

Abstract
Hypertension is one of the leading causes of health morbidity and mortality which are linked to many life threatening diseases such as stroke heart problems and renal dysfunction.
The integrity of renal microcirculation is crucial to maintaining the clearance and the excretory function in the normotensive and hypertensive conditions. Furthermore, any alteration in the renal function is involved in the pathophysiology of hypertension.
The aim of this review is to provide a brief discussion of some factors that regulate renal haemodynamics in spontaneously hypertensive rats, an animal model of hypertension, and how these factors are linked to the disease.
Keywords: Renal circulation, Spontaneously hypertensive rats, Reactive oxygen species, Nitric oxide
1. Introduction
Hypertension is a state of chronically elevated blood pressure (BP) that leads to functional and structural changes of the blood vessels. It is a very common disorder in man, affecting about 25% of the general population (Schoen and Cotran, 1994). Hypertension is associated with an increased incidence of coronary heart disease and cerebrovascular incidents. The disease is currently believed to be multifactorial and there is some evidence of familial inheritance which increases susceptibility to the condition, however, it is also recognised that lifestyle and environment are important factors in the development of the disease. Ninety percent of hypertensive patients suffer from essential hypertension in which there is no known underlying cause (Ballinger et al., 2007). In the remaining 10% of patients, hypertension is secondary to renal disease or less frequent adrenal disorders (Schoen and Cotran, 1994). Table 1 shows the common types of hypertension with a brief description for each type.
Table 1.
Common types of hypertension.
| Type of hypertension | Description | Blood pressure range |
|---|---|---|
| Essential hypertension (major type) | Chronic elevation in blood pressure with no underlying disease | Both systolic and diastolic blood pressure are elevated more than 140/90 mmHg |
| Secondary hypertension (second common type) | Chronic elevation in blood pressure due to underlying pathology (mostly due to renal problems) | Both systolic and diastolic blood pressure are elevated more than 140/90 mmHg |
| Malignant hypertension | When blood pressure is severely elevated and causes an organ damage | Both systolic and diastolic blood pressure are elevated but the diagnosis made mainly when the diastolic blood pressure is higher than 130 mmHg |
| Isolated systolic hypertension | Common in elderly due to the loss of elasticity of major blood arteries | The systolic blood pressure is higher than 140 mmHg while the diastolic blood pressure is close to the normal range |
| Resistant hypertension | When more than three different antihypertensive agents are prescribed and blood pressure is still elevated | Both systolic and diastolic blood pressure are elevated more than 140/90 mmHg |
The kidney plays an important role in the control of BP by way of the rennin–angiotensin system, sodium homoeostasis and production of vasodepressor substances such as prostaglandins. Abnormalities in any of these systems can lead to BP changes and they are all thought to play a role in essential hypertension (Schoen and Cotran, 1994). Hypertension is often accompanied by further complications such as impaired renal function and structural changes to the vasculature (Soergel and Schaefer, 2002). Whether renal dysfunction is as a result of continued hypertension or the primary cause of the hypertensive state is still under discussion. There is a view that the changes in kidney function are a result of structural and functional alterations caused by exposure of the kidney to increased perfusion pressure (Cowley et al., 1995). It has been shown that increased basal BP can exacerbate existing structural changes in the kidney of the hypertensive patient resulting in secondary hypertension (Rettig et al., 1990). Hypertension can develop as a result of changes within the brain or endocrine organs or alteration in the neural control of salt and water excretion, which then increase BP thereby affecting the kidney (Denton et al., 2006).
In patients with essential hypertension endothelium-dependent relaxation is diminished (Linder et al., 1990, Panza et al., 1990), and impaired endothelial vasodilation can be improved by the antioxidant vitamin C, as shown by forearm blood flow measurements. This effect can be reversed by the nitric oxide synthase (NOS) inhibitor (Taddei et al., 1998).
In this review we considered the experimental animal as a model of hypertension when both systolic and diastolic blood pressure are elevated above the level of 140/90 mmHg. Furthermore, the animal model of hypertension that we explored is considered as a model of essential hypertension.
2. Renal microvasculature
The kidney is divided into two major regions, the cortex and the medulla and can be grossly divided into four zones: the cortex, the outer stripe of the outer medulla, the inner stripe of the outer medulla, and the inner medulla (Mattson, 2003, Navar et al., 2008). The perfusion of these different regions is highly heterogeneous. Total tissue blood flow averages 700 ml min−1/100 g−1 of tissue in the renal cortex, is 300 ml/min−1/100 g−1 near the junction of the cortex and the outer medulla, decreases to 200 ml min−1/100 g−1 in the inner stripe of the outer medulla, and ranges from 50 to 100 ml/min−1/100 g−1 in the inner medulla (Navar et al., 2008, Pallone et al., 2012).
An adequate blood supply is crucial for the production of an ultrafiltrate from plasma and the ability to modify the filtrate through reabsorption and secretion into the tubule enables the kidneys to effectively carry out these functions. The kidneys receive approximately 25% of total cardiac output however the distribution is not uniform within the kidney. The renal cortex receives approximately 90% of the total renal blood flow (RBF) and plays a major role in ultrafiltration within the kidney. A dense peritubular capillary plexus arising from efferent arterioles within the cortex surrounds the proximal and distal convoluted tubules to facilitate the reabsorption of the glomerular filtrate (Navar et al., 2008, Pallone et al., 2012). The remaining 10% of the RBF perfuses the medulla through vessels arising from the postglomerular vasculature (efferent arterioles) of the inner cortical or juxtamedullary nephrons (Fig.1).
Figure 1.
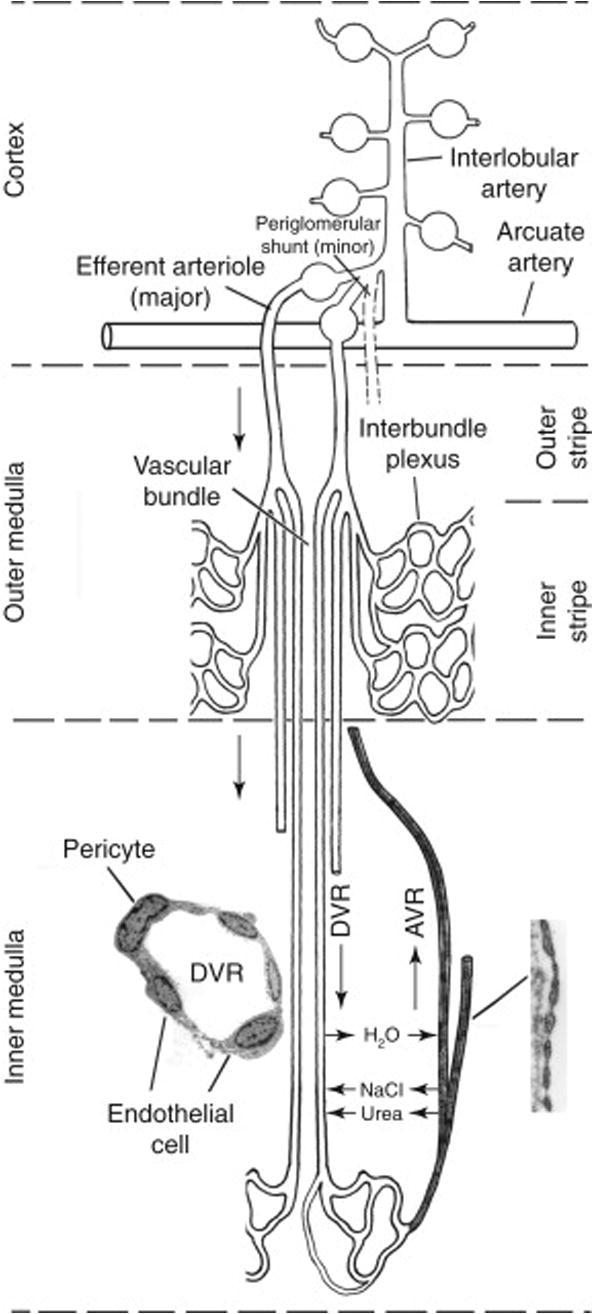
Medullary microcirculation. Juxtamedullary glomeruli originate at an acute angle from the interlobular artery. Most blood flow into the medulla arises from juxtamedullary efferent arterioles. At the outer stripe of the outer medulla, juxtamedullary efferent arterioles progress into descending vasa recta [DVR] and form vascular bundles at the inner stripe of the outer medulla. DVR around the exterior of the vascular bundles perfuse the interbundle capillary plexus that supplies nephrons. DVR in the centre of the bundles continue across the inner–outer medullary junction to perfuse the inner medulla. The vascular bundles disappear in the inner medulla with the vasa recta becoming dispersed between thin loops of Henle, collecting ducts, and ascending vasa recta [AVR]. AVR ascend to the cortex through outer medullary vascular bundles. DVR have a continuous endothelium (left inset) and are surrounded by contractile pericytes. AVR are highly fenestrated vessels (right inset). Taken from Navar et al. (2008), 550–683. Fig. 13.11, p. 567.
The efferent arterioles of the juxtamedullary nephrons enter the outer stripe of the outer medulla and divide into a series of vascular loops called vasa recta. The vasa recta descend into the inner stripe of the outer medulla and form vascular bundles. The descending vasa recta (DVR) in the centre of the bundles continue into the inner medulla whereas the DVR on the outer margins of the bundles give rise to a capillary plexus between the vascular bundles in the outer medulla. The DVR found in either the outer or inner medulla divide and eventually merge to form ascending vasa recta (AVR) that carry metabolic substances that enter the medulla in the DVR blood back to the cortex. To avoid the possibility of medullary hypoxia resulting from this process the kidney has adapted to exert a subtle control over regional perfusion of the outer and inner medulla (Mattson, 2003, Navar et al., 2008, Johns and Ahmeda, 2014).
3. Renal medullary blood flow
Although the kidney strives to maintain its perfusion within tight boundaries, considerable blood flow fluctuations do occur under normal conditions. However should these fluctuations be prolonged, target organ damage can occur (Persson, 2002). The renal medulla is renowned for its low rate of blood perfusion (Cowley, 1997) and a minimal reserve of oxygen (less than 40%) (Heyman et al., 1997), therefore the maintenance of adequate perfusion of this zone is critically important for its survival, for the functional integrity of the kidney and for the appropriate renal regulation of body fluid balance and arterial pressure (Badzynska et al., 2003). The regulation of blood flow in the renal medulla has been a subject of interest for many years. Previous reports by Persson (2002) indicate that the renal medulla has control over its own blood flow. It has also been suggested that the renal medulla has an impaired autoregulatory capacity, or may even exhibit a complete passive blood flow response to renal perfusion pressure changes however more recent findings have challenged this hypothesis (Persson, 2002). The vascular architecture of the kidney appears to be arranged in a way that protects the renal medulla from ischaemic insults (Evans et al., 2004). The details of this process are far from clear and much experimental evidence suggests there may be a complex interaction between paracrine agents and autocoids in modulating vasomotor tone at various locations along the micro-vascular circuit (Pallone et al., 2012).
Given the importance of medullary perfusion in influencing water and salt balance, it was necessary to determine which location(s) along the micro-vascular circuit provides control of medullary blood perfusion (MBP) (Pallone et al., 2012). Pallone (1994) first demonstrated that when DVR are isolated from outer medullary vascular bundles in the rat kidney they are capable of constricting at various foci after exposure to contractile agonists such as Angiotensin II. Based on anatomic considerations it is probable that DVR are an important site of regulation. It has since been reported that this regulation is mediated by contraction of the vasa recta pericytes (Pallone, 1994, Park et al., 1997, Pallone et al., 2012). The vasa recta capillaries therefore appear to be capable of exerting regional control of blood perfusion to the renal medulla. Similar studies using vascular casting methodology suggest that juxtamedullary glomerular arterioles are not the chief regulators of MBP, which is consistent with the idea that outer medullary descending vasa recta play a key role in MBP control (Evans et al., 2004).
4. Kidney and blood pressure control
Guyton et al. (1972) reported that raised arterial pressure was accompanied by increased urine flow and sodium excretion by way of the pressure-diuresis mechanism (Guyton et al., 1972). By increasing urine flow the effective circulating volume is reduced and thereby normalises arterial BP. This process enables the kidneys to control long-term arterial pressure by monitoring blood volume. In genetic rat models of hypertension and using micropuncture techniques, Arendshorst and Beierwaltes (1979) reported that higher BP than in the normotensive model were necessary to excrete a given amount of sodium (Arendshorst and Beierwaltes, 1979). Therefore, hypertension can occur as a result of a malfunction of the pressure-natriuresis system in the kidney, for example impaired excretory ability of the kidney. This hypothesis is supported by the fact that with anti-hypertensive treatment, for example angiotensin converting enzyme (ACE) blockers, there is an alteration in the pressure-natriuresis balance and the return sodium and water excretion back to control levels (Cowley et al., 1995).
The renal medulla specifically plays a crucial role in the long-term control of arterial BP and water and salt homoeostasis. Cowley et al. (1995) reported that the pressure-natriuresis mechanism observed in the rat kidney was related to renal blood flow and BP in the vasa recta vessels of the inner medulla. The medullary blood perfusion (MBP) and its pressure natriuresis response is regulated by angiotensin II, prostaglandins and nitric oxide (Zou et al., 1998, Liu, 2009), amongst other paracrine and humoral mediators, which are all known to play important roles in the control of renal function. Factors which are known to reduce blood flow in the renal medulla such as sympathetic nerve activity, angiotensin II, Cyclooxygenase (COX) and nitric oxide synthase (NOS) inhibition, are associated with increasing BP. Conversely factors related to increased medullary flow such as prostaglandins and angiotensin converting enzyme (ACE) inhibitors are linked to reductions in BP. This supports the argument that it is the renal medulla, specifically, which is important in the regulation of BP and is therefore a vital region involved the pathogenesis of hypertension (Cowley et al., 1995).
5. Animal models of hypertension
The kidney plays a crucial long-term role in the pathophysiology of hypertension by way of structural changes and the increased retention of fluid and sodium. Animal models of hypertension have been used to show the central importance of the kidney in its pathogenesis. There are some important similarities between animal models of hypertension and the human condition. For example, there is a genetic component of the disease that is believed to be polygenic (Ganten, 1987). There are a number of animal models of hypertension that have been used in order to further our understanding of the condition. Tigerstedt & Bergman in 1898 developed the concept of a relationship between kidney and BP when they injected renin, extracted from kidneys, into the systemic circulation and observed a sustained increase in BP (Tigerstedt and Bergman, 1898). Another animal model of hypertension is the rat renovascular model, otherwise known as the Goldblatt one-kidney one-clip model (Goldblatt et al., 1934), this model is characterised by a unilateral nephrectomy and an arterial clip placed on the renal artery of the remaining kidney. Levy et al. (2001) reported a mean arterial pressure (MAP) of 174 ± 11 mmHg 4 weeks after arterial clipping in the one-kidney one-clip model (Levy et al., 2001). A similar one-kidney one-clip model has been described by Wiesel et al. in 1997, in which MAP was reported to be 20 mmHg higher than controls 4 weeks after surgery (Wiesel et al., 1997). Another popular animal model of hypertension is the deoxycorticosterone acetate (DOCA)-salt hypertensive rat. This model is reported to have increased Reactive Oxygen Species (ROS) production and reduced angiotensin II activity (Beswick et al., 2001).
Spontaneously hypertensive rats (SHR), a genetic hypertensive rat model, develop consistently higher BP compared with other strains, have increased activity of fluid retention mechanisms, sodium reabsorption and increased vascular resistance (Grisk et al., 2002). It is possible to identify structural changes in the genetically hypertensive rat kidney even before hypertension is apparent. The genetic model, SHR, is probably the most comparable with the human form of the condition. The preglomerular arterioles have a reduced lumen diameter and increased maximal resistance by 14–36% compared to Wistar rats (Skov et al., 1992).
Since the development of the first animal models of hypertension, major advances have been made in the development of genetically hypertensive rats (Ganten, 1987). Okamoto et al. (1974) produced a colony of rats by selectively inbreeding Wistar rats with higher than normal BP and produced the SHR colony (Okamoto et al., 1974). One hundred percent of SHR develop hypertension and a MAP of 184 ± 17 at the age of 10 weeks. The SHR develop complications associated with hypertension similar to those occurring in human hypertension, such as cerebral and myocardial lesions. Hypertension in the SHR model develops as a result of increased peripheral resistance, first produced by neurogenic factors and later structural vascular changes associated with increased vascular protein synthesis (Okamoto et al., 1974). Enhanced vasoconstrictor prostaglandin production in the SHR was reported by Armstrong et al. in 1976 (Armstrong et al., 1976), where they found elevated levels of Prostaglandin F2alpha (PGF2α). A sub-strain of SHR has been developed, the stroke-prone SHR (SHRSP) that is characterised by severe hypertension, MAP above 230 mmHg, and 80% incidence of stroke by 9–13 months of age (Okamoto et al., 1974). In a previous review about the animal models of hypertension, it was suggested that the results are greatly influenced by the duration of exposure to the causal factors, difference in animal species, gender and the age of animals (Dornas and Silva, 2011).
6. Stroke-prone SHR (SHRSP)
The SHRSP characteristically displays a more severe hypertension than the SHR. The SHRSP also demonstrates increase vascular reactivity to vasoconstrictor substances such as angiotensin II and noradrenaline, in comparison to normotensive Wistar rats (Berecek et al., 1980). The SHRSP have a higher renovascular resistance than the Wistar, which is apparent from an early age but becomes more pronounced after prolonged hypertension. An increased salt diet causes accelerated development of hypertension in both the SHR and SHRSP, however BP measurements have shown that SHRSP are more salt-sensitive than SHR and that may cause greater renal damage in SHRSP (Griffin et al., 2001). Nagaoka et al. (1981) reported that the SHRSP had autoregulatory control of renal blood flow (RBF) and glomerular filtration rate (GFR), which was operative over a higher BP range than that in Wistar rats (Nagaoka et al., 1981). When perfusion pressure was reduced so was the sodium and water excretion of the SHRSP, therefore it appears that SHRSP requires higher than normal BP in order to efficiently excrete electrolytes and water. As well as the increased severity of hypertension in the SHRSP in comparison to the SHR, Churchill et al. (2002) showed that the SHRSP displayed other differences, namely increased intrinsic susceptibility to renal damage (Churchill et al., 2002). They proposed that there were genetic differences between SHR and SHRSP that accounted for the increased incidence of hypertensive lesions and malignant nephrosclerosis after only 4–6 weeks on a high-salt diet, the SHR showed little tubular damage under similar conditions. The salt-induced kidney damage was thought to be not only linked to the increase in BP but also due to the vasculotoxic effects of salt (Griffin et al., 2001). In summary, the SHRSP have a higher BP level, increased tendency for renal damage and nephrosclerosis and are more salt-sensitive than SHR.
7. SHR and renal circulation
The SHR have characteristic differences in renal blood flow and renal function to the normotensive Wistar rat. These differences are due in part to structural changes namely, vascular remodelling caused by extracellular matrix deposition, hypertrophy and impaired vascular endothelium-dependent vasodilatation (Park et al., 2002). These factors cause a reduction in lumen diameter and increased peripheral resistance and therefore increased MAP. SHR display increased renal vascular resistance and increased reactivity to vasoactive substances such as noradrenaline and angiotensin II (Berecek et al., 1980). Stankevicius et al. (2002) showed that the arteries from hypertensive animals, contracted with noradrenaline, have decreased relaxation to acetylcholine (Stankevicius et al., 2002). This effect was proposed to be due to exposure of the arteries to the increased intraluminal pressure, because the extraluminal endothelium was found to be normal. The SHR have higher haematocrit levels and display increased platelet aggregation when exposed to thrombotic stimuli in comparison to normotensive Wistar rats (Lominadze et al., 1997).
Cowley et al. (1995) showed that in SHR compared to normotensive Wistar controls, renal MBP and sodium excretion were lower while cortical flow was similar (Cowley et al., 1995). These measurements were carried out using a laser Doppler flowmeter and indicated that MBP in SHR was reduced even before the appearance of hypertension. This apparent lower blood flow was accompanied by a raised vascular tone in the afferent arterioles of the juxtamedullary nephrons. With prolonged hypertension the cortex was also reported to become involved in the development of glomerular lesions and renal disease. In summary, the SHR have characteristic differences in renal blood flow and renal function to the normotensive Wistar rat. SHR display increased renal vascular resistance and increased reactivity to vasoactive substances. Also in SHR renal MBP is lower than that observed in normotensive control rats.
8. SHR and the renin–angiotensin system (RAS)
After over a century of studies the RAS is now recognised as an endocrine, paracrine and intracrine system which plays important roles in aldosterone synthesis, blood pressure control and body fluid balance (Zhuo and Li, 2011, Achard et al., 2001).The renin angiotensin system is a network of hormonal cascades which regulates cardiovascular, adrenal and renal function predominantly through the use of the angiotensin hormones. This system has been the target for many drugs in order to combat hypertension and other cardiovascular related events (Nguyen Dinh Cat and Touyz, 2011).
RAS can be described as a dual function system which balances between the vasoconstrictor, proliferative actions of Ang II and the vasodilator, anti-proliferative function of Ang-(1–7) (Pinheiro, 2012).
Components of the RAS have been implicated in contributing to the development of hypertension and increased expressions of some of the constituents of the system have been found in the SHR (Obata et al., 2000). With regard to the endothelial dysfunction observed in hypertension, angiotensin II-stimulated NADH/NADPH oxidase activity and subsequent superoxide anion production appears to mediate the development of hypertension. Indeed, in rats made hypertensive by infusions of angiotensin II, endothelium-dependent vascular relaxations are impaired. The vessels show an increased NADH/NADPH oxidase activity and produce increased amounts of superoxide anions (Rajagopalan et al., 1996).
Two types of Ang II receptors were pharmacologically described in 1993 and classified as the AT1 and AT2 receptors (Meister et al., 1993, Miyata et al., 1999). To date the AT1 receptor has been shown to mediate most of the physiological actions of Ang II (Sadoshima, 1998) and was localised to the outer medullary proximal tubules using in situ hybridisation in rat kidneys by (Meister et al., 1993). Zhuo et al., 1992 also reported high levels of AT1 receptor binding in glomerular mesangial cells and in renal interstitial cells located between the tubules and vasa recta bundles within in the inner stripe of the outer medulla of rats (Zhuo et al., 1992). Ang II directly constricts vascular smooth muscle cells and it is this vasoconstrictor effect that contributes to the regulation of renal function and BP (Rajagopalan et al., 1996). Ang II plays a very important role to increase the intra-glomerular pressure by constricting the efferent arterioles at the glomerulus.
Angiotensin II stimulates NADPH oxidase and therefore the production of oxygen radicals via AT1 receptors in the juxtaglomerular apparatus, diminishing NO signalling in the SHR by shortening its half-life (Schnackenberg, 2002). Inhibition of angiotensin II activity by way of AT1 receptor blockade, reduces ROS mediated NO inactivation (Vaziri et al., 2002). However, there is excessive vasoconstriction in the renal vessels of the SHR that cannot be solely attributed to angiotensin II action on the AT1 receptors (Welch and Wilcox, 2001). ACE inhibitors are an effective therapy for hypertension as they lower BP and have further beneficial effects on both systemic vasculature and the kidney (Griffin et al., 2003). Harrap et al. (1990) showed that the treatment of young SHR with the ACE inhibitor perindopril resulted in a marked reduction in MAP. They reported that after the withdrawal of the treatment MAP increased but plateaued at a reduced level in comparison to normal SHR. They also proposed that permanent structural changes occurred within the kidney causing the reduction of MAP to persist even when ACE inhibitor was stopped (Harrap et al., 1990).
It was postulated by Brenner et al. (1988) that increased BP in the SHRSP was caused by a decreased filtration surface area in the glomerulus or a reduced number of nephrons causing sodium retention and the resulting rise in the BP (Brenner et al., 1988). Hypertension, therefore, could exacerbate the situation by causing further damage to the existing glomeruli. Iversen et al., 1998a, Iversen et al., 1998b reported that increased glomerular capillary pressure in the SHR resulted in increased glomerulosclerosis (Iversen et al., 1998a). It is thought that ACE inhibitors stimulate glomerular growth and so may account for its permanent beneficial effect on hypertension. In contrast, Black et al. (2001) showed that ACE inhibitors had no effect on the glomerular number or size in the SHR which were lower than in the Wistar (Black et al., 2001). In summary, angiotensin II stimulates NADPH oxidase activity, produces increased amounts of oxygen radicals (superoxide) and diminishes NO signalling in the SHR. Treatment of SHR with ACE inhibitor decreases blood pressure, and so it can be concluded that the renin–angiotensin system plays an important role in maintaining the high BP levels in the SHR.
9. SHR and autoregulation of blood flow
Iversen et al., 1998a, Iversen et al., 1998b had previously shown that despite increased cortical perfusion pressure in the SHR, their ability to autoregulate the RBF and GFR was unaltered (Iversen et al., 1998b). The autoregulatory system adapts to the increased BP of the SHR, which develops with age, and the pressure limits around which the autoregulation mechanism works are reset to a higher level. If the MAP level of the SHR is reduced the autoregulatory mechanism compensates and resets to a lower pressure. This phenomenon of resetting could be as a result of interaction with the renal nerves or changes in plasma renin levels (Iversen et al., 1998b).
Tubuloglomerular feedback (TGF) is effective in both normotensive and hypertensive rat strains and there is little difference in feedback response kinetics between strains, indicating that TGF does not play a role in the pathogenesis of hypertension (Daniels and Arendshorst, 1990). Ren et al. (2002) suggested that intracellular superoxide in the macula densa enhanced TGF (Ren et al., 2002). This is due to superoxide scavenging NO, which effectively decreases its availability. Thus, superoxide may exert an enhanced influence on TGF under normal conditions. It may also act as an important factor under pathological conditions associated with high levels of superoxide and impaired endothelial function.
Renal prostaglandins are necessary for the normal functioning of the kidney, as indicated previously. There is evidence that there is increased prostaglandin synthesis in the kidney of the SHR, which becomes more severe with age (Schirner and Taube, 1993). Iversen et al. (1992) showed that prostaglandins are necessary for RBF autoregulation in the SHR and their effect was dependent on the renal nerves. In the same study, it was found that the inhibition of prostaglandin production by a cyclooxygenase inhibitor indomethacin attenuated RBF autoregulation in the SHR, provided that the kidney was innervated (Iversen et al., 1992). Renal tissue damage such as that caused by genetic hypertension in rats is generally not evenly distributed. There is evidence of glomerulosclerosis and tubular atrophy in the juxtamedullary cortex of the SHR at a time when there is much less in the superficial cortex (Roald et al., 2002).
10. SHR and reactive oxygen species
Oxidative stress in the vasculature has been implicated in the development of increased BP of the SHR. It has been argued that increased ROS production in the kidney is important in renal dysfunction associated with hypertension. ROS can affect the vascular endothelium of the kidney by way of reducing NO availability, or by oxidising arachidonic acid resulting in the generation of vasoactive products. ROS are present in low concentrations in all cell types and their levels are strictly regulated by constitutive scavenging systems, for example, superoxide dismutase enzyme (SOD) (Makino et al., 2002). Increased levels of ROS in the kidney of SHR have been implicated in the pathogenesis of hypertension. Acute and long term studies indicate that increased production of ROS play a part in the development of increased renal vascular resistance and increased sensitivity of TGF and therefore increased MAP in the SHRSP model. It has been demonstrated that intravenous infusion of SOD can lower BP in hypertensive rats (Nakazono et al., 1991).
Studies in which SOD-like compounds, such as Tempol or Vitamin C, were administrated in vivo resulted in a reduction in BP and increased the diameter of renal blood vessels (Makino et al., 2002, de Richelieu et al., 2005). Makino et al. (2002) also demonstrated that in the normotensive Sprague–Dawley rats, acute intramedullary infusion of DETC, a SOD inhibitor, resulted in increased medullary superoxide anion concentrations, reduced renal blood flow and a sustained increase in BP. Intravenous infusion of DETC resulted in no change in BP, which suggested that the increased oxidative stress in the renal medulla was solely responsible for the observed increase in BP (Ahmeda and Johns, 2012). The role of superoxide anions in the progression of hypertension is further supported by work carried out by Park et al. (2002) in which Tempol, a SOD mimetic, was infused into the renal medulla of SHRSP, resulting in decreased levels of ROS and attenuation of the progression of hypertension (Park et al., 2002).
11. SHR and nitric oxide
Impaired release of NO occurs in most animal and human models of hypertension, contributing to the increased peripheral resistance and most likely to the development of cardiovascular complications. Many in vitro studies have reported endothelial dysfunction in thoracic aortae of SHR (Konishi and Su, 1983, Luscher and Vanhoutte, 1986, Carvalho et al., 1987, Shirasaki et al., 1988). Nitrite and Nitrate levels in the SHR however are increased due to enhanced expression of constitutive NOS, despite impaired endothelium-dependent relaxation in the kidney of SHRSP (Stankevicius et al., 2002). NO seems to play a smaller role in tubular and vascular regulation than in the Wistar rats, which may contribute to the SHR having an impaired pressure natriuretic response and increased vascular resistance.
There is a high concentration of constitutive NOS (endothelial NOS (eNOS) and neuronal NOS (nNOS)) present in the endothelium of the renal tubules of SHRSP that is perhaps due to a compensatory up regulation of expression. Increased ROS may cause an increase in NOS enzyme production, particularly inducible NOS (iNOS), in order to counteract the negative effects of ROS on renal haemodynamics (Vaziri et al., 2002).
Hypertension has often been linked to changes in blood vessel properties. There is increased whole blood viscosity due to increased plasma protein concentrations including fibrinogen in the SHR. There is also evidence that endothelial cell alteration and changes in platelet activity in hypertension are crucial in the development of thrombotic lesions. Lominadze et al. (1997) reported that the SHR displayed increased platelet activation in the presence of thrombotic stimuli and an increased haematocrit compared to Wistar rats, which contribute to the hypertensive state of the SHR (Lominadze et al., 1997). This was supported by data presented by Yamashita et al. (2002), who found that platelet activation of SHRSP was decreased in vitro but also showed thrombus formation was increased in vivo (Yamashita et al., 2002). NO generation by the vascular endothelium causes vasodilatation and prevents platelet aggregation in the rat. It was shown that the vascular endothelium of the SHRSP had impaired NO generation compared with the Wistar, which could have occurred as a result of vascular damage due to increases in BP and could account for its increased thrombus formation (Yamashita et al., 2002). Vaziri et al. (1998) have published data in which they reported that there is an increase in NO production and eNOS and iNOS protein concentration in the young SHR in comparison to the Wistar (Vaziri et al., 2002).
Ichihara et al. (2001) reported that despite normal NOS activity NO, actions in the renal vasculature of the SHR are impaired, and this phenomenon was contributed to by the presence of oxygen radicals (Ichihara et al., 2001). NOS activity in the renal vessels of SHR has been shown to be equal to that in normotensive Wistar rats and NO is produced by the endothelium of SHR and Wistar rats in equal amounts. However, the vascular relaxing action of NO was diminished. Superoxide ions scavenge NO to form peroxynitrate, which is a less potent vasodilator than NO and has a shorter half-life (Ichihara et al., 2001). It has been shown by Grunfeld et al. (1995) that the increased production of superoxide radicals in the SHR is responsible for scavenging endothelial NO and thereby causing increased vascular resistance and the characteristic increase in BP (Grunfeld et al., 1995). However in another study they found that in vitro superoxide scavengers or increased NOS substrate failed to restore NO release from endothelial cells of the SHR to levels similar to those of the Wistar endothelium (Stankevicius et al., 2002). The evidence suggested a link between the impaired renal activity of NO on the vascular endothelium and the characteristic hypertension of the SHR. In relation to l-arginine pathway whether that normal or enhanced in the SHR and SHRSP a recent study suggested that the renal medullary uptake of l-arginine is impaired in SHR which mechanistically explaining reduced NO production (Rajapakse et al., 2012).
Cowley et al. (1995) showed that in the SHR renal MBP was lower in comparison to Wistar rats, but cortical flows were the same. These authors used laser-Doppler flowmetry to measure renal blood flow, and they indicated that MBP in the SHR was reduced in comparison to the Wistar rat even before the appearance of hypertension. This apparent reduction in MBP was accompanied by increased vascular tone in the afferent arterioles of juxtaglomerular nephrons (Cowley et al., 1995). With prolonged hypertension the cortex can also become involved with the development of glomerular lesions and renal vascular degeneration.
Vascular differences between the SHRSP and the Wistar rat, such as increased vascular resistance, may be due to the alteration in reactive oxygen species and NO availability. It has been reported that in a rat model of hypertension (SHR), the concentration of superoxide anion is at a high level in the renal medulla compared to the normotensive rats. Indeed, Kerr et al., 1999 confirmed that NO production was greater in SHR compared with normotensive rats. In relation to the SHRSP, it could be argued that superoxide anion production overrides the enhanced NO generation and counteracts its action (Kerr et al., 1999).
In summary, it has been shown that the increased production of superoxide radicals in the SHR is responsible for scavenging endothelial NO and thereby causing increased vascular resistance and the characteristic increase in BP. The evidence suggests a link between the impaired renal activity of NO on the vascular endothelium and the characteristic hypertension of the SHR.
12. Hypertension and NOS
Genetic manipulation to the gene that codes for eNOS in mice has been shown to lead to the development of hypertension (Huang et al., 1995). Therefore impaired NO mediated vasodilatation is believed to contribute to the hypertensive state. NO plays a major role in reducing the systemic vascular resistance. It is therefore considered that endothelial vasodilator dysfunction could precipitate hypertension. Indeed, an endothelial abnormality is associated with hypertension in animal models (Luscher et al., 1987). Depending on the experimental model, the reduction in endothelium-dependent relaxation is due to attenuation of endothelium mediated NO-dependent activity or it is due to the augmented elaboration of an endothelium-derived contracting factor (possibly a prostanoid). Whether endothelial dysfunction is primary in the initiation of hypertension or merely an epiphenomenon is not clear. Treatment of the elevated BP normalises endothelium-dependent relaxation suggesting that the endothelial abnormality is secondary in the hypertensive process (Cooke and Dzau, 1997a, Cooke and Dzau, 1997b, Luscher et al., 1987, Shultz and Raij, 1989). Conversely, infusion of NOS antagonists produces a marked increase in BP in experimental animals (Ribeiro et al., 1992). These inhibitors have been considered non-specific, and the effect on BP could conceivably be due to an effect on nNOS. However, more definitive data for a primary role of NO in the regulation of BP was provided by Huang et al. in 1995 (Huang et al., 1995). They found that inactivation of the mouse endothelial NOS gene by homologous recombination produced mice that were significantly hypertensive.
There is extensive evidence for endothelial vasodilator dysfunction in hypertensive humans (Linder et al., 1990, Panza et al., 1990). The most direct evidence has come from measurements of forearm blood flow by strain-gauge plethysmography in response to intra-arterial infusions of endothelium-dependent and independent vasodilators. In young patients with mild essential hypertension, endothelium-independent vasodilation is relatively undisturbed. In contrast, cholinergic vasodilation (presumably endothelium-dependent) is attenuated. Whether this is a primary or secondary phenomenon is not known. The impairment of endothelium-dependent vasodilation is likely multifactorial and may involve abnormalities of signal transduction, NO activity, or NO biosynthesis. There is preliminary evidence that, at least in some cases, the endothelial deficit may precede the appearance of essential hypertension (Panza et al., 1990). In young normotensive individuals with hypertensive parents cholinergic forearm vasodilation is impaired; in contrast, endothelium-independent vasodilation is normal (Cooke and Dzau, 1997b).
13. SHR and endothelin-1 (ET-1)
Endothelins are a paracrine regulator, and are a family of 21-amino acid peptides produced by endothelial cells which act at vascular smooth muscle to produce vasoconstriction. Three pharmacologically distinct iso-peptides of endothelin arising from three different genes have been identified in mammalian species; in humans, endothelin-1 appears to be functionally the most important in both health and disease (Yanagisawa et al., 1988, Inoue et al., 1989).
Two main endothelin receptors have been identified in humans. The ETA receptor is present on vascular smooth muscle cells, and stimulation of this receptor leads to a vasoconstrictor response. In contrast, activation of the endothelial ETB receptor results in vasodilatation through NO release. The ET-1 stimulated the release of nitric oxide, which modulate the ETA receptor-mediated contraction of the vascular smooth muscle. When ET-1 is administered intravenously, there is a transient hypotensive (vasodilator) response followed by a prolonged hypertensive (vasoconstrictor) response. The initial vasodilation is due to NO release, whereas the prolonged vasoconstrictor response is due to ET-1 acting on smooth muscle ETA and ETB receptors. NO also inhibits the formation of ET-1. Therefore, there are complex interactions between NO and ET-1 actions on blood vessels. A third Endothelin receptor has been identified in Xenopus laevis and shown to have a high affinity for endothelin-3; however, the human homologue of this receptor has yet to be identified (Schwerte et al., 2002).
Activation of the ETA receptor by endothelin-1 stimulates the release of arachidonic acid via the activation of cytosolic phospholipase A2 (Trevisi et al., 2002). This may in turn result in the synthesis of other endothelium-dependent vasoactive factors such as PGI2 and thromboxane A2. Additionally, endothelin-1 has been reported to promote COX-2 expression as well as prostaglandin synthesis in vitro. Thus, besides acting as a potent vasoconstrictor in its own right, endothelin-1 also may promote release of other endothelium-dependent contracting factors (Molnar and Hertelendy, 1995).
Activation of endothelin system is a characteristic for SHR and that the SHR have an endothelin-dependent component (Sharifi et al., 1998, Schiffrin, 1999).
Infusion of ET-1 into renal artery exerts an effect on renal microvasculature which leads to vasoconstriction. That effect confirmed to be through the ETA and ETB receptors which found in the renal blood vessels. Interestingly when ETA receptor blocked, the vasoconstriction response remains the same due to the fact that the ET-1 exerts the vasoconstrictor action on ETB receptor (Gellai et al., 1994, Just et al., 2004, Matsuura et al., 1996, Wellings et al., 1994).
In previous studies that investigated the role of ET-1 in the SHR model and the overall results of those studies were not similar. The endothelin system does not seem to play a vital role in SHR. (Lariviere et al., 1993, Lariviere et al., 1995, Schiffrin et al., 1995, Li and Schiffrin, 1995). However, some studies have confirmed that increased vasoconstrictor responses to ET-1 (Miyauchi et al., 1989, Cargnelli et al., 1990) and that the role of endothelin is exist in SHR.
Other reports supported the later findings and provided further evidence for the important role of Endothelin in regulating the renal function in SHR, after administration of ET-1 antagonist the renal function correspondingly improved (Kato et al., 1995, Bird et al., 1995).
In a different study where the effect of intrarenal administration of Endothelin receptors blockade, in both SHR and Wistar rats, was investigated and the result showed no changes on renal hemodynamic or excretory function in Wistar rats. However, Endothelin receptors blocker within the kidney improves renal hemodynamic and excretory function in the SHR (Kato et al., 1995).
14. Conclusion
This review article provided a summary for the factors that influencing the renal hemodynamic in SHR and explored briefly the mechanisms by which those factors can affect the renal circulation.
In conclusion, it is apparent that the development of hypertension in SHR is a result of complex mechanisms which involve many factors and molecules that may act directly on the renal blood vessels or interacting with other autocrine and paracrine factors, whether locally or systematically, that resulted in a modification of the tone of the renal blood vessels hence affect the renal function and blood pressure.
Hopefully this review will provide the basis for the subsequent articles to explore the other factors that may contribute in the development of hypertension due to their local effect on renal circulation.
Disclosures
Conflict of interest statement: no conflict of interest, financial or otherwise, are declared by the authors.
Acknowledgments
The authors are thankful to the College of Medicine Research Centre (CMRC), Deanship of Scientific Research, King Saud University, Riyadh, Saudi Arabia for supporting the research.
Footnotes
Peer review under responsibility of King Saud University.
References
- Achard J.-M., Fournier A., Mazouz H., Caride V.J., Penar P.L., Fernandez L.A. Protection against ischemia: a physiological function of the renin-angiotensin system. Biochem. Pharmacol. 2001;62:261–271. doi: 10.1016/s0006-2952(01)00687-6. [DOI] [PubMed] [Google Scholar]
- Ahmeda A.F., Johns E.J. The regulation of blood perfusion in the renal cortex and medulla by reactive oxygen species and nitric oxide in the anaesthetised rat. Acta Physiol. (Oxf.) 2012;204:443–450. doi: 10.1111/j.1748-1716.2011.02346.x. [DOI] [PubMed] [Google Scholar]
- Arendshorst W.J., Beierwaltes W.H. Renal tubular reabsorption in spontaneously hypertensive rats. Am. J. Physiol. 1979;237:F38–F47. doi: 10.1152/ajprenal.1979.237.1.F38. [DOI] [PubMed] [Google Scholar]
- Armstrong J.M., Blackwell G.J., Flower R.J., McGiff J.C., Mullane K.M., Vane J.R. Genetic hypertension in rats is accompanied by a defect in renal prostaglandin catabolism. Nature. 1976;260:582–586. doi: 10.1038/260582a0. [DOI] [PubMed] [Google Scholar]
- Badzynska B., Grzelec-Mojzesowicz M., Sadowski J. Prostaglandins but not nitric oxide protect renal medullary perfusion in anaesthetised rats receiving angiotensin II. J. Physiol. 2003;548:875–880. doi: 10.1113/jphysiol.2002.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger A., Patchett S., Kumar P.J. Saunders/Elsevier; 2007. Pocket Essentials of Clinical Medicine. [Google Scholar]
- Berecek K.H., Schwertschlag U., Gross F. Alterations in renal vascular resistance and reactivity in spontaneous hypertension of rats. Am. J. Physiol. 1980;238:H287–H293. doi: 10.1152/ajpheart.1980.238.3.H287. [DOI] [PubMed] [Google Scholar]
- Beswick R.A., Dorrance A.M., Leite R., Webb R.C. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension. 2001;38:1107–1111. doi: 10.1161/hy1101.093423. [DOI] [PubMed] [Google Scholar]
- Bird J.E., Webb M.L., Giancarli M.R., Chao C., Dorso C.R., Asaad M.M. A role for endothelin ETA receptors in regulation of renal function in spontaneously hypertensive rats. Eur. J. Pharmacol. 1995;294:183–189. doi: 10.1016/0014-2999(95)00537-4. [DOI] [PubMed] [Google Scholar]
- Black M.J., Briscoe T.A., Dunstan H.J., Bertram J.F., Johnston C.I. Effect of angiotensin-converting enzyme inhibition on renal filtration surface area in hypertensive rats. Kidney Int. 2001;60:1837–1843. doi: 10.1046/j.1523-1755.2001.00996.x. [DOI] [PubMed] [Google Scholar]
- Brenner B.M., Garcia D.L., Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am. J. Hypertens. 1988;1:335–347. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- Cargnelli G., Rossi G., Bova S., Pessina A.C. In vitro vascular reactivity to endothelin: a comparison between young and old normotensive and hypertensive rats. Clin. Exp. Hypertens. A. 1990;12:1437–1451. doi: 10.3109/10641969009073529. [DOI] [PubMed] [Google Scholar]
- Carvalho M.H., Scivoletto R., Fortes Z.B., Nigro D., Cordellini S. Reactivity of aorta and mesenteric microvessels to drugs in spontaneously hypertensive rats: role of the endothelium. J. Hypertens. 1987;5:377–382. doi: 10.1097/00004872-198706000-00019. [DOI] [PubMed] [Google Scholar]
- Churchill P.C., Churchill M.C., Griffin K.A., Picken M., Webb R.C., Kurtz T.W., Bidani A.K. Increased genetic susceptibility to renal damage in the stroke-prone spontaneously hypertensive rat. Kidney Int. 2002;61:1794–1800. doi: 10.1046/j.1523-1755.2002.00321.x. [DOI] [PubMed] [Google Scholar]
- Cooke J.P., Dzau V.J. Derangements of the nitric oxide synthase pathway, l-arginine, and cardiovascular diseases. Circulation. 1997;96:379–382. [PubMed] [Google Scholar]
- Cooke J.P., Dzau V.J. Nitric oxide synthase: role in the genesis of vascular disease. Annu. Rev. Med. 1997;48:489–509. doi: 10.1146/annurev.med.48.1.489. [DOI] [PubMed] [Google Scholar]
- Cowley A.W., Jr. Role of the renal medulla in volume and arterial pressure regulation. Am. J. Physiol. 1997;273:R1–R15. doi: 10.1152/ajpregu.1997.273.1.R1. [DOI] [PubMed] [Google Scholar]
- Cowley A.W., Jr., Mattson D.L., Lu S., Roman R.J. The renal medulla and hypertension. Hypertension. 1995;25:663–673. doi: 10.1161/01.hyp.25.4.663. [DOI] [PubMed] [Google Scholar]
- Daniels F.H., Arendshorst W.J. Tubuloglomerular feedback kinetics in spontaneously hypertensive and Wistar-Kyoto rats. Am. J. Physiol. 1990;259:F529–F534. doi: 10.1152/ajprenal.1990.259.3.F529. [DOI] [PubMed] [Google Scholar]
- de Richelieu L.T., Sorensen C.M., Holstein-Rathlou N.H., Salomonsson M. NO-independent mechanism mediates Tempol-induced renal vasodilation in SHR. Am. J. Physiol. Renal Physiol. 2005;289:F1227–F1234. doi: 10.1152/ajprenal.00116.2005. [DOI] [PubMed] [Google Scholar]
- Denton K.M., Kett M.M., Dodic M. Early Life Origins of Health and Disease. Springer; 2006. Programming hypertension–animal models. [Google Scholar]
- Dornas W.C., Silva M.E. Animal models for the study of arterial hypertension. J. Biosci. 2011;36:731–737. doi: 10.1007/s12038-011-9097-y. [DOI] [PubMed] [Google Scholar]
- Evans R.G., Eppel G.A., Anderson W.P., Denton K.M. Mechanisms underlying the differential control of blood flow in the renal medulla and cortex. J. Hypertens. 2004;22:1439–1451. doi: 10.1097/01.hjh.0000133744.85490.9d. [DOI] [PubMed] [Google Scholar]
- Ganten D. Role of animal models in hypertension research. Hypertension. 1987;9:1–2. [Google Scholar]
- Gellai M., Dewolf R., Pullen M., Nambi P. Distribution and functional role of renal ET receptor subtypes in normotensive and hypertensive rats. Kidney Int. 1994;46:1287–1294. doi: 10.1038/ki.1994.396. [DOI] [PubMed] [Google Scholar]
- Goldblatt H., Lynch J., Hanzal R.F., Summerville W.W. Studies on experimental hypertension I. The production of persistent elevation of systolic blood pressure by means of renal ischemia. J. Exp. Med. 1934;59:347–379. doi: 10.1084/jem.59.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin K.A., Churchill P.C., Picken M., Webb R.C., Kurtz T.W., Bidani A.K. Differential salt-sensitivity in the pathogenesis of renal damage in SHR and stroke prone SHR. Am. J. Hypertens. 2001;14:311–320. doi: 10.1016/s0895-7061(00)01282-6. [DOI] [PubMed] [Google Scholar]
- Griffin K.A., Abu-Amarah I., Picken M., Bidani A.K. Renoprotection by ACE inhibition or aldosterone blockade is blood pressure-dependent. Hypertension. 2003;41:201–206. doi: 10.1161/01.hyp.0000049881.25304.73. [DOI] [PubMed] [Google Scholar]
- Grisk O., Kloting I., Exner J., Spiess S., Schmidt R., Junghans D., Lorenz G., Rettig R. Long-term arterial pressure in spontaneously hypertensive rats is set by the kidney. J. Hypertens. 2002;20:131–138. doi: 10.1097/00004872-200201000-00019. [DOI] [PubMed] [Google Scholar]
- Grunfeld S., Hamilton C.A., Mesaros S., McClain S.W., Dominiczak A.F., Bohr D.F., Malinski T. Role of superoxide in the depressed nitric oxide production by the endothelium of genetically hypertensive rats. Hypertension. 1995;26:854–857. doi: 10.1161/01.hyp.26.6.854. [DOI] [PubMed] [Google Scholar]
- Guyton A.C., Coleman T.G., Cowley A.W., Jr., Liard J.F., Liard J.F., MANNING R.D., Jr. Systems analysis of arterial pressure regulation and hypertension. Ann. Biomed. Eng. 1972;1:254–281. doi: 10.1007/BF02584211. [DOI] [PubMed] [Google Scholar]
- Harrap S.B., van der Merwe W.M., Griffin S.A., Macpherson F., Lever A.F. Brief angiotensin converting enzyme inhibitor treatment in young spontaneously hypertensive rats reduces blood pressure long-term. Hypertension. 1990;16:603–614. doi: 10.1161/01.hyp.16.6.603. [DOI] [PubMed] [Google Scholar]
- Heyman S.N., Rosen S., Brezis M. The renal medulla: life at the edge of anoxia. Blood Purif. 1997;15:232–242. doi: 10.1159/000170341. [DOI] [PubMed] [Google Scholar]
- Huang P.L., Huang Z., Mashimo H., Bloch K.D., Moskowitz M.A., Bevan J.A., Fishman M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- Ichihara A., Hayashi M., Hirota N., Saruta T. Superoxide inhibits neuronal nitric oxide synthase influences on afferent arterioles in spontaneously hypertensive rats. Hypertension. 2001;37:630–634. doi: 10.1161/01.hyp.37.2.630. [DOI] [PubMed] [Google Scholar]
- Inoue A., Yanagisawa M., Kimura S., Kasuya Y., Miyauchi T., Goto K., Masaki T. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. U.S.A. 1989;86:2863–2867. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen B.M., Kvam F.I., Morkrid L., Sekse I., Ofstad J. Effect of cyclooxygenase inhibition on renal blood flow autoregulation in SHR. Am. J. Physiol. 1992;263:F534–F539. doi: 10.1152/ajprenal.1992.263.3.F534. [DOI] [PubMed] [Google Scholar]
- Iversen B.M., Amann K., Kvam F.I., Wang X., Ofstad J. Increased glomerular capillary pressure and size mediate glomerulosclerosis in SHR juxtamedullary cortex. Am. J. Physiol. 1998;274:F365–F373. doi: 10.1152/ajprenal.1998.274.2.F365. [DOI] [PubMed] [Google Scholar]
- Iversen B.M., Kvam F.I., Matre K., Ofstad J. Resetting of renal blood autoregulation during acute blood pressure reduction in hypertensive rats. Am. J. Physiol. 1998;275:R343–R349. doi: 10.1152/ajpregu.1998.275.2.R343. [DOI] [PubMed] [Google Scholar]
- Johns E.J., Ahmeda A.F. Reference Module in Biomedical Sciences. Elsevier; 2014. Renal circulation; p. Elsevier. [Google Scholar]
- Just A., Olson A.J., Arendshorst W.J. Dual constrictor and dilator actions of ET(B) receptors in the rat renal microcirculation: interactions with ET(A) receptors. Am. J. Physiol. Renal Physiol. 2004;286:F660–F668. doi: 10.1152/ajprenal.00368.2003. [DOI] [PubMed] [Google Scholar]
- Kato T., Kassab S., Wilkins F.C., Jr., Kirchner K.A., Keiser J., GRANGER J.P. Endothelin antagonists improve renal function in spontaneously hypertensive rats. Hypertension. 1995;25:883–887. doi: 10.1161/01.hyp.25.4.883. [DOI] [PubMed] [Google Scholar]
- Kerr S., Brosnan M.J., McIntyre M., Reid J.L., Dominiczak A.F., Hamilton C.A. Superoxide anion production is increased in a model of genetic hypertension: role of the endothelium. Hypertension. 1999;33:1353–1358. doi: 10.1161/01.hyp.33.6.1353. [DOI] [PubMed] [Google Scholar]
- Konishi M., Su C. Role of endothelium in dilator responses of spontaneously hypertensive rat arteries. Hypertension. 1983;5:881–886. doi: 10.1161/01.hyp.5.6.881. [DOI] [PubMed] [Google Scholar]
- Lariviere R., Thibault G., Schiffrin E.L. Increased endothelin-1 content in blood vessels of deoxycorticosterone acetate-salt hypertensive but not in spontaneously hypertensive rats. Hypertension. 1993;21:294–300. doi: 10.1161/01.hyp.21.3.294. [DOI] [PubMed] [Google Scholar]
- Lariviere R., Sventek P., Schiffrin E.L. Expression of endothelin-1 gene in blood vessels of adult spontaneously hypertensive rats. Life Sci. 1995;56:1889–1896. doi: 10.1016/0024-3205(95)00163-z. [DOI] [PubMed] [Google Scholar]
- Levy B.I., Duriez M., Samuel J.L. Coronary microvasculature alteration in hypertensive rats. Effect of treatment with a diuretic and an ACE inhibitor. Am. J. Hypertens. 2001;14:7–13. doi: 10.1016/s0895-7061(00)01212-7. [DOI] [PubMed] [Google Scholar]
- Li J.S., Schiffrin E.L. Effect of chronic treatment of adult spontaneously hypertensive rats with an endothelin receptor antagonist. Hypertension. 1995;25:495–500. doi: 10.1161/01.hyp.25.4.495. [DOI] [PubMed] [Google Scholar]
- Linder L., Kiowski W., Buhler F.R., Luscher T.F. Indirect evidence for release of endothelium-derived relaxing factor in human forearm circulation in vivo. Blunted response in essential hypertension. Circulation. 1990;81:1762–1767. doi: 10.1161/01.cir.81.6.1762. [DOI] [PubMed] [Google Scholar]
- Liu K.L. Regulation of renal medullary circulation by the renin-angiotensin system in genetically hypertensive rats. Clin. Exp. Pharmacol. Physiol. 2009;36:455–461. doi: 10.1111/j.1440-1681.2009.05153.x. [DOI] [PubMed] [Google Scholar]
- Lominadze D., Joshua I.G., Schuschke D.A. In vivo platelet thrombus formation in microvessels of spontaneously hypertensive rats. Am. J. Hypertens. 1997;10:1140–1146. doi: 10.1016/s0895-7061(97)00214-8. [DOI] [PubMed] [Google Scholar]
- Luscher T.F., Vanhoutte P.M. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension. 1986;8:344–348. doi: 10.1161/01.hyp.8.4.344. [DOI] [PubMed] [Google Scholar]
- Luscher T.F., Vanhoutte P.M., Raij L. Antihypertensive treatment normalizes decreased endothelium-dependent relaxations in rats with salt-induced hypertension. Hypertension. 1987;9 doi: 10.1161/01.hyp.9.6_pt_2.iii193. III193-7. [DOI] [PubMed] [Google Scholar]
- Makino A., Skelton M.M., Zou A.P., Roman R.J., Cowley A.W., Jr. Increased renal medullary oxidative stress produces hypertension. Hypertension. 2002;39:667–672. doi: 10.1161/hy0202.103469. [DOI] [PubMed] [Google Scholar]
- Matsuura T., Yukimura T., Kim S., Miura K., Iwao H. Selective blockade of endothelin receptor subtypes on systemic and renal vascular responses to endothelin-1 and IRL1620, a selective endothelin ETB-receptor agonist, in anesthetized rats. Jpn. J. Pharmacol. 1996;71:213–222. doi: 10.1254/jjp.71.213. [DOI] [PubMed] [Google Scholar]
- Mattson D.L. Importance of the renal medullary circulation in the control of sodium excretion and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;284:R13–R27. doi: 10.1152/ajpregu.00321.2002. [DOI] [PubMed] [Google Scholar]
- Meister B., Lippoldt A., Bunnemann B., Inagami T., Ganten D., Fuxe K. Cellular expression of angiotensin type-1 receptor mRNA in the kidney. Kidney Int. 1993;44:331–336. doi: 10.1038/ki.1993.248. [DOI] [PubMed] [Google Scholar]
- Miyata N., Park F., Li X.F., Cowley A.W., Jr. Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am. J. Physiol. 1999;277:F437–F446. doi: 10.1152/ajprenal.1999.277.3.F437. [DOI] [PubMed] [Google Scholar]
- Miyauchi T., Ishikawa T., Tomobe Y., Yanagisawa M., Kimura S., Sugishita Y., Ito I., Goto K., Masaki T. Characteristics of pressor response to endothelin in spontaneously hypertensive and Wistar-Kyoto rats. Hypertension. 1989;14:427–434. doi: 10.1161/01.hyp.14.4.427. [DOI] [PubMed] [Google Scholar]
- Molnar M., Hertelendy F. Signal transduction in rat myometrial cells: comparison of the actions of endothelin-1, oxytocin and prostaglandin F2 alpha. Eur. J. Endocrinol. 1995;133:467–474. doi: 10.1530/eje.0.1330467. [DOI] [PubMed] [Google Scholar]
- Nagaoka A., Kakihana M., Suno M., Hamajo K. Renal hemodynamics and sodium excretion in stroke-prone spontaneously hypertensive rats. Am. J. Physiol. 1981;241:F244–F249. doi: 10.1152/ajprenal.1981.241.3.F244. [DOI] [PubMed] [Google Scholar]
- Nakazono K., Watanabe N., Matsuno K., Sasaki J., Sato T., Inoue M. Does superoxide underlie the pathogenesis of hypertension? Proc. Natl. Acad. Sci. U.S.A. 1991;88:10045–10048. doi: 10.1073/pnas.88.22.10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navar L.G., Arendshorst W.J., Pallone T.L., Inscho E.W., Imig J.D., Bell P.D. The renal microcirculation. In: Ley R F.T.N.D., editor. Microcirculation. second ed. Academic Press; San Diego: 2008. (Chapter 13) [Google Scholar]
- Nguyen Dinh Cat A., Touyz R.M. A new look at the renin–angiotensin system—focusing on the vascular system. Peptides. 2011;32:2141–2150. doi: 10.1016/j.peptides.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Obata J., Nakamura T., Takano H., Naito A., Kimura H., Yoshida Y., Shimizu F., Guo D.F., Inagami T. Increased gene expression of components of the renin-angiotensin system in glomeruli of genetically hypertensive rats. J. Hypertens. 2000;18:1247–1255. doi: 10.1097/00004872-200018090-00011. [DOI] [PubMed] [Google Scholar]
- Okamoto K., Yamori Y., Nagaoka A. Establishment of the stroke-prone spontaneously hypertensive rat (SHR) Circ. Res. 1974;34:143–153. [Google Scholar]
- Pallone T.L. Extravascular protein in the renal medulla: analysis by two methods. Am. J. Physiol. 1994;266:R1429–R1436. doi: 10.1152/ajpregu.1994.266.5.R1429. [DOI] [PubMed] [Google Scholar]
- Pallone T.L., Edwards A., Mattson D.L. Renal medullary circulation. Compr. Physiol. 2012;2:97–140. doi: 10.1002/cphy.c100036. [DOI] [PubMed] [Google Scholar]
- Panza J.A., Quyyumi A.A., Brush J.E., Jr., Epstein S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990;323:22–27. doi: 10.1056/NEJM199007053230105. [DOI] [PubMed] [Google Scholar]
- Park F., Mattson D.L., Roberts L.A., Cowley A.W., Jr. Evidence for the presence of smooth muscle alpha-actin within pericytes of the renal medulla. Am. J. Physiol. 1997;273:R1742–R1748. doi: 10.1152/ajpregu.1997.273.5.R1742. [DOI] [PubMed] [Google Scholar]
- Park J.B., Touyz R.M., Chen X., Schiffrin E.L. Chronic treatment with a superoxide dismutase mimetic prevents vascular remodeling and progression of hypertension in salt-loaded stroke-prone spontaneously hypertensive rats. Am. J. Hypertens. 2002;15:78–84. doi: 10.1016/s0895-7061(01)02233-6. [DOI] [PubMed] [Google Scholar]
- Persson P.B. Renal blood flow autoregulation in blood pressure control. Curr. Opin. Nephrol. Hypertens. 2002;11:67–72. doi: 10.1097/00041552-200201000-00010. [DOI] [PubMed] [Google Scholar]
- Pinheiro S.V.B. Angiotensin converting enzyme 2, angiotensin-(1–7), and receptor mas axis in the kidney. Int. J. Hypertens. 2012;2012 doi: 10.1155/2012/414128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S., Kurz S., Munzel T., Tarpey M., Freeman B.A., Griendling K.K., Harrison D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse N.W., Kuruppu S., Hanchapola I., Venardos K., Mattson D.L., Smith A.I., Kaye D.M., Evans R.G. Evidence that renal arginine transport is impaired in spontaneously hypertensive rats. Am. J. Physiol. Renal Physiol. 2012;302:F1554–F1562. doi: 10.1152/ajprenal.00084.2011. [DOI] [PubMed] [Google Scholar]
- Ren Y., Carretero O.A., Garvin J.L. Mechanism by which superoxide potentiates tubuloglomerular feedback. Hypertension. 2002;39:624–628. doi: 10.1161/hy0202.103299. [DOI] [PubMed] [Google Scholar]
- Rettig R., Folberth C., Stauss H., Kopf D., Waldherr R., Unger T. Role of the kidney in primary hypertension: a renal transplantation study in rats. Am. J. Physiol. 1990;258:F606–F611. doi: 10.1152/ajprenal.1990.258.3.F606. [DOI] [PubMed] [Google Scholar]
- Ribeiro M.O., Antunes E., de Nucci G., Lovisolo S.M., Zatz R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension. 1992;20:298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- Roald A.B., Ofstad J., Iversen B.M. Attenuated buffering of renal perfusion pressure variation in juxtamedullary cortex in SHR. Am. J. Physiol. Renal Physiol. 2002;282:F506–F511. doi: 10.1152/ajprenal.00199.2001. [DOI] [PubMed] [Google Scholar]
- Sadoshima J. Versatility of the angiotensin II type 1 receptor. Circ. Res. 1998;82:1352–1355. doi: 10.1161/01.res.82.12.1352. [DOI] [PubMed] [Google Scholar]
- Schiffrin E.L. State-of-the-Art lecture. Role of endothelin-1 in hypertension. Hypertension. 1999;34:876–881. doi: 10.1161/01.hyp.34.4.876. [DOI] [PubMed] [Google Scholar]
- Schiffrin E.L., Lariviere R., Li J.S., Sventek P., Touyz R.M. Deoxycorticosterone acetate plus salt induces overexpression of vascular endothelin-1 and severe vascular hypertrophy in spontaneously hypertensive rats. Hypertension. 1995;25:769–773. doi: 10.1161/01.hyp.25.4.769. [DOI] [PubMed] [Google Scholar]
- Schirner M., Taube C. Different effects of aspirin on blood pressure of spontaneously hypertensive rats (SHR) with high and spontaneously low levels of blood pressure. Br. J. Pharmacol. 1993;109:900–901. doi: 10.1111/j.1476-5381.1993.tb13704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnackenberg C.G. Oxygen radicals in cardiovascular-renal disease. Curr. Opin. Pharmacol. 2002;2:121–125. doi: 10.1016/s1471-4892(02)00133-9. [DOI] [PubMed] [Google Scholar]
- Schoen F.J., Cotran R.S. Saunders; 1994. Robbins Pathologic Basis of Disease. [Google Scholar]
- Schwerte T., Printz E., Fritsche R. Vascular control in larval Xenopus laevis: the role of endothelial-derived factors. J. Exp. Biol. 2002;205:225–232. doi: 10.1242/jeb.205.2.225. [DOI] [PubMed] [Google Scholar]
- Sharifi A.M., He G., Touyz R.M., Schiffrin E.L. Vascular endothelin-1 expression and effect of an endothelin ETA antagonist on structure and function of small arteries from stroke-prone spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 1998;31(Suppl 1):S309–F312. doi: 10.1097/00005344-199800001-00086. [DOI] [PubMed] [Google Scholar]
- Shirasaki Y., Kolm P., Nickols G.A., Lee T.J. Endothelial regulation of cyclic GMP and vascular responses in hypertension. J. Pharmacol. Exp. Ther. 1988;245:53–58. [PubMed] [Google Scholar]
- Shultz P.J., Raij L. Effects of antihypertensive agents on endothelium-dependent and endothelium-independent relaxations. Br. J. Clin. Pharmacol. 1989;28(Suppl 2):151S–157S. doi: 10.1111/j.1365-2125.1989.tb03590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skov K., Mulvany M.J., Korsgaard N. Morphology of renal afferent arterioles in spontaneously hypertensive rats. Hypertension. 1992;20:821–827. doi: 10.1161/01.hyp.20.6.821. [DOI] [PubMed] [Google Scholar]
- Soergel M., Schaefer F. Effect of hypertension on the progression of chronic renal failure in children. Am. J. Hypertens. 2002;15:53S–56S. doi: 10.1016/s0895-7061(01)02296-8. [DOI] [PubMed] [Google Scholar]
- Stankevicius E., Martinez A.C., Mulvany M.J., Simonsen U. Blunted acetylcholine relaxation and nitric oxide release in arteries from renal hypertensive rats. J. Hypertens. 2002;20:1571–1579. doi: 10.1097/00004872-200208000-00020. [DOI] [PubMed] [Google Scholar]
- Taddei S., Virdis A., Ghiadoni L., Magagna A., Salvetti A. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation. 1998;97:2222–2229. doi: 10.1161/01.cir.97.22.2222. [DOI] [PubMed] [Google Scholar]
- Tigerstedt R., Bergman P. Kidney and circulation. Scand. Arch. Physiol. 1898;8:223–271. [Google Scholar]
- Trevisi L., Bova S., Cargnelli G., Ceolotto G., Luciani S. Endothelin-1-induced arachidonic acid release by cytosolic phospholipase A2 activation in rat vascular smooth muscle via extracellular signal-regulated kinases pathway. Biochem. Pharmacol. 2002;64:425–431. doi: 10.1016/s0006-2952(02)01066-3. [DOI] [PubMed] [Google Scholar]
- Vaziri N.D., Ni Z., Oveisi F. Upregulation of renal and vascular nitric oxide synthase in young spontaneously hypertensive rats. Hypertension. 1998;31:1248–1254. doi: 10.1161/01.hyp.31.6.1248. [DOI] [PubMed] [Google Scholar]
- Vaziri N.D., Wang X.Q., Ni Z.N., Kivlighn S., Shahinfar S. Effects of aging and AT-1 receptor blockade on NO synthase expression and renal function in SHR. Biochim. Biophys. Acta. 2002;1592:153–161. doi: 10.1016/s0167-4889(02)00309-9. [DOI] [PubMed] [Google Scholar]
- Welch W.J., Wilcox C.S. AT1 receptor antagonist combats oxidative stress and restores nitric oxide signaling in the SHR. Kidney Int. 2001;59:1257–1263. doi: 10.1046/j.1523-1755.2001.0590041257.x. [DOI] [PubMed] [Google Scholar]
- Wellings R.P., Corder R., Warner T.D., Cristol J.P., Thiemermann C., Vane J.R. Evidence from receptor antagonists of an important role for ETB receptor-mediated vasoconstrictor effects of endothelin-1 in the rat kidney. Br. J. Pharmacol. 1994;111:515–520. doi: 10.1111/j.1476-5381.1994.tb14767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel P., Mazzolai L., Nussberger J., Pedrazzini T. Two-kidney, one clip and one-kidney, one clip hypertension in mice. Hypertension. 1997;29:1025–1030. doi: 10.1161/01.hyp.29.4.1025. [DOI] [PubMed] [Google Scholar]
- Yamashita T., Taka T., Nojima R., Ohta Y., Seki J., Yamamoto J. There is no valid evidence presented as to an impaired endothelial NO system in the stroke-prone spontaneously hypertensive rats. Thromb. Res. 2002;105:507–511. doi: 10.1016/s0049-3848(02)00069-5. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M., Kurihara H., Kimura S., Tomobe Y., Kobayashi M., Mitsui Y., Yazaki Y., Goto K., Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- Zhuo J.L., Li X.C. New insights and perspectives on intrarenal renin-angiotensin system: focus on intracrine/intracellular angiotensin II. Peptides. 2011;32:1551–1565. doi: 10.1016/j.peptides.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo J., Alcorn D., Allen A.M., Mendelsohn F.A. High resolution localization of angiotensin II receptors in rat renal medulla. Kidney Int. 1992;42:1372–1380. doi: 10.1038/ki.1992.429. [DOI] [PubMed] [Google Scholar]
- Zou A.P., Wu F., Cowley A.W., Jr. Protective effect of angiotensin II-induced increase in nitric oxide in the renal medullary circulation. Hypertension. 1998;31:271–276. doi: 10.1161/01.hyp.31.1.271. [DOI] [PubMed] [Google Scholar]