, this study was adjusted for many important confounders, which makes our findings less biased.
This work also demonstrated some limitations. It included only 115 small-vessel VaD cases and may be underpowered to detect the genetic effect on VaD. Similarly, the joint effect of CHRNA7 polymorphisms and smoking on LOAD risk needs to be interpreted with caution and should be regarded as exploratory and hypotheses generating, due to a relatively small number of smokers.
In summary, the variants of CHRNA7 rs7179008 and GT haplotype in block4 were associated with reduced LOAD risk among APOE ε4 non-carriers. The association between CHRNA7 polymorphisms and LOAD was substantially modified by APOE ε4 and smoking status. Future large studies are warranted to confirm our findings.
Additional Information
How to cite this article: Weng, P.-H. et al. CHRNA7 Polymorphisms and Dementia Risk: Interactions with Apolipoprotein ε4 and Cigarette Smoking. Sci. Rep. 6, 27231; doi: 10.1038/srep27231 (2016).
Supplementary Material
Acknowledgments
Funding for the study was provided by National Science Council grants 96-2314-B-002-197 and 97-2314-B-002-168-MY3.
Footnotes
Author Contributions P.-H.W. and Y.-C.C. wrote the manuscript and analyzed the data. J.-H.C., T.-F.C., Y.S. and P.K.Y. collected the data. L.-L.W. and Y.-M.C. contributed reagents and materials.
References
- London E. D., Ball M. J. & Waller S. B. Nicotinic binding sites in cerebral cortex and hippocampus in Alzheimer’s dementia. Neurochem. Res. 14, 745–750 (1989). [DOI] [PubMed] [Google Scholar]
- Selkoe D. J., Bell D. S., Podlisny M. B., Price D. L. & Cork L. C. Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer’s disease. Science. 235, 873–877 (1987). [DOI] [PubMed] [Google Scholar]
- Colloby S. J. et al. Alterations in nicotinic alpha4beta2 receptor binding in vascular dementia using (1)(2)(3)I-5IA-85380 SPECT: comparison with regional cerebral blood flow. Neurobiol. Aging. 32, 293–301 (2011). [DOI] [PubMed] [Google Scholar]
- Wang H. Y. et al. Beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 275, 5626–5632 (2000). [DOI] [PubMed] [Google Scholar]
- Frazier C. J., Buhler A. V., Weiner J. L. & Dunwiddie T. V. Synaptic potentials mediated via alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J. Neurosci. 18, 8228–8235 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter B. E., de Fiebre C. M., Papke R. L., Kem W. R. & Meyer E. M. A novel nicotinic agonist facilitates induction of long-term potentiation in the rat hippocampus. Neurosci. Lett. 168, 130–134 (1994). [DOI] [PubMed] [Google Scholar]
- Kihara T. et al. Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J. Biol. Chem. 276, 13541–13546 (2001). [DOI] [PubMed] [Google Scholar]
- Dineley K. T. Beta-amyloid peptide–nicotinic acetylcholine receptor interaction: the two faces of health and disease. Front Biosci. 12, 5030–5038 (2007). [DOI] [PubMed] [Google Scholar]
- Cataldo J. K., Prochaska J. J. & Glantz S. A. Cigarette smoking is a risk factor for Alzheimer’s Disease: an analysis controlling for tobacco industry affiliation. J. Alzheimers Dis. 19, 465–480 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman A. et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet. 349, 151–154 (1997). [DOI] [PubMed] [Google Scholar]
- Akaike A., Takada-Takatori Y., Kume T. & Izumi Y. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J. Mol. Neurosci. 40, 211–216 (2010). [DOI] [PubMed] [Google Scholar]
- Gotti C., Zoli M. & Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol. Sci. 27, 482–491 (2006). [DOI] [PubMed] [Google Scholar]
- Carson R. et al. Alpha7 nicotinic acetylcholine receptor gene and reduced risk of Alzheimer’s disease. J. Med. Genet. 45, 244–248 (2008). [DOI] [PubMed] [Google Scholar]
- Barabash A. et al. APOE, ACT and CHRNA7 genes in the conversion from amnestic mild cognitive impairment to Alzheimer’s disease. Neurobiol. Aging. 30, 1254–1264 (2009). [DOI] [PubMed] [Google Scholar]
- Swaminathan S. et al. Genomic Copy Number Analysis in Alzheimer’s Disease and Mild Cognitive Impairment: An ADNI Study. Int J Alzheimers Dis. 2011, 729478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen E. L. et al. Genome-wide scan of copy number variation in late-onset Alzheimer’s disease. J. Alzheimers Dis. 19, 69–77 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 65, 45–53 (2008). [DOI] [PubMed] [Google Scholar]
- Reiman E. M. et al. GAB2 alleles modify Alzheimer’s risk in APOE epsilon4 carriers. Neuron. 54, 713–720 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook L. J. et al. Candidate gene association studies of genes involved in neuronal cholinergic transmission in Alzheimer’s disease suggests choline acetyltransferase as a candidate deserving further study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 132B, 5–8 (2005). [DOI] [PubMed] [Google Scholar]
- Kawamata J. & Shimohama S. Association of novel and established polymorphisms in neuronal nicotinic acetylcholine receptors with sporadic Alzheimer’s disease. J. Alzheimers Dis. 4, 71–76 (2002). [DOI] [PubMed] [Google Scholar]
- Schmechel D. E. et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 90, 9649–9653 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager D. S. & Krosnick J. A. The validity of self-reported nicotine product use in the 2001–2008 National Health and Nutrition Examination Survey. Medical care. 48, 1128–1132 (2010). [DOI] [PubMed] [Google Scholar]
- McKhann G. et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 34, 939–944 (1984). [DOI] [PubMed] [Google Scholar]
- Roman G. C. et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 43, 250–260 (1993). [DOI] [PubMed] [Google Scholar]
- Folstein M. F., Folstein S. E. & McHugh P. R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 12, 189–198 (1975). [DOI] [PubMed] [Google Scholar]
- Pfeiffer E. A short portable mental status questionnaire for the assessment of organic brain deficit in elderly patients. J Am Geriatr Soc. 23, 433–441 (1975). [DOI] [PubMed] [Google Scholar]
- Chen Y. C. et al. Sequence variants of Toll-like receptor 4 and susceptibility to prostate cancer. Cancer Res. 65, 11771–11778 (2005). [DOI] [PubMed] [Google Scholar]
- Gabriel S. B. et al. The structure of haplotype blocks in the human genome. Science. 296, 2225–2229 (2002). [DOI] [PubMed] [Google Scholar]
- Stram D. O. et al. Modeling and E-M estimation of haplotype-specific relative risks from genotype data for a case-control study of unrelated individuals. Hum Hered 55, 179–190 (2003). [DOI] [PubMed] [Google Scholar]
- Chapman J., Estupinan J., Asherov A. & Goldfarb L. G. A simple and efficient method for apolipoprotein E genotype determination. Neurology. 46, 1484–1485 (1996). [DOI] [PubMed] [Google Scholar]
- Ghebranious N., Ivacic L., Mallum J. & Dokken C. Detection of ApoE E2, E3 and E4 alleles using MALDI-TOF mass spectrometry and the homogeneous mass-extend technology. Nucleic Acids Res. 33, e149 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. & Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 57, 289–300 (1995). [Google Scholar]
- Gillentine M. A. & Schaaf C. P. The human clinical phenotypes of altered CHRNA7 copy number. Biochem Pharmacol. 97, 352–362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J. et al. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic 40 acetylcholine receptor gene (CHRNA7). Genomics. 52, 173–185 (1998). [DOI] [PubMed] [Google Scholar]
- Szafranski P. et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum. Mutat. 31, 840–850 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash Y. et al. Deciphering the splicing code. Nature. 465, 53–59 (2010). [DOI] [PubMed] [Google Scholar]
- Murcray C. E., Lewinger J. P. & Gauderman W. J. Gene-environment interaction in genome-wide association studies. Am J Epidemiol. 169, 219–226 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnacott S., Barik J., Dickinson J. & Jones I. W. Nicotinic receptors modulate transmitter cross talk in the CNS: nicotinic modulation of transmitters. J. Mol. Neurosci. 30, 137–140 (2006). [DOI] [PubMed] [Google Scholar]
- Weng P. H. et al. CHRNA7 polymorphisms and response to cholinesterase inhibitors in Alzheimer’s disease. PLoS One. 8, e84059 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L., Yamada, K., Nabeshima, T. & Sokabe, M. alpha7 Nicotinic acetylcholine receptor as a target to rescue deficit in hippocampal LTP induction in beta-amyloid infused rats. Neuropharmacology 50, 254–268(2006). [DOI] [PubMed] [Google Scholar]
- Gay E. A., Klein R. C. & Yakel J. L. Apolipoprotein E-derived peptides block alpha7 neuronal nicotinic acetylcholine receptors expressed in xenopus oocytes. J. Pharmacol. Exp. Ther. 316, 835–842 (2006). [DOI] [PubMed] [Google Scholar]
- Poirier J. et al. Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc. Natl. Acad. Sci. USA 92, 12260–12264 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhin A. G. et al. Greater nicotinic acetylcholine receptor density in smokers than in nonsmokers: a PET study with 2-18F-FA-85380. bre 49, 1628–1635 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi M. et al. Protein and mRNA levels of nicotinic receptors in brain of tobacco using controls and patients with Alzheimer’s disease. Neuroscience. 122, 515–520 (2003). [DOI] [PubMed] [Google Scholar]
- Inestrosa N. C. et al. Nicotine prevents synaptic impairment induced by amyloid-beta oligomers through alpha7-nicotinic acetylcholine receptor activation. Neuromolecular Med. 15, 549–569 (2013). [DOI] [PubMed] [Google Scholar]
- Breese C. R. et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 23, 351–364 (2000). [DOI] [PubMed] [Google Scholar]
- Pimlott S. L. et al. Nicotinic acetylcholine receptor distribution in Alzheimer’s disease, dementia with Lewy bodies, Parkinson’s disease, and vascular dementia: in vitro binding study using 5-[(125)i]-a-85380. Neuropsychopharmacology. 29, 108–116 (2004). [DOI] [PubMed] [Google Scholar]
- Kim Y. & Lee C. The gene encoding transforming growth factor beta 1 confers risk of ischemic stroke and vascular dementia. Stroke. 37, 2843–2845 (2006). [DOI] [PubMed] [Google Scholar]
- Lee C. & Kong M. An interactive association of common sequence variants in the neuropeptide Y gene with susceptibility to ischemic stroke. Stroke. 38, 2663–2669 (2007). [DOI] [PubMed] [Google Scholar]
- Kim Y., Kong M., An J., Ryu J. & Lee C. Genetic dissection of susceptibility to vascular dementia. Psychiatr. Genet. 21, 69–76 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.